
Abstract
The quorum-sensing (QS) machinery in disease-causing microorganisms is critical in developing antibiotic resistance. In Pseudomonas aeruginosa, QS is involved in biofilm formation, virulence factors production, and general tolerance to antimicrobials. Owing to the major role QS plays, interference in the process is probably a facile route to overcome antimicrobial resistance. Some furanone-derived compounds from marine sources have shown promising anti-QS activity. However, their protein targets and potential mechanisms of action have not been explored. To elucidate their potential protein targets in this study, marine metabolites with furanone backbones similar to their cognitive autoinducers (AIs) were screened against various QS receptors (LasR, RhlR, and PqsR) using molecular docking and molecular dynamics (MD) simulation techniques. The order by which the compounds bind to the receptors follows LasR > RhlR > PqsR. Compounds exhibited remarkable stability against LasR and RhlR, likely because the AIs of these receptors are structural analogs of furanones. Furanones with shorter alkyl side chains bound strongly against RhlR. The presence of halogens improved binding against various receptors. PqsR, with its hydrophobic-binding site and structurally different AIs, showed weaker binding. This study provides a molecular basis for the design of potent antagonists against QS receptors using marine-derived furanones.
Furanones attenuate the binding of autoinducers and interfere with the expression, and hence, the production of virulence factors.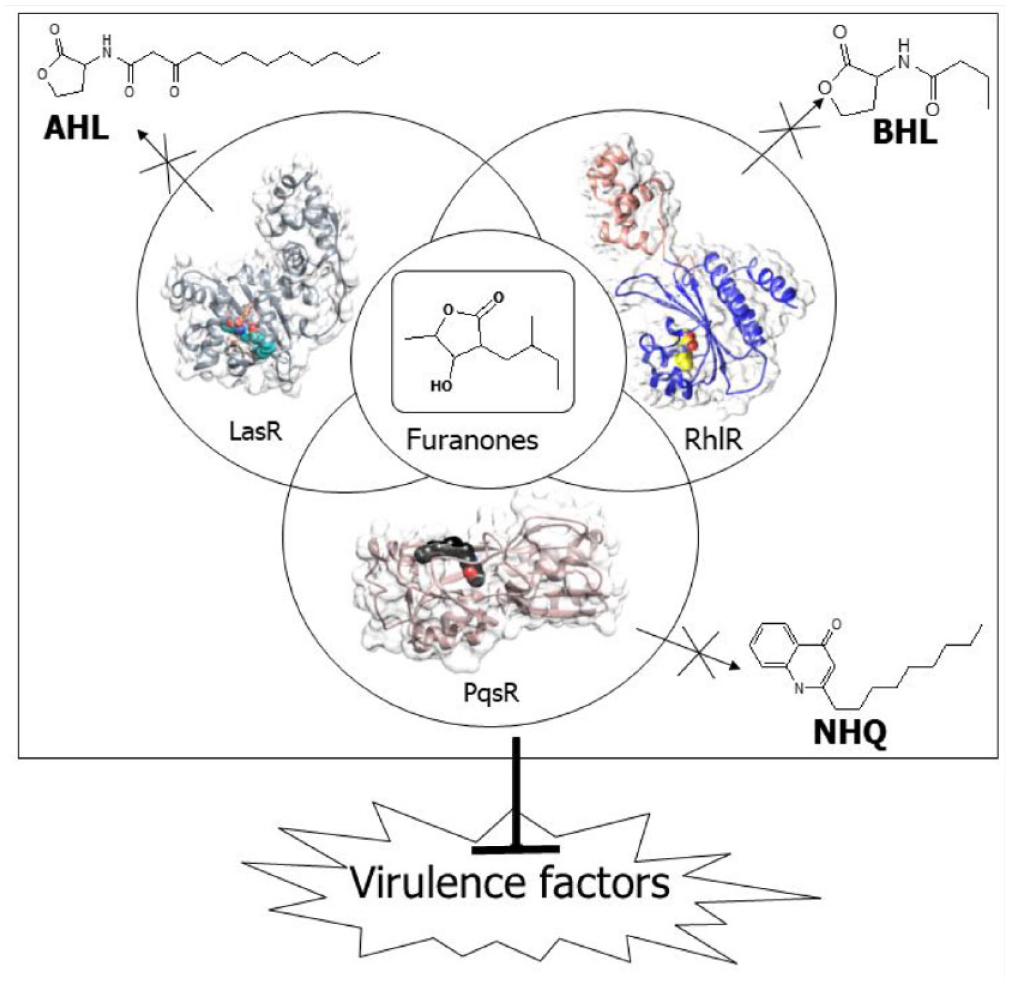
Keywords
Introduction
The global emergence of multidrug-resistant bacteria and the paucity of new clinically effective antibiotics have led to an interest in developing new agents that can control infections by attenuating bacterial virulence rather than growth. 1 Several cases of antimicrobial resistance (AMR) have been reported in disease-causing microorganisms such as Escherichia coli, Staphylococcus aureus, Acinetobacter baumannii, Klebsiella pneumoniae, and Streptococcus pneumoniae, 2 which have resulted in high health costs and fatalities. Antimicrobial resistance has increased rapidly due to prolonged and inappropriate use of antibiotics, evolution, horizontal gene transfer, and innate processes.
Pseudomonas aeruginosa is a ubiquitous gram-negative bacterium that can become opportunistic, particularly in individuals with cystic fibrosis (CF) and compromised immune systems. 3 It is well known for causing nosocomial infections. The gradual shift from acute infection to chronic disease is facilitated by virulence factors such as elastase, rhamnolipids, lipopolysaccharide (LPS), and alginate. One of the key resistance mechanisms observed in P. aeruginosa is attributed to its quorum-sensing (QS) machinery. Quorum sensing is a major regulator of pathogenicity factors in P. aeruginosa, giving a selective advantage to this pathogen over the host immune system by coordinating the expression of several virulence genes.3,4 Quorum sensing is crucial in regulating bacterial genes that help P aeruginosa adapt to harsh environmental conditions. 5 It coordinates activities such as biofilm formation, virulence, antibiotic production, plasmid transfer, pigmentation, and exopolysaccharide (EPS) production. 6
Targeting QS machinery and inhibiting the production of virulence factors provides druggable targets in the search for potential leads to fight against AMR. This approach maintains the potency of antibiotics without tempering bacterial survival and growth, reducing selective pressure on pathogens.7-9 Quorum-sensing inhibitors (QSIs) do not inhibit bacterial growth, but they interfere with virulence factor production, reducing selective pressure on bacterial resistance development to the treatment.10-12
In P aeruginosa, QS exists in a hierarchy consisting of the las, rhl, pqs, and iqs systems that are interconnected.8,13 The synthases (LasI, RhlI, PqsA-D) produce QS signaling molecules—N-acyl homoserine lactones (AHLs), N-butyryl homoserine lactone (BHL), and Pseudomonas quinolone signaling molecule (PQS), respectively. The receptors (LasR, RhlR, and PqsR) act as response regulators, binding to QS signaling molecules and coordinating downstream responses (Figure 1). 7 The las system is at the top of the hierarchy. The transcriptional regulator, LasR, in the las system, together with its cognate autoinducer (AI) 3-oxo-C12-HSL, induces the expression of the rhl, pqs, and iqs systems in a cell density-dependent manner. 14 LasR has 2 binding domains: the N-terminal ligand-binding domain (LBD) and the C-terminal DNA-binding domain. LasR dimerizes when AHL binds to it. This allows the dimer complex to attach to the DNA promoter to stimulate gene transcription. The binding of AHL is stabilized by hydrophobic interactions that shield the ligand-binding pocket from the bulk solvent. 15 The Rhl system comprises of 2 components: RhlI, which produces BHL, and RhlR, which is the regulatory protein that acts as the cognate receptor of BHL. 16 The expression of RhlR restores the production of several exoproducts such as elastase, pyocyanin, hemolysin, and rhamnolipids. 17 The RhlR active site contains a hydrophobic pocket, an H-bond acceptor, and an H-bond donor zones. 18 The quinolone-based pqs system regulates the production of pyocyanin, which is highly cytotoxic to mammalian cells, and extracellular DNA release for biofilm formation.1,7,19 The crystal structure of PqsR shows that the protein possesses a highly conserved hydrophobic pocket, and the bicyclic ring of the 2-nonyl-4-hydroxyquinoline (NHQ) is enclosed in pocket B by hydrophobic contacts.1,19

Crystal structures of AHL-LasR, NHQ-PqsR, and BHL-RhlR complexes. Ligands and receptor surfaces have been color-coded. Receptors—LasR (gray), PqsR (tan), and RhlR (sienna and navy blue). Ligands—AHL (cyan), NHQ (black), and BHL (yellow).
Microorganisms in the marine environment develop signal interference mechanisms to adapt to different environments and compete for nutrients and ecological niches.20,21 Owing to marine biodiversity, marine species have been identified as a source of new chemical compounds for various biological functions. Recently, QSIs have been identified in some microbial species, such as marine bacteria, actinomycetes, and fungi. 22 For instance, 2 phenethylamide metabolites isolated from a Halobacillus salinus strain obtained from a seagrass sample inhibited bioluminescence production by Vibrio harveyi in co-cultivation experiments. 23 Similarly, malyngolide isolated from the cyanobacterium, Lyngbya majuscula, has been reported to interfere with the QS circuitry in Chromobacterium violaceum CV017 at concentrations ranging from 3.57 to 57 µM. 24 Some furanone compounds (Figure 2 and Supplemental Table S1) that are similar in structure to AIs have also been identified to attenuate the QS mechanism in bacterial cell-to-cell communication in the marine environment.25-27

Antiquorum-sensing metabolites from marine sources with furanone backbone.
