
Abstract
Prostate cancer is the second most common disease in men and the sixth leading cause of death from cancer globally, with 20 million men expected to be affected by 2024 thus considered as chronic illness which requires immediate attention. As an androgen-dependent illness that relies on the androgen receptor for development and progression, inhibition of the androgen receptor can lead to a therapeutic solution, hence serving as a vital therapeutic target. This study focused on the computational analysis of the inhibitory potentials of Vitis vinifera, a reported plant with anti-cancer properties, against androgen receptor employing molecular docking, ADMET studies, Binding energy study, pharmacophore modeling, and molecular dynamics simulation approaches. After the investigation, it was determined that 5 compounds: cis-piceid, cis-astrigin, gallocatechin, phlorizin, and trans-polydatin, might be possible androgen receptor inhibitors since they had higher docking scores and ADMET qualities than compared standards, with cis-piceid being the best-predicted inhibitor.
Background
Prostate cancer (PCa) is the second most frequent malignancy in men and the sixth greatest cause of death from cancer worldwide. 1 According to GLOBOCAN 2018, 1 276 106 new cases of prostate cancer were recorded globally in 2018, with a greater frequency in industrialized nations, resulting in 358 989 fatalities. GLOBOCAN (2018) also reported that by 2024, approximately 20 million males would have PCa. 1 This poses a serious concern and considered a chronic illness that needs a rapid pharmacological solution. 2
According to research, prostate cancer begins as an androgen-dependent disease that relies on the androgen receptor (AR) for growth and progression, 3 thus, through the use of AR antagonists or combined androgen blockade therapy, AR has become a potential and appealing target for PCa therapy over the years.4,5 Several major mutation processes in AR have been discovered within the last 2 decades. According to laboratory observations, AR mutations have changed the growth-inhibitory impact of anti-androgens into a growth-promoting effect in the castration-resistant type. 6 Inhibiting the synthetic pathway and the participating enzymes of androgen synthesis and developing novel androgen receptor antagonists are 2 popular methods of disrupting the signaling pathway that propagates prostate cancer. However, the latter has been poorly exploited. 7
The use of phytomedicine has been popular amongst the general population in regions worldwide. This represents remedies from plant secondary metabolites, which possess significant pharmacological effects. The anti-cancer potential of Vitis vinifera targeting the Androgen receptor was evaluated in this study using computational approaches employing molecular docking, binding energy calculation, ADMET study, pharmacophore modeling, and molecular dynamics simulation, respectively. The secondary metabolites with specific potential inhibitory activity against prostate cancer propagation were reported. V. vinifera has been investigated for its anti-cancer potential for a long time, and different studies have reported its anti-cancer potential.8,9
Methods
Virtual Screening and Docking Platform
Using Maestro 11.1, 10 in silico screening was performed to predict compounds with the strongest inhibitory potential, using an online generated library of 84 compounds (Supplemental Material) that have been characterized with V. vinifera and docked them to the active region of AR.
The docking of the molecules was carried out following standard procedures.
Ligand Library and Target Generation and Preparation
Secondary metabolites recovered from V. vinifera were retrieved in Standard Format (SDF) from the Pubchem web database. 11 The mined structures were transformed into a three-dimensional structure using the ligprep tool 12 by ionizing at pH (7.2 ± 0.2) and eliminating salt with Epik.13,14 The OPLS3 force field 15 was employed for ionization and tautomeric state formation. The X-ray crystalline structure of AR ligand binding domain coupled with an inhibitor (PDB ID: 2AXA) 16 was downloaded from Protein Data Bank. 17 The protein was prepared using the Protein preparation wizard software of Maestro, Schrodinger Suite. The protein was modified by optimizing the H-bond assignment and minimizing it with the OPLS3e force field.
Receptor Grid Generation
The receptor grid displays the receptor region where the ligand and protein combine. The designed protein grid on the binding domain was created using the Receptor Grid Generation tool (Glide Grid). The binding site was discovered by selecting the co-crystallized ligand at the active site of 2AXA. A cubic grid box containing all amino acid residues at the active site was automatically created using the coordinates X = −0.360, Y = 14.401, and Z = 47.310, respectively.