The molecular bases for the QS interfering actions of these compounds are largely underexplored. As furanones are structural analogs of the AIs, it has been hypothesized that these compounds target any of the QS receptors in eliciting their mechanism of action. This study aims to provide a molecular basis for the antiQS activities of marine-derived furanones using molecular docking and molecular dynamic simulation approaches. The role of substituents on the furanone backbone and how they affect selective binding to their respective receptors were also explored. From the results obtained, furanones with short-chain alkyl substitution allowed compounds to fit perfectly within the binding pocket of RhlR and may compete with BHL. The presence of halogens improved the binding of compounds against the LasR, PqsR and RhlR. Against LasR, compounds were seen to bind at the acyl homoserine lactone (AHL), AHL-binding domain, establishing extensive hydrophilic interactions that may lead to protein inactivation and aggregation. For PqsR, binding of compounds showed little effect on protein dynamics. Compounds bound relatively weaker against PqsR. As the AI of PqsR, NHQ is structurally different from AHL and BHL, that do not allow perfect fit within the PqsR-binding pocket. This study provides a molecular basis for designing potent antagonists against QS receptors using marine-derived furanones.
Methods
Protein selection
The crystal structures of the LasR (PDB ID: 6V7X) and PqsR (PDB ID: 4JVD) receptors of P aeruginosa were obtained from the Protein Data Bank (https://www.rcsb.org). LasR was observed in a complex with an AI, AHL, and an antiactivator, Aqs1, with a resolution of 2.90 Å. PqsR was observed to complex with an agonist, 2-NHQ, with a resolution of 2.95 Å. All missing residues were modeled and refined using the modeler suite in UCSF Chimera. Polar hydrogens and partial charges were added to the protein structure to ensure charge balance on the protein structures.
As the crystal structure of RhlR has not been solved, a homology model was used. Nam and co-workers generated a homology model of RhlR of P aeruginosa from the SWISS model server using SdiA (PDB ID: 4Y15) as a template. 28 The homology model was downloaded from their Supplementary Material. The model was simulated for 200 ns under dynamic conditions to obtain the energy-minimized conformation. A frame from the most stable plot was extracted for further docking study. To identify BHL binding site on the RhlR model, Rex et al 18 performed a sequence alignment of RhlR using LasR as a template. From the sequence alignment, the major active site residues of LasR (Tyr 56, Trp 60, Asp 73, and Ser 129) were mapped onto that of RhlR (Tyr 64, Trp 68, Asp 81, and Ser 135), confirming the binding site of RhlR.
A global docking of the natural substrate, N-BHL, was performed on the protein. To further screen compounds against RhlR, residues Ser 135, Trp 68, and Asp 81 were used as references to map out the binding site of BHL.
Ligand preparation
Compounds were modeled with Spartan ‘14 (Wavefunction, Inc. Irvine, California). Structural optimizations and energy minimization were performed using the density functional theory (DFT) basis set, as described in previous works.15,29 The optimized compounds were saved in their PDB and SDF file formats for further analysis.
Molecular docking
Global and precision docking approaches were employed for molecular docking studies. Initially, global docking was performed on all receptors. The grid dimensions were set to cover the entire receptors and allow the compounds to identify their preferential binding site on the receptors. Docking was performed using the AutoDock vina tool 30 in PyRx. For LasR, PqsR, and RhlR, compounds that bind at the AHL, NHQ, and BHL-binding domains, respectively, were considered for precision docking studies. Before the precision docking of compounds, native ligands of selected receptors were redocked in their respective binding sites to validate the precision docking protocol employed for this study. The redocked pose of the native ligands and their co-crystallized structures were superimposed to determine the extent of the deviation. A root mean square deviation (RMSD) value of <2 Å confirmed that the procedure was valid for subsequent docking runs. Selected compounds were then subjected to precision docking to identify their binding affinity against receptors. For LasR, a grid size of 19.2 Å and grid dimensions of X = −22.9073, Y = −3.38932, and Z = −4.83075 were used. For PqsR, a grid size of 17.8 Å and grid dimensions of X = −33.3486, Y = 57.8889, and Z = 9.39797 were used. The grid dimensions obtained for BHL bound to RhlR were X = 77.7799, Y = 76.5451, Z = 21.3231, and R = 23 Å. The docking scores were compared with the native ligand of the various receptors. Binding conformations predicted from molecular docking were further validated using the molecular dynamic simulation model.
Molecular dynamics simulation study
All-atom molecular dynamics (MD) simulations were performed for 200 ns on selected complexes. GROMACS v.2018.6 was used for the MD simulations on the Lengau cluster, provided by the Center for High-Performance Computing in Cape Town, South Africa. Ligand topologies were generated using the CGENFF web server, 31 whereas receptor topologies were generated using the CHARM36 all-atom force field. The systems were solvated in a dodecahedron boundary box of 1 nm using the TIP3P water model. Sodium and chloride ions were added to the complexes to neutralize the system. Energy minimization of the complexes was performed using the steepest descent algorithm for 50 000 steps, followed by Number of particles, Pressure and Temperature (NPT) and Number of particles, Volume and Temperature (NVT) equilibration at 300 K and 1 bar pressure for 100 ps. The equilibrated system was further simulated for 200 ns in a production run. The Particle Mesh Ewald (PME) approach was used to calculate long-range electrostatic interactions, and cut-offs for the Coulomb and van der Waals interactions were set to 1.2 nm. The production run was defined by a time step of 2 fs, with coordinate trajectories written every 10 ps. The simulations were performed with 3-dimensional (3D) periodic boundary conditions (PBCs) applied in all instances.32,33
Stability and trajectory analysis
Output files from the MD production were examined to analyze the stability of the complexes. Stability was assessed using RMSD, RMSF, and radius of gyration (RG) values. Visual Molecular Dynamics (VMD) 1.9.345 was used to visualize systems, using the structural (.gro) and pbc compressed trajectory (.xtc) files obtained for all complexes after MD production. 34
Binding free energy calculations
The g_mmpbsa tool in GROMACS 35 was employed to determine the binding free energy of the complexes. The last 50 ns of the most stable complexes were analyzed to calculate the average binding energies and the individual energetic contributions to the overall binding energy. The binding energy was determined by taking into account several energetic terms such as bond angle, torsional energies, van der Waals and electrostatic interactions, desolvation of various species, including polar and nonpolar solvation energy using the implicit solvation model, and the configurational entropy-related to the complex formation. The polar (Gpol), apolar (Gapol), and molecular mechanical potential energy (EMM) were included in the computed binding energies. Python scripts (MmPbSaDecomp.py and MmPSaStat.py) were used for these computations.
Hydrogen bond analysis
The calculation of hydrogen bonding occupancies and total number of hydrogen bonding interactions during the simulation involved determining the hydrogen bonds with bond angles of 180° and bond lengths of 3 Å.
In silico ADME analysis
The Swiss ADME web server (http://www.swissadme.ch) and ADMET lab 2.0 webserver (https://admetmesh.scbdd.com) were used to calculate the drug-likeness of the molecules. The physicochemical parameters, including molecular weight, number of hydrogen bond acceptors (HBAs), hydrogen bond donors (HBDs), number of rotatable bonds, lipophilicity, solubility, and topological surface area, were computed. Lipinski’s rule of 5 was applied to estimate the oral bioavailability of the compounds. Caco-2-permeability and human intestinal absorptions (HIAs) were used to determine the absorption parameters. The excretion properties, such as clearance level and half-life, were also estimated. The SMILES of the compounds were generated from PubChem (https://pubchem.ncbi.nlm.nih.gov), and the ADME web server was used to determine the pharmacokinetic properties.
Results
In this study, we focused on 11 furanone derivatives that were isolated from marine organisms. These compounds were assigned identification numbers (CMP) for convenience. In general, compounds were categorized into 2 groups (halogenated and nonhalogenated). However, they were further grouped into 3 main categories. Among them, the first compound (CMP1) contained halogen atoms, whereas CMP2 to CMP4 lacked a double bond in the furanone ring and had shorter alkyl chain substitutions. These 3 compounds (CMP2-4) did not contain halogen atoms. On the contrary, CMP5 to CMP9 did not have halogen atoms but exhibited longer alkyl chain substitutions on the furanone head group. As compounds were generally categorized into halogenated and nonhalogenated derivatives, furanone (CMP10) and bromofuranone (CMP11) were included in this study. This was done to examine the importance of substitution in the ring system and how it influences receptor binding. Previous studies have reported anti-QS activities for CMP 1 to CMP9 (Figure 2). To investigate the preferential binding receptors of compounds, we employed global and precision docking analyses.
Molecular docking
We investigated the binding interactions between native ligands and their respective receptors from the crystal structures. In the case of AHL bound to LasR, hydrogen bonds were observed between the ligand and Trp60, Ser129, and Asp73, whereas hydrophobic interactions occurred with Ile52, Leu40, Ala50, Val76, Ala127, Cys79, and Leu125 (Supplemental Figure S1a). Similarly, for NHQ against PqsR, hydrophobic interactions were observed with Ala 168, Ile 149, Leu 207, Ile 263, Ala 102, Ile 236, Val 170, Leu 189, Ile 186, and Tyr 258 (Supplemental Figure S1b). The BHL bound to RhlR showed hydrogen bond interactions with Ser135, Trp68, and Asp81, and hydrophobic interactions with Trp108, Phe101, Trp96, and Ala111 (Supplemental Figure S1c). These residues served as a guide to explore the binding interactions of our selected compounds.