Docking
Docking was accomplished using the Glide tool on maestro 11.1 (Schrödinger Release 2017). 18 The derived crystal structure of AR was used to screen the synthesized compounds virtually to determine compounds with the lowest docking score using Standard Precision (SP) and Extra Precision (XP) docking algorithms. The docking research was conducted with the protein considered a rigid body and the rotatable bonds of the ligand set to be free. The co-crystallized ligand with the target (Supplemental Material) and bicalutamide an androgen receptor inhibitor was also docked using the same procedures to serve as the standard for a comparative study.
ADMET/Tox Screening
The swissADME (http://www.swissadme.ch) and Pro-Tox II online servers (https://tox-new.charite.de/protoII) online servers were used to determine the lead compounds’ pharmacokinetics, drug-likeness, and toxicity.
Pharmacophore Modeling
The receptor-ligand complexes of the lead compounds were investigated, and a hypothesis (E-pharmacophore) was created using the phase interface of the Schrodinger suite to illustrate the major properties that make significant contributions to the lead ligands’ characteristic binding to the active sites of the target proteins.
Binding Energy Calculations
For the relative binding energy calculations, the one-average molecular mechanics generalized Born surface area (MM/GBSA)19,20 methods developed in the MOLAICAL code 21 were employed, in which the ligand (L) combines with the protein receptor (R) to generate the complex (RL).
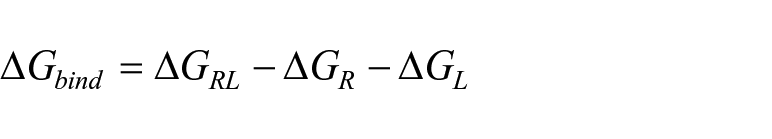
Which can be expressed by the contribution of various interactions,

Where the changes in the gas phase molecular mechanics
Molecular Dynamics (MD) Simulations
The NAMD 2.13 package22,23 and the CHARMM36 24 force field were used for all simulations. The TIP3P explicit solvation approach was utilized, and the periodic boundary conditions had dimensions of 75.33, 96.17, and 87.57 (x, y, z in Å). The system was then neutralized with 3 sodium (Na+) ions. Minimization, annealing, equilibration, and manufacturing were part of the MD protocols. In the minimization and annealing simulations, the protein backbone atoms were confined, while the Ca atoms of the protein were impeded in the 1 ns equilibration simulation. Also, no atoms were constrained in the 100 ns MD manufacturing simulation. The isothermal–isobaric (NPT) ensemble and a 2 ns simulation integration time were used for all MD simulations. During the 100 ns of MD generation, the pressure was held at 1 atm by employing the Nose’–Hoover Langevin piston barostat25,26 with a Langevin piston decay of 0.2 ps and a period of 0.4 ps. The Langevin thermostat 27 was used to regulate the temperature to 298.15 K. Short-range nonbonded interactions with a pair list distance of 12 Å were given a distance cutoff of 10.0 Å.
In contrast, Lennard Jones interactions were trimmed smoothly at 8.0 Å. The particle-mesh Ewald (PME) method28,29 was employed to treat long-range electrostatic interactions, with a grid spacing of 1.0 Å utilized for all simulation cells. The SHAKE technique was used to limit all covalent bonds that involved hydrogen atoms. 30
Results and Discussions
In the light of this, the phyto-compounds in V. vinifera were docked against the androgen receptor binding domain guided by the generated grid box and automated by glide script. Five compounds, namely cis-piceid, cis-astrigin, gallocatechin, phlorizin, and trans-polydatin, had docking scores of −14.565, −13.734, 13.350, −13.030, and 12.956 respectively (Figure 1). Androgen receptor signaling has a pivotal function in the growth and initiation of prostate cancer tumors, and androgens initiate the survival of prostate cancer. 31 Developing an antagonist that suitably binds to the receptor’s ligand-binding domain is a proven therapeutic target in treating prostate cancer.

The docking scores and binding energy of the test compounds.