Molecular docking was performed to explore the binding conformations and affinities of the AIs with their respective receptors. In the redocking process, NHQ, AHL, and BHL exhibited binding affinities of −7.4, −8.4, and −6.5 kcal/mol, respectively. On redocking, AHL formed hydrogen bonds with Trp 60 and Tyr 64, interacting hydrophobically with Leu40, Ala50, Val76, and Ala70. Redocked NHQ displayed hydrophobic interactions with Leu208, Leu9, Val211, Ala168, Trp234, Tyr258, Ile236, Ile263, and Val170. When docked with RhlR, BHL formed hydrogen bonds with Ser135, Trp68, and Asp81, while engaging in hydrophobic interactions with Trp108, Phe101, Trp96, and Ala111.
To assess the accuracy of the docking procedure, the crystallographic and redocked poses of the ligands were compared, and their RMSD was calculated. The superimposition resulted in RMSD values of 1.860 Å for AHL and 1.090 Å for NHQ (Supplemental Figure S1). These findings confirmed the reliability of the docking procedure for subsequent studies. The compounds were then docked against LasR, PqsR, and RhlR to determine their preferred binding receptors in the P aeruginosa QS machinery. Initially, global docking was performed, and this was followed by precision docking.
LasR
All 11 compounds were found to bind to the AHL-binding domains of LasR (Figure 3A). Precision docking was further performed on compounds. The binding affinities of the compounds against LasR obtained ranged from −4.8 to −8.0 kcal/mol (Table 1). Of the compounds under investigation, CMP7 recorded the highest binding score, whereas CMP10 recorded the lowest. Generally, the halogenated compound showed higher binding affinity than the nonhalogenated furanones. Overall, the compounds displayed lower binding affinities to LasR compared with AHL. The interactions observed between the compounds and LasR were mostly similar to those observed with AHL. Docking analysis revealed that most compounds interacted with Trp60, Tyr64, and Ser129 through hydrogen bonds. Hydrophobic interactions were observed with Ala 50, Val 76, Ala 127, Leu 40, and Ile 52. Interactions with other residues, such as Asp73, Tyr52, Leu36, and Ala105, were also observed. The CMP7, with the highest binding affinity, formed a hydrogen bond with Thr75 and displayed hydrophobic interactions with Trp88 and Tyr56. The CMP11, which recorded the lowest binding affinity, established hydrophobic interaction with Leu 110 (see Supplemental Figure S4).

Global docking of furanones against quorum-sensing receptors in P aeruginosa. (A) LasR-furanone complexes, (B) PqsR-furanone complexes, (C) RhlR-furanone complexes. All compounds bound at AHL, NHQ, and BHL-binding domains.
Precision docking of selected furanones against LasR.

PqsR
In a similar manner, all 11 compounds investigated in this study were found to bind to the NHQ binding site (Figure 3B). The binding affinities of the compounds against PqsR ranged from −4.1 to −6.5 kcal/mol (Table 2 and Supplemental Figure S5). The CMP7 exhibited the strongest binding affinity, whereas the furanone compound (CMP10) displayed the weakest binding. Most compounds interacted with Gln194 through hydrogen bonds, and hydrophobic interactions were observed with Ile236, Ile263, Leu208, and Ala168. These interactions were also observed with NHQ. The CMP7, with the highest binding affinity, formed a hydrogen bond with Gln194 and displayed hydrophobic interactions with Ile236, Ala168, Leu208, Leu197, Phe221, Thr265, and Ile149. The CMP10, with the lowest binding affinity, formed hydrogen bonds with Ser196, Gln194, and Ile236, and also exhibited a hydrophobic interaction with Leu208. Generally, hydrophobic interactions were observed for most compounds, which also interacted with other residues such as Leu208, Leu197, Phe221, and Gln194.
Precision docking of selected furanones against PqsR.

RhlR
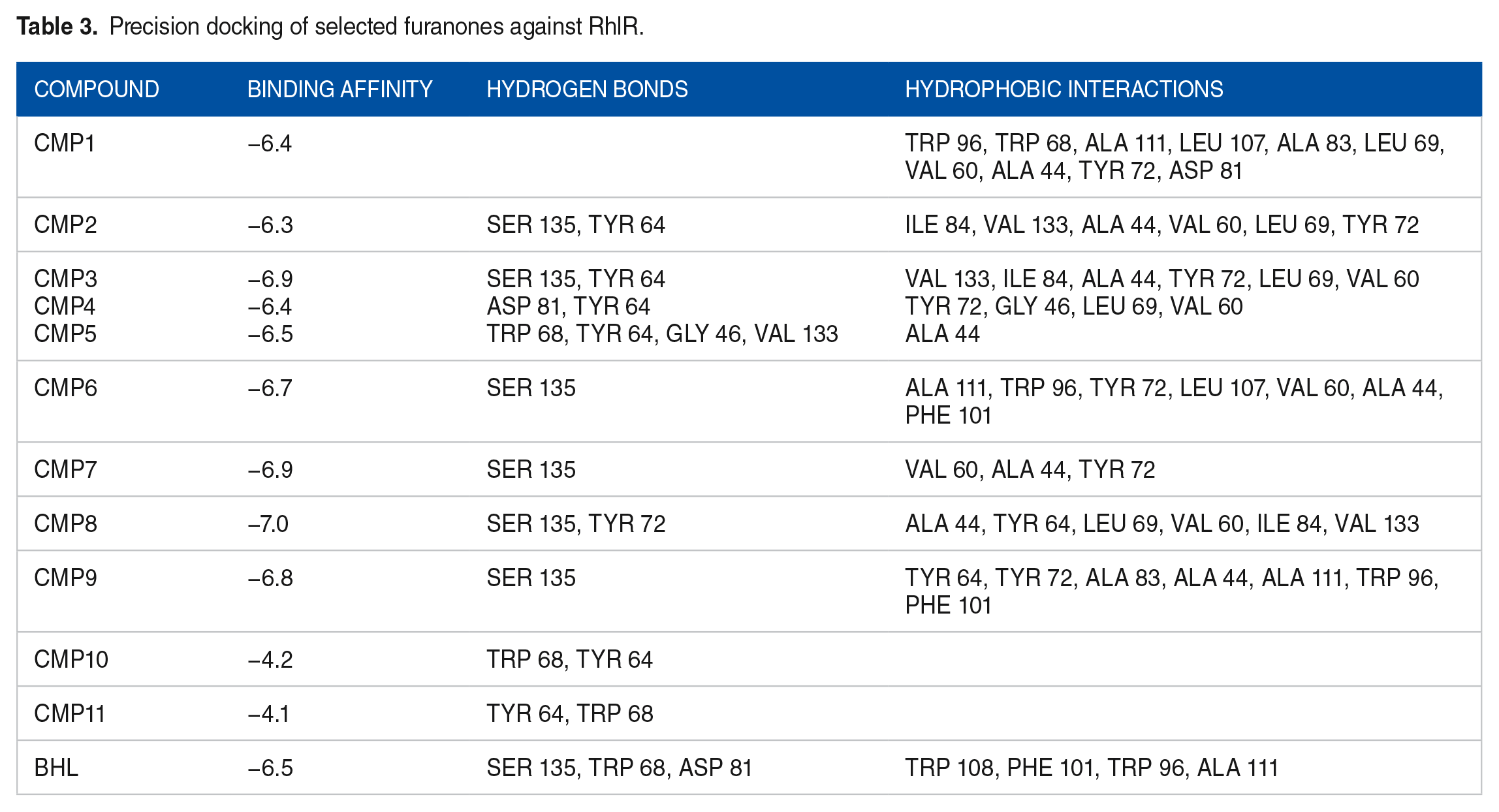
In the case of RhlR, all 11 compounds were found to bind at the BHL binding site (Figure 3C), and after a precision docking study, their binding affinities against RhlR were determined to range from −4.1 to −7.0 kcal/mol (Table 3). Interestingly, the compounds exhibited similar binding affinities to BHL, with the highest binding score observed for CMP8 and the least for CMP11. Hydrogen bonding interactions with Ser 135 were observed for most compounds, whereas bromofuranone (CMP11) exhibited hydrogen bond interactions with Trp 68. Hydrophobic interactions with Tyr64, Trp96, Trp68, Ala111, Asp81, and Phe101 were commonly observed. Visual inspection indicated that most compounds interacted with Trp68, Ser135, and Tyr64 via hydrogen bonds, whereas hydrophobic contacts were observed with Trp96, Ala111, and Phe101. Similar interactions were also observed for BHL (see Figure 4C).