Interestingly, all the compounds had higher docking scores than the co-crystallized ligand and bicalutamide (Figure 1). The MM/GBSA method is a much more precise method of calculating the free binding energies (dG) of protein-ligand complexes. 32 It’s one of the most promising methods for improving virtual screening outcomes. Just like docking scores, a negative dG value indicates that the complexes formed in the binding pocket of the target were stable. 33 All lead compounds show a negative dG value. However, while the co-crystalized ligand show more binding energy than all other compounds, cis-piecid also show a higher binding energy than the androgen receptor inhibitor as shown in Figure 1.
Different forces contribute to binding a small molecular weight compound to protein targets. These interactions include hydrogen bonding, salt bridges, pi stacking, etc. The major interactions observed in the binding of the lead compounds to the Androgen receptor are hydrogen bonding and pi-stacking. Hydrogen bonding is one of the most vital and specific interactions in biological systems and plays a major role in the recognition, binding, and affinity of a ligand to the complementary protein target.
Cis-piceid, while interacting with the binding site of 2AXA, formed 5 hydrogen bonds (LEU 704, GLN 711, ASN 705, GLN 738, MET 742) and pi-stacking with the aromatic amino acid residue, namely Tryptophan (TRP 741) (Figure 2). In the same vein, Cis-Astrigin formed a single hydrogen bond with GLN 711 and MET 742 but 2 with GLN 738. Similarly, Gallocatechin had 3 amino acid interactions with the substrate-binding site of the receptor (GLN 711 and 2 others with ASN 705) (Figure 2). Phlorizin and Trans-polydatin had 3 and 4 hydrogen bonds interacting with the receptor’s binding site, and both formed a pi stacking interaction with TRP741. Hydrogen bond and pi stacking are peculiar to all interactions (Figure 2).
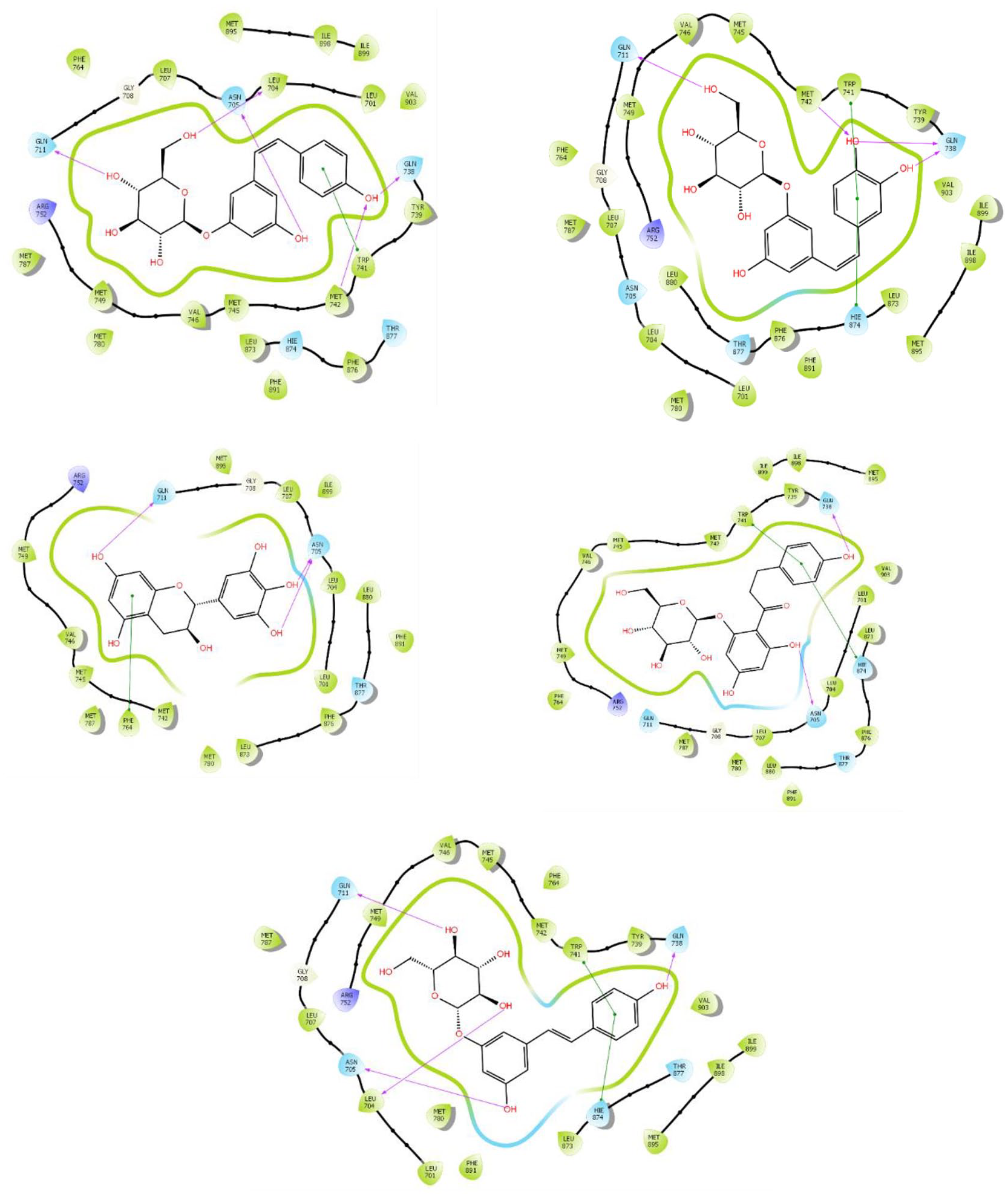
Interactions of the compounds with the ligand-binding domain of androgen receptor.
The receptor-based pharmacophore models of the compounds showed that aromatic rings, hydrogen bond acceptors, and hydrogen bond donors are the fundamental properties that contributed to the binding of the ligands to the ligand-binding domain of the androgen receptor (Figure 3). In accordance with the predictive model, potential receptor antagonists require these peculiar physicochemical properties to enable bind effectively to the ligand-binding domain.

Receptor-based pharmcophore models of the top-scoring compounds.