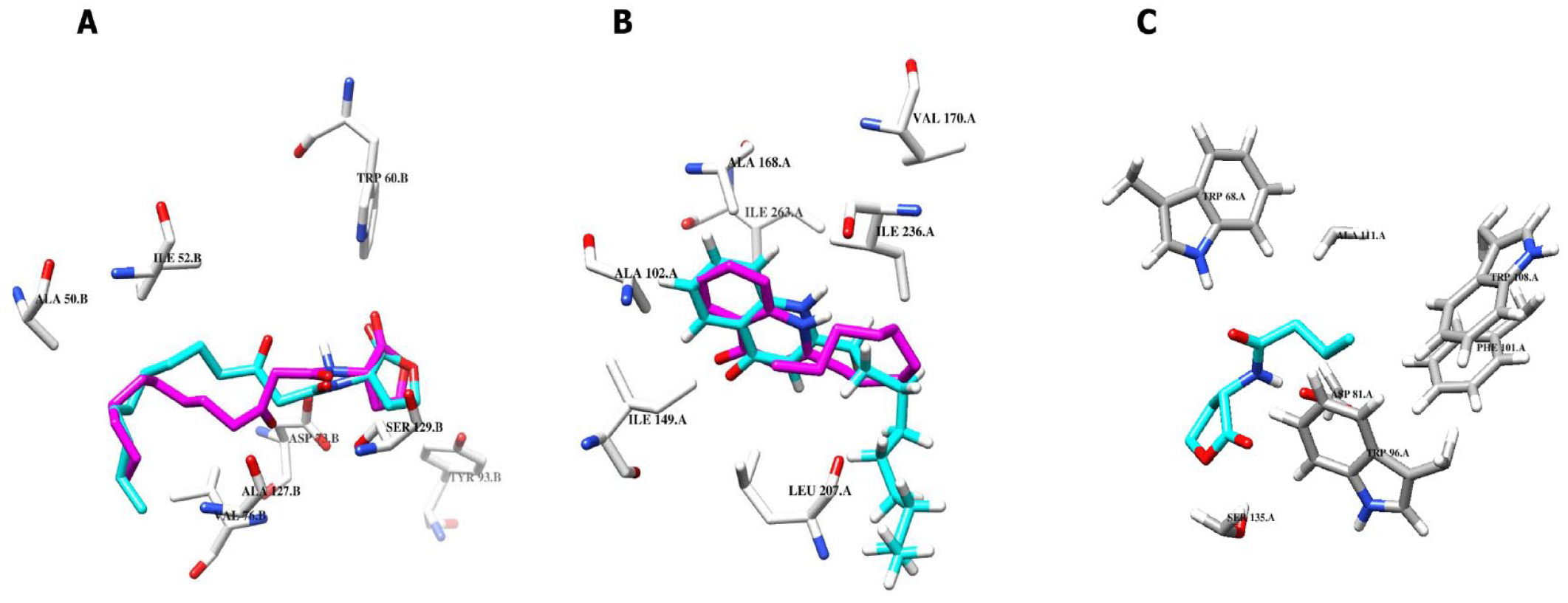
(A) Interactions of AHL with LasR from redocked pose (magenta) and co-crystallized pose (cyan). (B) Interactions of NHQ with PqsR from the redocked pose (magenta) and co-crystallized pose (cyan). (C) Interactions of BHL with RhlR from the redocked pose (cyan). Superimposition of the redocked pose (cyan) and crystallographic pose (green) of AHL and NHQ produced RMSD of 1.860 and 1.090 Å, respectively.
Precision docking of selected furanones against RhlR.
Overall, all compounds preferentially bind at the binding sites of each receptor. However, the binding affinities were higher for LasR, followed by RhlR, and PqsR. Most compounds exhibited good binding to all 3 receptors, but their affinity was stronger for LasR. Furthermore, halogenated furanones consistently demonstrated better binding affinities compared with nonhalogenated furanones.
Molecular dynamics simulation study
A 200-ns simulation was performed, and various parameters such as stability analysis, binding free energies, hydrogen bonding occupancies, residual contributions, and trajectory analysis were analyzed to gain insights into the behavior and interactions of the ligand complexes in detail. This approach allowed for a comprehensive understanding of the stability and dynamic behavior of the ligand complexes during the simulation period.
Stability analysis
To assess the stability of the ligand complexes, various metrics were employed, including RMSD, RMSF, and RG. These analyses provided valuable information about the structural changes, fluctuations, and overall compactness of the ligand complexes throughout the MD simulation.
LasR complexes
A total of 12 complexes, namely CMP1, CMP2, CMP3, CMP4, CMP5, CMP6, CMP7, CMP8, CMP9, CMP14, CMP11, and AHL, were subjected to MD simulations against LasR. The RMSD values of these complexes were calculated to assess the stability of both the ligands and proteins over the course of the simulations. The results indicated that all compounds, except CMP10, exhibited marked stability with RMSD values ranging from approximately 0.1 to 0.5 nm. In contrast, CMP10 was unstable, with an RMSD greater than 2 nm (Figure 5A). Among the compounds, CMP2 to CMP4, which featured shorter alkyl chain substitutions and lacked a double bond within the furanone ring, demonstrated higher stability compared to those with longer alkyl substitutions. To evaluate the protein backbone stability, the RMSD values were computed by comparing the average structures relative to the first frame after post-MD equilibration. The RMSD values of the protein backbones in the complexes varied from approximately 0.25 to 0.5 nm. The AHL and the apoprotein exhibited an RMSD of approximately 0.25 nm (Figure 5B).

Stability analysis of furanones against LasR in P aeruginosa. (A) RMSD of ligands relative to their starting conformation. (B) RMSD of protein backbone atoms. (C) Radius of gyration of apo and bound complexes. (D) RMSF of side-chain residues in apo and protein-ligand complexes.
To assess the variability of side-chain positions in amino acid residues, RMSF analysis was conducted, following the method described by Martínez. 36 Notably, larger fluctuations were observed in residues 40 to 51, 172 to 175, and 180 to 215 (Figure 5D). On visual inspection, it was observed that residues 170 to 180 comprised short loop regions that linked the AHL-binding domain to the DNA-binding domain. Furthermore, the RG values for the bound complexes ranged from approximately 1.9 to 2.05 nm. In contrast, the apoprotein and AHL complex displayed an RG value of approximately 1.97 nm (Figure 5C).
PqsR complexes
The MD simulations were performed on 11 complexes, namely CMP1, CMP2, CMP3, CMP4, CMP5, CMP6, CMP7, CMP8, CMP9, CMP10, CMP11, and NHQ, against PqsR. Among these complexes, CMP6 and CMP10 exhibited instability, as indicated by RMSD values exceeding 2 nm. The NHQ and CMP11 remained stable throughout the simulation, with RMSD values around 1 nm. However, at approximately 80 ns, NHQ and CMP11 underwent a pose flip when visually inspected. This indicates that the docking poses of these compounds readjusted within the binding pocket to assume more stable conformations. On the contrary, CMP1, CMP2, CMP3, CMP4, CMP5, CMP8, CMP9, and CMP11 demonstrated stability, with RMSD values below 1 nm (Figure 6A).

Stability analysis of furanones against PqsR in P aeruginosa. (A) RMSD of ligands relative to their starting conformation. (B) RMSD of protein backbone atoms. (C) Radius of gyration of apo and bound complexes. (D) RMSF of side-chain residues in apo and protein-ligand complexes.
The RMSD values for the apoprotein and NHQ-bound complexes were approximately 0.3 nm. The remaining ligand-bound complexes displayed RMSD values ranging from approximately 0.2 to 0.7 nm (Figure 6B). Fluctuations were observed in specific residue regions, namely residues 150 to 160, 200 to 220, and 250 to 270. Unstable complexes, such as CMP6 and CMP10, exhibited higher fluctuations. In addition, a residue-specific analysis of RMSF for NHQ binding residues was conducted. In the apoprotein, fluctuations increased in the following order: Ala 168, Trp 234, Val 170, Ile 263, Leu 189, Tyr 258, Leu 208, and Val 211 (Figure 6C). The RG values for the apo and NHQ-bound complexes were approximately 1.85 nm. The RG values for the other ligand-bound complexes ranged from approximately 1.80 to 1.95 nm (Figure 6D).
RhlR complexes
Molecular dynamics simulations were conducted on CMP 1 to CMP11 and BHL complexes against RhlR. All complexes except CMP11 exhibited stability, with RMSD values around 1.25 nm. Notably, CMP10 demonstrated even greater stability, with RMSD values of approximately 0.25 nm (Figure 7A). The RMSD values were analyzed to compare the protein backbones with the apoprotein. Among the complexes, CMP3, CMP4, and CMP7 displayed the highest fluctuations, with RMSD ranging from approximately 0.25 to 1.25 nm, compared to the apoprotein with an RMSD of around 0.5 nm. On the contrary, CMP6 and CMP10 exhibited the least fluctuations, with RMSD values below 0.5 nm, compared with the apoprotein (Figure 7B). Significant fluctuations were observed in specific residue regions, namely residues 224 to 227, which represent a short loop connecting 2 helices, residues 130 to 150, which correspond to a beta-sheet and loop structure in the BHL-binding domain, and residues 200 to 225, which are located in the DNA-binding region (Figure 7D). Furthermore, the RMSF values for the BHL binding residues were compared between the bound and unbound complexes. In the apoprotein, fluctuations increased in the following order: Ser135, Trp96, Asp81, Phe101, Trp68, Trp108, and Ala111. The RG values for the bound complexes ranged from approximately 1.93 to 2.4 nm. The apoprotein exhibited an RG value of approximately 2.2 nm, whereas the BHL-bound protein displayed RG values similar to the other bound complexes (Figure 7C).
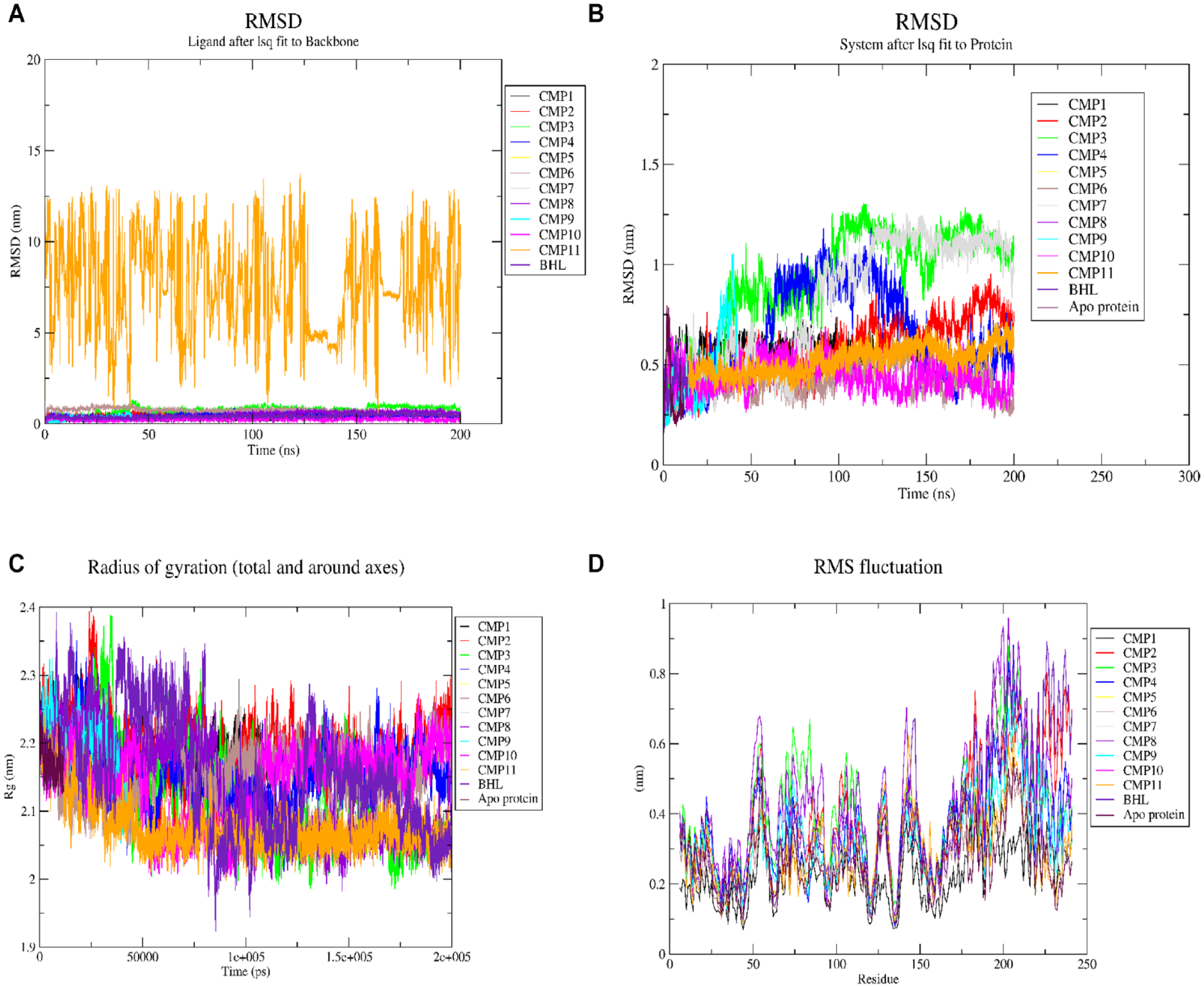
Stability analysis of furanones against RhlR in P aeruginosa. (A) RMSD of ligands relative to their starting conformation. (B) RMSD of protein backbone atoms. (C) Radius of gyration of apo and bound complexes. (D) RMSF of side-chain residues in apo and protein-ligand complexes.
Binding energy estimation, residual contributions, and hydrogen bonding estimations
Figure 8A to C (Supplemental Table S4 to S6) presents the computed total binding energies and energetic contributions of the stable compounds to LasR, PqsR, and RhlR. These calculations provide insights into the overall strength of binding between the compounds and their respective receptors. The energetic contributions shed light on specific interactions and factors influencing the binding affinity of the compounds.
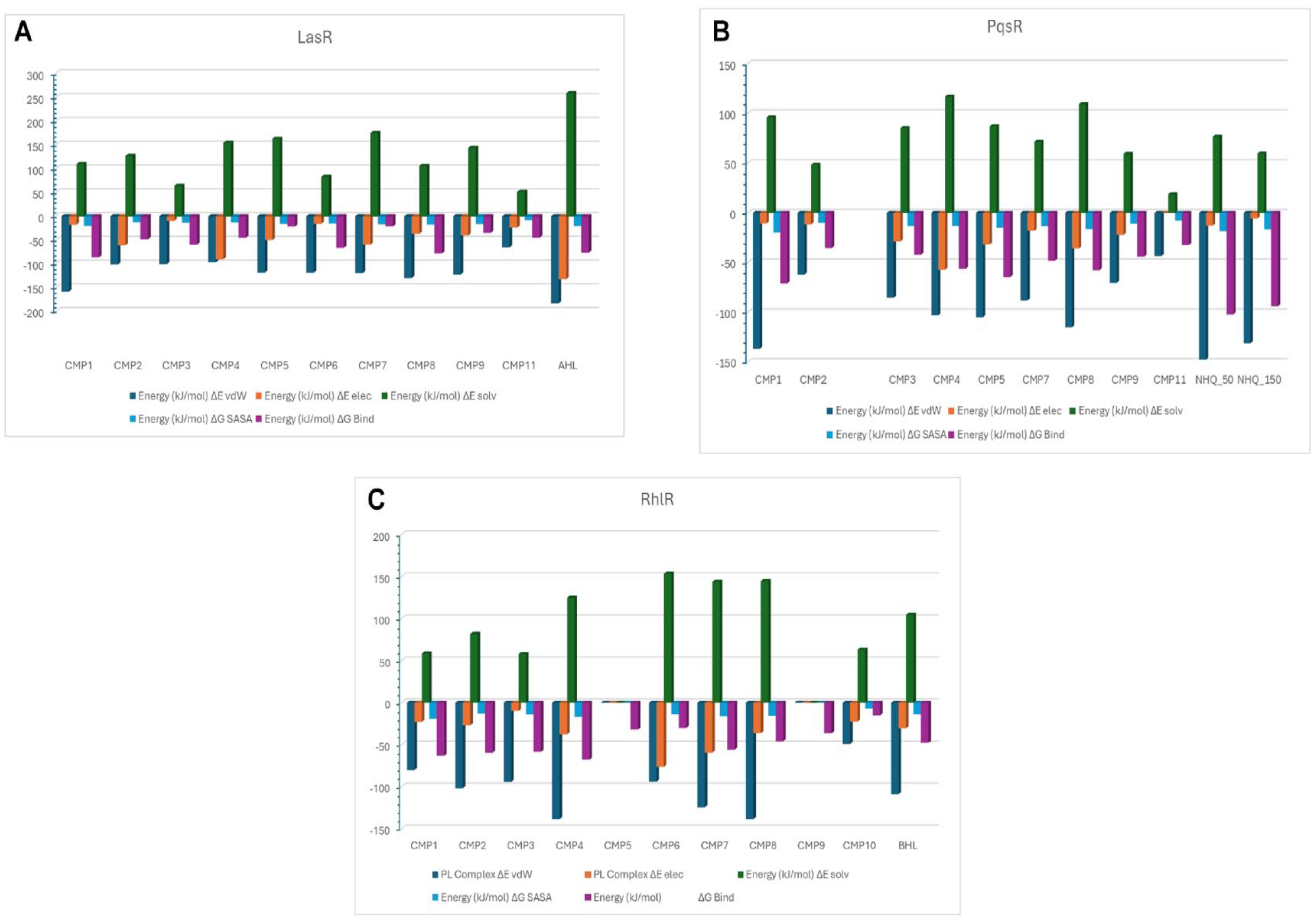
Binding energy estimation of compounds against receptors: (A) LasR, (B) PqsR, and (C) RhlR.
The LasR complexes
From Figure 8A and Supplemental Table S4, the binding energies of various compounds, such as CMP1 (−86.15 kJ/mol) and CMP8 (−78.06 kJ/mol), were found to be higher than that of AHL. Conversely, CMP11 exhibited the weakest binding energy. The nonpolar contributions, including solvent accessible surface area (SASA) and the van der Waals interactions, ranged from −17.26 to −12.68 kJ/mol and −158.3 to −64.96 kJ/mol, respectively, showing higher values for all compounds. The polar solvation contributions varied from 51.59 to 174.879 kJ/mol. However, the net solvation energies (ΔESOLV) were generally less favorable across all systems. Comparatively, the electrostatic contributions were relatively lower than the other energetic factors. Furthermore, we explored the residual contributions to the total binding energies when the compounds interacted with the different receptors. Notably, positive binding energies were predominantly associated with Arg61, Arg71, and Asp73, whereas negative binding energy contributions were observed primarily with Leu36, Tyr64, Val76, Ile52, Leu125, and Trp88 (see Supplemental Figure S1).