Some physicochemical properties of the test compounds, including molar refractivity, number of hydrogen bond acceptors, number of hydrogen bond donors, and topological surface area, are presented (Table 1). The number of hydrogen bond donors and acceptors present in a compound is one of the physicochemical properties that contribute substantially to the accurate prediction of its bioavailability as a drug candidate. Also, it has been reported that the increase in enthalpy during the formation of internal hydrogen bond facilitates the movement of compounds across cell membranes despite the formation being entropically unfavorable. 34
Physicochemical Properties of the Top-Scoring Compounds.

As per SwissADME pharmacokinetic profiling (Table 2), all the test compounds are considered soluble. The Silicos-IT model of water solubility showed the solubility level in the range of −1.61 to – 1.02, phlorizin being the least soluble and cis-Astrigin, the most. Water solubility is significant for drug candidates, and poor water solubility may result in inefficient absorption even though the rate of permeation of the drug is considered high. 35
SWISSADME Predicted Lipophilicity (Log P) and Water Solubility (Log Sw).

The lipophilicity of the test compounds was predicted using the consensus log P-value, which is in the mean of 5 different predictive models. The lipophilicity of a compound is experimentally the partition coefficient of n-octanol to water. Trans-polydatin was predicted to be the most lipophilic with a Log P value of .64 and has a better chance of being absorbed than the other compounds. However, the gastrointestinal absorption rate must also be considered to accurately determine how efficiently a compound is absorbed into the systemic circulation. Low lipid solubility levels equate to a high water solubility level, an integral factor for drug-likeness. However, the ability of a drug to reach its therapeutic target may be compromised if the lipophilicity is too low. Contrarily, compounds with high water solubility return low lipophilicity and poorly permeated membranes and substantially reduce the absorption mechanism. Phlorizin was the least lipophilic, with a log P value of .06.
The drug-likeness and bioavailability profiling of the lead (Table 3) compounds returned a favorable bioavailability score of 0.55. The drug-likeness predictions using 3 rule-based filters, namely Lipinski, Ghose, and Verber filters, showed positive results. In detail, all compounds violated just one of the filters of the Lipinski rule of 5 and none in the Ghose filter. However, one violation each was recorded for Cis-Astrigin and Phlorizin in Verber’s rule-based filter. Computer-aided drug-likeness prediction is essential in the preliminary stages of drug development and is also a cost-effective approach. It predicts the extent to which a small molecular weight compound is drug-like.
Drug-Likeness and Bioavailability.

A structural orientation that aids absorbance in the gastrointestinal tract is an essential pharmacokinetic descriptor that contributes to the likelihood of a compound being an oral drug candidate. Based on the SWISSAdme predicted pharmacokinetic profile (Table 4), Cis-piceid, Gallocatechin, and trans-polydatin have the structure architecture that gives them a high permeation into the systemic circulation. In addition, the lipophilicity of compounds also contributes substantially to this property. Therefore, the high GI absorption of these compounds (Cis-Piceid, gallocatechin, and trans-polydatin) could be linked to the relatively high lipophilic property. Phlorizin was the least lipophilic and unsurprisingly had a low GI absorption.
Predicted Pharmacokinetic Properties of Test Compounds.

Similarly, Cis-Astrigin was found to have the same absorption profile. Furthermore, none of the lead compounds can permeate the Blood-brain barrier. This is considered a suitable property for a drug candidate whose target site is not the brain primarily because permeating the brain could cause adverse drug reactions.
Cis-Astrigin and gallocatechin are predicted to be non-substrate of Permeability glycoprotein while the transporter can act upon the rest. P-glycoprotein belongs to the family of ATP-binding cassette transporters, which is mainly expressed in organs that function in excretion and provide a barrier. The organs include but are not limited to the liver, kidney, and blood-brain barrier. 36 Consequentially, P-glycoprotein substrates are effluxed out of the cell before bio-accumulating up to a significant or therapeutic level. This leaves cis-Astrigin and gallocatechin with an increased probability of accumulation up to significant concentration in the cell.
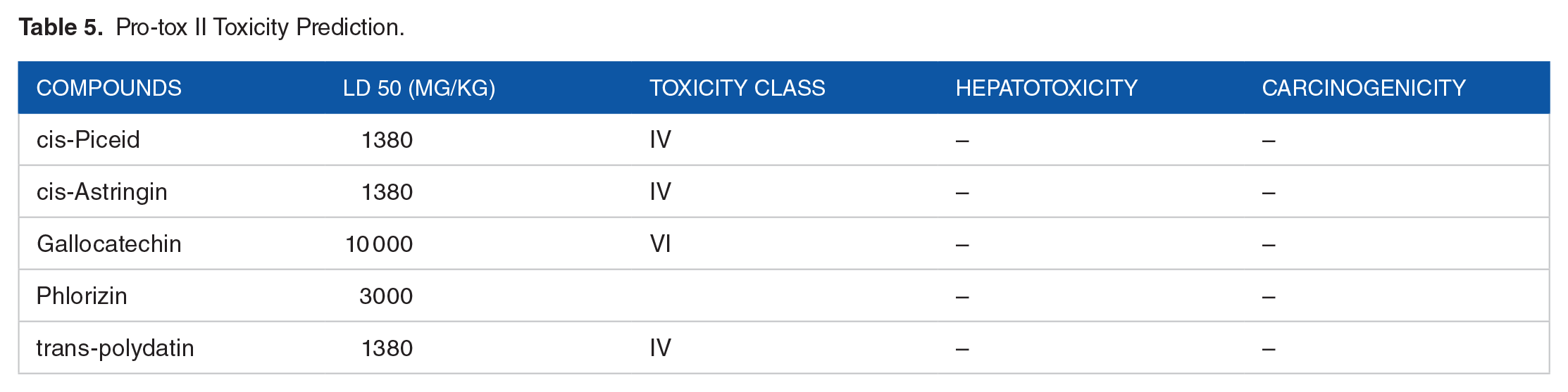
Interestingly, all compounds are non-substrates of the Cytochrome p450 isoforms analyzed. Cytochrome P450 enzymes are phase 1 reaction specialists; inhibiting these enzymes may induce a drug-drug reaction and ultimately lead to toxic accumulation of the drug or its metabolites due to inefficient processing and clearance. 37 In the same vein, none of the compounds are predicted to have hepatotoxic and carcinogenic activity as per Pro-tox II (Table 5).
Pro-tox II Toxicity Prediction.