The PqsR complexes
The total binding energies of all PqsR-furanone complexes were lower than that of the PqsR-NHQ complex, as indicated in Figure 8B and Supplemental Table S5. The van der Waals contribution exhibited a greater magnitude, ranging from −147.51 to −61.98 kJ/mol. On the contrary, the polar solvation contributions ranged from 18.69 to 116.91 kJ/mol. Among the furanones, CMP1 displayed the highest total binding energy, amounting to −70.89 kJ/mol. However, the binding spontaneity of the compounds to PqsR was strongly impeded by their interaction with several hydrophilic residues, including Asp100, Asp150, Arg209, and Thr265. Conversely, residues such as Ile263, Ile236, Leu208, and Ile149 played a favorable role in contributing to the binding of the compounds, as illustrated in Supplemental Figure S2.
The RhlR complexes
The binding energies of some of the compounds were found to be higher against RhlR than BHL in general (Figure 8C and Supplemental Table S6). Specifically, CMP2, CMP3, CMP4, CMP7, and CMP1 demonstrated a greater binding affinity for RhlR, with binding energies greater than −47.68 kJ/mol as shown in the table. The van der Waals contributions ranged from −48.95 to −138.49 kJ/mol, indicating that nonpolar forces played a critical role in binding. On the contrary, polar solvation potentials ranged from 57.94 to 162.42 kJ/mol, indicating a lower contribution to binding. Electrostatic contributions varied significantly among all compounds. Certain residues such as Trp68, Tyr72, Val60, Val44, and Phe101 were found to contribute favorably to the stabilization of compounds at the BHL-binding domains, whereas hydrophilic residues like Asp81 and Arg46 were found to contribute unfavorably to the binding of the compounds as shown in Supplemental Figure S3.
Analysis of ADME properties
The ADME properties of selected furanones were analyzed using the ADMETlab 2.0 web servers. The results are explained based on the criteria set by the ADMET lab servers (Table 4). The oral bioavailability of molecules was evaluated based on Lipinski’s rule of 5 (molecular weight [MW] ⩽ 500, lipophilicity [log P] ⩽ 5, number of HBDs ⩽ 5, and number of HBAs ⩽ 10). The absorption parameters included HIA, solubility class, and caco-2-permeability (C2P). Excretion parameters included clearance level and half-life. If 2 of the set criteria are violated, poor absorption might be observed. Optimum solubility values range from −4 to 0.5 logmol/L. Acceptable C2P values are higher, if values are greater than −5.15 log unit. Clearance levels are higher when values >15 mL/min/kg; moderate when values range from 5 to 15 mL/min/kg, and lower when values <5 mL/min/kg. Optimum values of Log P ranged from 0 to 3. From the results, all compounds obeyed the Lipinski’s rule. The CMP1 violated the optimum Log P values. All compounds produced acceptable C2P values with high HIA. In all, nonhalogenated furanones showed the moderate clearance level, whereas halogenated furanone showed the low clearance level. Half-life probabilities were greater for nonhalogenated furanones than halogenated furanones.
ADME properties of furanone compounds.
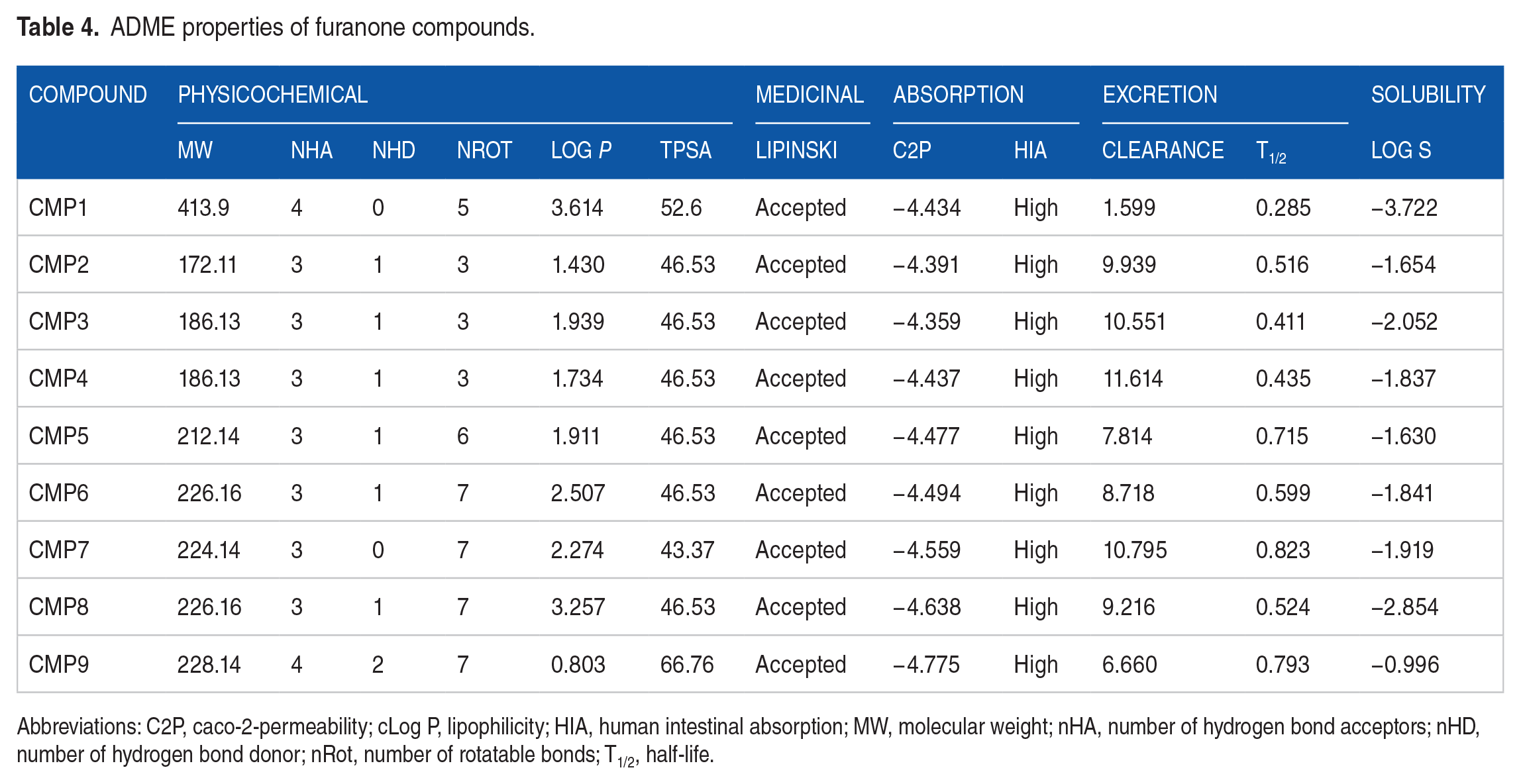
Abbreviations: C2P, caco-2-permeability; cLog P, lipophilicity; HIA, human intestinal absorption; MW, molecular weight; nHA, number of hydrogen bond acceptors; nHD, number of hydrogen bond donor; nRot, number of rotatable bonds; T1/2, half-life.
Discussion
Various virulence factors, including elastase, rhamnolipids, LPS, and alginate 37 facilitate the transition from acute infection to chronic disease. In P aeruginosa, the shift from planktonic growth to biofilm formation is regulated by multiple signaling systems. The QS machinery plays a crucial role in producing virulence factors and contributes to AMR. 38 This process involves the synthesis of QS signaling molecules by synthases (LasI, RhlI, PqsA-D), such as N-AHLs, N-BHL, and PQS, respectively. The receptors (LasR, RhlR, and PqsR) act as response regulators, binding to these signaling molecules and coordinating downstream responses. 7 Interfering with this regulatory system presents an interesting target for developing strategies to control diseases. 24 Lactone-based molecules, including furanones and their analogues, have shown promise in mimicking signaling molecules and regulating QS and biofilm formation. 39 Previous studies have also demonstrated the inhibitory effect of synthetic furanone compounds on the development of microbial biofilms. 24 In addition, certain furanone compounds with structural similarities to AIs have been shown to attenuate cell-to-cell communication in marine bacteria.25-27 Therefore, this study aims to explore the molecular basis of the anti-QS activities of marine-derived furanones using molecular docking and molecular dynamic simulation approaches.
Initially, a molecular docking approach was employed to predict the binding conformations and energies of the compounds with their respective biological targets.40,41 From the molecular docking results, it was observed that all 11 compounds bound to the AHL, NHQ, and BHL-binding domains of LasR, PqsR, and RhlR, respectively (Figure 3). The interactions between the compounds and LasR were like those observed with AHL. Hydrogen bonds were formed with Trp60, Tyr64, and Ser129, whereas hydrophobic interactions occurred with Ala50, Val76, Ala127, Leu40, and Ile52. Interactions with other residues such as Asp73, Tyr52, Leu36, and Ala105 were also observed (Table 1 and Supplemental Figure S4). These interactions align with previous findings where phenolic compounds with anti-QS activity interacted with LasR, forming hydrogen bonds with Ser129 and Trp60, and hydrophobic interactions with Ala127, Val76, and Asp73. 15 Hydrophobic interactions observed in the AHL-LasR complex contribute to shielding the ligand-binding pocket from the solvent and creating a favorable hydrophobic environment for nonpolar residues.
Overall, the binding affinities were higher for LasR, followed by RhlR and PqsR. Most compounds exhibited good binding to all 3 receptors, but their affinity was stronger for LasR. It is worth mentioning that Nicholas Mok reported that vanillin, with a binding affinity of −4.0 kcal/mol against PqsR, exhibited better experimental inhibition. 7 Therefore, even compounds with seemingly low-docking score may possess interesting PqsR inhibitory capabilities. Hydrophobic interactions were observed for most compounds with PqsR residues such as Leu208, Leu197, Phe221, and Gln194 (Table 2 and Supplemental Figure S5). Visual inspection revealed hydrogen bond formation with Gln194. Protein engineering studies have highlighted the importance of hydrogen bonding interactions with Gln194 and pi-stacking interaction with Tyr258 in antagonizing PqsR. 42
LasR and RhlR, unlike PqsR, possess AIs with furanone backbones, making their active site architecture more suitable for binding furanone metabolites, resulting in stronger affinities. Furthermore, halogenated furanones consistently demonstrated better binding affinities compared with nonhalogenated furanones. It is worth noting that the presence of heavy halogen atoms can create an anisotropic electron distribution and electron-withdrawing ability, resulting in the formation of a σ-hole in halogen bonding. 43 This σ-hole effect poses a challenge in accurately simulating the binding conformations of halogenated compounds. 44 For RhlR, the compounds exhibited binding affinities similar to those of BHL. Visual inspection revealed hydrogen bond interactions with Trp68, Ser135, and Tyr64, along with hydrophobic contacts with Trp96, Ala111, and Phe101, similar to the interactions observed with BHL (Table 3 and Supplemental Figure S6). A quantitative structure activity relationship (QSAR) study conducted by Nam et al 28 reported that an alkynyl ketone compound acting as an RhlR antagonist interacted with active site residues such as Trp68, Tyr72, Asp81, and Ser135.
The limitations of molecular docking tools arise from their inability to consider natural conformational changes in proteins, solvation effects, ionic interactions, and entropic contributions to binding free energies.32,34,45 To address these limitations and obtain a more realistic understanding of protein-ligand interactions, MD simulations are employed. The MD simulations enable exploring dynamic interactions between proteins and ligands by simulating their behavior over time. This approach provides valuable insights into the stability, energetics, and conformational dynamics of protein-ligand complexes. Several analyses, including stability assessment, estimation of binding free energies, evaluation of hydrogen bonding patterns, identification of residue contributions, and trajectory analysis, are conducted to investigate the dynamic behavior of ligands within the protein. 46 By incorporating MD simulations, a more comprehensive understanding of ligand binding can be achieved, accounting for the dynamic nature of the system. In this study, MD simulations were employed to evaluate the conformational stability and dynamics of the ligand complexes initially generated through molecular docking.
The binding pocket of LasR can be divided into 2 distinct sites: a hydrophobic site that accommodates the hydrophobic tail of the pocket and a hydrophilic region that binds the polar group of the AI.15,47 Therefore, it is crucial to investigate the conformational stability of the protein both with and without ligand binding. The dynamic stability of the compounds was assessed by analyzing their RMSD profiles. The CMP10 exhibited high instability with an RMSD greater than 2 nm (Figure 5A). This instability could be attributed to the absence of substituents on the furanone head group, limiting extensive interactions with the receptor. These findings suggest that alkyl substitutions on the ring system are critical in ligand binding. Interestingly, LasR was found to accommodate most of the compounds and form hydrogen bonds with hydrophilic residues without significantly altering the structural dynamics of the protein at the AHL binding site. The RMSD analysis was further supported by evaluating residue fluctuations using RMSF (Figure 5B). The fluctuations of the AHL-binding residues were examined, and it was observed that in the apoprotein, the fluctuations increased in the following order: Ser129 < Val76 < Tyr64 < Ala70 < Ala50 < Leu40. On binding of CMP1, lower fluctuations were observed with Leu40, Ala50, Trp60, and Ala70. However, CMP6, CMP7, and CMP8 binding resulted in higher fluctuations in Leu40, Ala50, Trp60, and Ala70. Among all the complexes, Ser129 exhibited the least fluctuations as it is located on a helix chain (Figure 5D). To compare the compactness of the bound and unbound proteins, RG values were calculated. Lower RG values indicate lower fluctuations in protein compactness, whereas higher values indicate greater fluctuations. 48 The CMP2, CMP8, and CMP11 showed lower compactness compared with the apoprotein (Figure 5C).
The stability of the furanone-PqsR complexes obtained from docking was assessed through MD simulations. Our results indicated that the halogenated furanones (CMP1 and CMP11) formed stable complexes with PqsR (Figure 6A). Interestingly, according to Shinada, although halogens on compounds typically do not engage in interactions, their presence enhances the binding of compounds to their targets. 43 Therefore, the enhanced stability of the CMP1 and CMP11 complexes may be due to the presence of halogens. To compare the stability of the bound and unbound complexes, the RMSD of the protein backbones was analyzed. The bound complexes exhibited relatively higher deviations in protein backbone than the apoprotein (Figure 6B). Furthermore, the fluctuations of side chains in the bound and unbound complexes were examined through RMSF analysis. In all the bound systems, lower fluctuations were observed for residues Ala168, Val170, and Trp234. However, CMP2 and CMP8 exhibited higher fluctuations for Tyr258 and Ile263. Generally, the bound complexes displayed significantly higher fluctuations than the apoprotein (Figure 6D).
For the furanone-RhlR complexes, all compounds were observed to form stable complexes with RhlR except CMP11 (Figure 7A). However, it was observed that CMP6 and CMP10 exhibited the highest fluctuations within the binding pocket when compared with the other compounds. Ligand binding had a noticeable effect on the protein’s movement on binding, particularly in the loop regions. The RMSF values of the residues involved in BHL binding were then compared between the bound and unbound complexes. Binding of CMP3 and CMP11 resulted in increased fluctuations, particularly with residues Trp68 and Asp81. Conversely, lower fluctuations were observed in the binding of CMP4, CMP6, and CMP10, compared with the apoprotein (Figure 7D). Notably, the DNA-binding domain displayed the highest level of fluctuations and was less compact compared with the BHL-binding domain (Figure 7C). Furthermore, stability analysis allowed for the computation of binding energies and assessing residue contributions in the different complexes. The binding energies of the stable complexes formed with LasR, PqsR, and RhlR were calculated using frames from the last 50 ns of the MD simulation.
The binding free energies of the stable docked complexes were calculated using the Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA) method. Frames from the last 50 ns were processed, and the net energies of the systems were calculated using the equations (1) to (3):
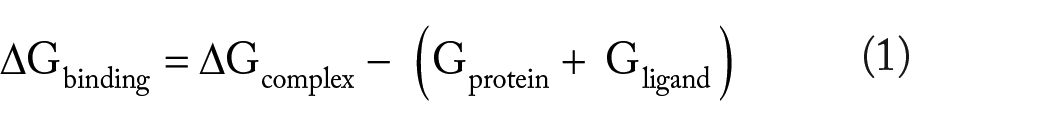
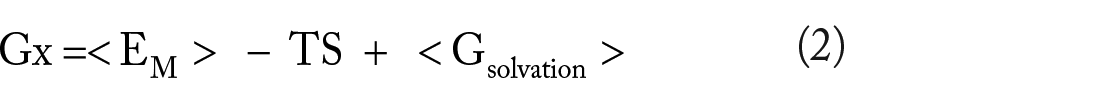
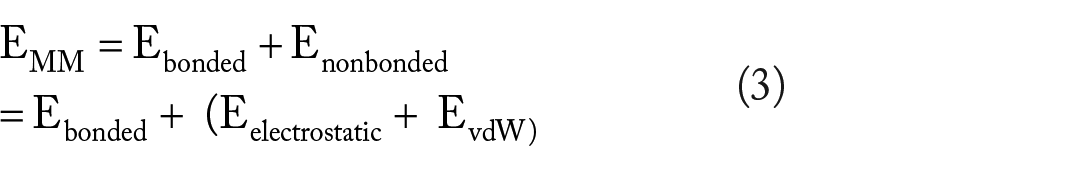

where Gcomplex is the total free energy of the protein-ligand complex. G protein and G ligand are the total free energies of the isolated protein and ligand in solvent, respectively. Gx is the protein or ligand or protein-ligand complex. <EMM> is the average molecular mechanics potential energy in a vacuum. The TS refers to the entropic contribution to the free energy in a vacuum, where T and S denote the temperature and entropy, respectively. The last term, <Gsolvation> is the free energy of solvation. Gpolar and Gnonpolar are the electrostatic and nonelectrostatic contributions to the solvation-free energy, respectively. 35
The MMPBSA computations provided insight into the binding energies, enabling predictions of the spontaneous binding of compounds to their target. In some cases, compounds exhibited stronger binding to their target than native compounds, indicating their potential inhibitory potency. However, even in cases where compounds did not bind strongly, their presence within the binding pocket influenced the dynamics and conformation of the target. This phenomenon could lead to misfolding or the adoption of different conformations, hindering efficient binding of the native ligand. Experimental observations have also reported that interaction with specific amino acid residues can contribute to inhibition.