The lead molecule was subjected to 100 ns MD simulation to study its interactions at the active site of the target. Root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), Radius of Gyration (ROG), and Hydrogen Bonding and ligand contact frequency with the amino acid residues at the binding site of the protein were evaluated (Figure 4)

MD simulation plot for cis-piceid/AR complex for 100 ns. (a) cis-piceid/AR complex RMSD plot. (b) cis-piceid/AR complex RMSF plot. (c) cis-piceid/AR complex radius of gyration plot. (d) cis-piceid/AR complex Contact frequency plot. (e) cis-piceid contact frequency with the amino acid residues at the active site of AR.
The RMSD measures the average distance between the atoms of the superimposed proteins. 38 The trajectory’s bounds of the complex RMSD over 100 ns were computed and shown in Figure 4A. Stability was observed in Ligand’s RMSD plot generated with a resolution less than 2.0 Å throughout the simulation. Stability is also seen throughout the simulation in the protein and the protein-ligand complex with a crystallographic resolution of less than 1.5 and 2.5 Å, respectively.
For the 100 ns simulation time, RMSF was used to track local changes along with the AR amino acid residues (Figure 4B). The alpha helices and beta strands of the docked complex oscillate between 0.8 and 1.6 Å. All simulated systems’ loop sections showed high swings of up to 2.2 Å.
The radius of gyration was also utilized in this study to estimate the structure’s compaction level. The mass-weighted root mean square distance of a cluster of atoms from their common center of mass is denoted as the radius of gyration. 39 Figure 4C shows the plot of the radius of gyration of AR for 100 ns simulation time. High fluctuation in the radius of the gyration plot shows less compatibility and might result in less stability of the protein. Figure 4C indicates low fluctuation between the protein with a resolution of 17.8 and less than 18.2 Å. This signifies that the protein compaction level is very tight with indicate stability of the protein during the simulation period.
In developing a ligand-protein complex, non-covalent interactions such as hydrogen bonds are crucial. Figure 4D shows that three hydrogen bonds were formed at the beginning of the simulation before dropping to 2 at around 2 ns. The hydrogen bonds fluctuated rapidly between 2 and 1 for about 35 ns before they dropped and fluctuated between 0 and 1 with few occurrences of 2 hydrogen bonds for the rest of the simulation time
It can be seen that ligand has contact with LEU 704, GLY 708, TRP 741, MET 742, MET 745, HSD 874, and THR 877 throughout the 100 ns simulation time, while other residues, which include ASN 705, LEU 707, GLN 711, GLN 738, TYR 739, VAL 746, MET 749, PHE 764, MET 787, LEU 874, LEU 880, MET 895, ILE 898, ILE 899, and VAL 903 show contact frequencies ranging from 20% - 90% for the 100 ns simulation time as shown in Figure 4E.
Conclusions
This study features a computational approach to study the inhibitory potentials of V. vinifera against AR. It was discovered that 5 compounds: cis-piceid, cis-astrigin, gallocatechin, phlorizin, and trans-polydatin, could be potential inhibitors of AR as they possess better docking scores and ADMET properties when compared to a standard compound, leaving cis-piceid as the best-predicted inhibitor.
The best compound was subjected to MD simulation, and good stability and interaction with the amino acid residues at the active site of the target were observed.
This study suggests that V.vinifera might be a good plant source for drug molecules that can treat prostate cancer by inhibiting the Androgen receptor. However, further validations using in-vivo and in-vitro analysis are recommended.
Supplemental Material
sj-docx-1-cix-10.1177_11769351221118556 – Supplemental material for Molecular Modeling Studies of Natural Inhibitors of Androgen Signaling in Prostate Cancer
Supplemental material, sj-docx-1-cix-10.1177_11769351221118556 for Molecular Modeling Studies of Natural Inhibitors of Androgen Signaling in Prostate Cancer by Samuel O Olubode, Mutolib O Bankole, Precious A Akinnusi, Olayinka S Adanlawo, Kehinde I Ojubola, Daniel O Nwankwo, Onome E Edjebah, Ayomide O Adebesin and Abigail O Ayodele in Cancer Informatics
Footnotes
Acknowledgements
Not applicable.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author contributions
SOO and PAA conceptualized the study and wrote the original draft of the article. MOB supervised and validated the experiment. OSA, KIO, DON, and OEE performed the experiments. AOA and AOA analyzed the result data. All authors read and approved the final manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.