The thermodynamic energies of LasR-furanone complexes were calculated to assess their binding characteristics (Figure 8A and Supplemental Table S4). The CMP1 (−86.15 kJ/mol) and CMP8 (−78.06 kJ/mol) demonstrated superior binding energies compared with AHL, indicating the enhanced binding of CMP1 in the presence of halogen atoms compared with PqsR. The contributions from nonpolar interactions, such as SASA and van der Waals forces, were prominent for all compounds, emphasizing the significance of hydrophobic interactions in stabilizing the compounds within the binding site. Key residues, including Leu36, Tyr64, Val76, Ile52, Leu125, and Trp88, played a crucial role in stabilizing the compounds, most of which are essential for AHL binding. However, unfavorable energy contributions were observed with hydrophilic residues, suggesting that the electrophilic substituents present in the compounds hindered the favorable binding through extensive electrostatic and polar solvation effects (Supplemental Figure S1). The interaction with catalytic residues further indicated that most compounds stabilized within the AHL-binding domain. It has been reported that AHL’s acyl chain facilitates the proper folding of LasR by promoting the wrapping of α-helices onto the central β-sheet through hydrogen bonding with Asp73 or Thr75.42,49 Thus, the compounds may inhibit LasR by binding to the AHL-binding domain and disrupting the formation of the hydrophobic core necessary for proper folding and function.
The stable compounds showed lower binding energies when interacting with PqsR compared with LasR complexes. However, the presence of halogen atoms in CMP1 improved its binding to PqsR (as shown in Figure 8B and Supplemental Table S5). In contrast to LasR complexes, the contributions from polar solvation were reduced for all compounds, indicating weaker polar interactions such as hydrogen bonding and electrostatics within the binding pockets. This can be attributed to the hydrophobic nature of the PqsR binding pocket. Another noteworthy observation is that the compounds exhibited lower hydrogen bonding interactions with PqsR. In a study by Kitao et al, 19 it was reported that the PqsR antagonist, M64, interacts with Tyr258 through pi-stacking and forms a hydrogen bond with Gln194, preventing the formation of the hydrophobic pocket responsible for NHQ interaction. However, due to the lower binding energies and electrostatic contributions observed for the furanones, it is unlikely that these compounds target PqsR effectively in their anti-QS activities. A comparison of the compound structures with the NHQ AI revealed structural differences. The furanone and quinoline backbones are 2 structurally distinct molecules. This may suggest that the compounds may not compete effectively with NHQ for binding to PqsR. Soheili and co-workers reported quinoline-based and quinazoline-based derivatives as the best candidates for developing PqsR inhibitors due to their structural similarities with PQS. 50
Among the compounds tested against RhlR, CMP2, CMP3, CMP4, CMP7, and CMP1 exhibited higher binding affinity (greater than −47.68 kJ/mol) (Figure 8C and Supplemental Table S6). The CMP2 to CMP4 had shorter alkyl side chains attached to the furanone backbone, whereas CMP1, despite having a halogen substituent, had a shorter alkyl chain. Interestingly, CMP1 showed higher binding energies compared with CMP10, highlighting the significance of the halogen substituent in the QS antagonist. Electrostatic contributions were relatively lower for CMP2 to CMP4, whereas the compounds displayed greater van der Waals contributions to the overall binding energies. This suggests that the hydrophobicity of the compounds and the presence of a secondary carbonyl group play crucial roles in influencing the binding affinity toward RhlR. 18 Analyzing their residue contributions, it was observed that Asp81 and Arg48 exhibited positive binding energy contributions that counteract spontaneous binding, which explains the relatively lower electrostatic contributions. These residues were found to be involved in hydrogen bonding events based on estimations. On the contrary, negative binding energy contributions were observed for residues Tyr64, Trp96, and Phe135 (see Supplemental Figure S3). In all cases, the compounds remained stable within the binding pocket throughout the simulations. Based on these observations, when comparing CMP2 to CMP4 with BHL, it is proposed that the shorter alkyl chains on the furanone compound enable CMP 2 to CMP4 to function as potential RhlR antagonists.
In general, it was observed that furanones exhibited higher binding energies against LasR and RhlR, while displaying weaker binding to the PqsR protein. In the case of LasR, compounds with longer alkyl chain substitutions (CMP5-9) demonstrated superior binding to those with short alkyl chain substitutions (CMP2-4). However, the binding of compounds to LasR was still weaker compared with AHL. An analysis of the residual contribution to the total binding energies revealed that the compounds interacted with hydrophilic residues, leading to the exposure of the binding pocket. This exposure hindered the formation of the hydrophobic core necessary for forming protein dimers, consequently inhibiting transcription. Regarding RhlR, compounds with short alkyl chain substitutions displayed better binding than BHL. This suggests that the compounds have the potential to compete with BHL, thereby influencing the dynamics of RhlR in the QS circuit. Like LasR, the compounds interacted with hydrophilic residues within the BHL-binding pocket, potentially exposing the hydrophobic pocket to solvent. This exposure could lead to protein inactivation. Overall, these findings highlight the capability of the compounds to bind to and interact with specific QS receptors, potentially affecting their functioning and transcriptional regulation.
In all cases, halogenated furanones showed greater binding compared with nonhalogenated furanones. These suggest that the presence of a halogen atom is critical for binding to the QS receptors. These observations suggest that furanones may be important scaffolds for designing QS inhibitors targeting the LasR and RhlR proteins.
The ADME properties of compounds were analyzed to evaluate their pharmacokinetic properties (Table 4) based on Lipinski’s rule of 5, HIA, caco-2-permeability, clearance level, and half-life. The in silico evaluation of ADME and toxicity parameters of compounds have become relevant in current drug research. 51 A molecule that violates 2 or more of the 4 Lipinski’s rules would likely not be orally bioavailable. All compounds did not violate Lipinski’s rule and are, therefore, likely to be orally bioavailable. Absorption determines how quickly a drug enters the bloodstream from an extravascular point of administration. 52 Solubility, physicochemical properties, and lipophilicity of the compound affect their absorption properties. When systemic effects are required, lower solubility may limit the HIA through the portal vein system to obtain a therapeutic impact. 53 High HIA suggests that compounds can be orally absorbed by the intestine. Caco-2-permeability measures the rate at which a compound accesses completely developed human epithelial cells. 54 Acceptable HIA and caco-2 properties indicate sufficient solubility, physicochemical properties, and lipophilicity of compounds, hence acceptable absorption properties. Excretion of compounds influences both the half-life and bioavailability, thus impacting the dose regimen and dose size of a drug. 52 The low clearance level observed for CMP1 suggests that it can stay for a longer time for absorption to occur.
Conclusion
This study aimed to investigate the potential of marine-derived furanones as anti-QS agents targeting QS receptors in P aeruginosa. Using molecular docking, MD simulations, MMPBSA calculations, and residual contribution estimations, the binding preferences of the compounds to LasR and RhlR, key components of the QS machinery, were elucidated. In P aeruginosa, the Las and Rhl systems play crucial roles in coordinating the production of virulence factors and forming biofilms. The structural similarity of furanone metabolites to AHL and BHL suggests that the compounds can effectively fit into the binding pocket, competing with the native ligands and causing unfavorable interactions within the pocket. The presence of short-chain alkyl substituents on the furanone backbone enables the compounds to effectively compete against BHL for binding to RhlR. In addition, the incorporation of halogens improves the binding affinity of the compounds to LasR, PqsR, and RhlR. When binding to LasR, the compounds interact extensively with the AHL-binding domain, leading to potential protein inactivation and aggregation through hydrophilic interactions. On the contrary, the binding of compounds to PqsR has a limited impact on protein dynamics, indicating relatively weaker binding. The structural difference between the AI of PqsR, NHQ, and AHL/BHL restricts the compounds’ ability to perfectly fit within the PqsR binding pocket. Overall, this study provides valuable insights into the molecular mechanisms underlying the design of potent QS receptor antagonists using furanones derived from marine sources. The study’s limitations include the static nature of molecular docking, simplified solvation models (implicit), and a limited range of compounds, which may not fully capture the dynamic interactions or reflect complex biological environments. Future research should incorporate advanced MD simulations with explicit solvent models and high-throughput virtual screening of a larger set of furanones to identify additional potent inhibitors. Experimental validation in vitro against the individual receptors will also be crucial to confirm the efficacy and specificity of the identified inhibitors, providing a more comprehensive strategy for developing effective anti-QS therapeutics.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322241275843 – Supplemental material for Marine-Derived Furanones Targeting Quorum-Sensing Receptors in Pseudomonas aeruginosa: Molecular Insights and Potential Mechanisms of Inhibition
Supplemental material, sj-docx-1-bbi-10.1177_11779322241275843 for Marine-Derived Furanones Targeting Quorum-Sensing Receptors in Pseudomonas aeruginosa: Molecular Insights and Potential Mechanisms of Inhibition by Aaron Boakye, Muntawakilu Padiga Seidu, Alice Adomako, Michael Konney Laryea and Lawrence Sheringham Borquaye in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
The authors are grateful to the Center for High-Performance Computing, Cape Town, South Africa, which granted generous access to the Lengau cluster for the MD simulations.
Author Contributions
LSB, AB, and MKL conceived the study. All experiments were designed by LSB, AB, MKL, MPS, and AA. Computations were made by AB, MPS, and AA. Data analysis was done by AB, MPS, AA, and LSB. The manuscript was prepared by AB, MPS, and AA and edited by all the authors. All authors read and approved the final manuscript.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Data Availability
All data generated or analyzed during this study are included in this published article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.