
Abstract
Brucellosis is a chronic and debilitating disease in humans, causing great economic losses in the livestock industry. Making an effective vaccine is one of the most important concerns for this disease. The new mRNA vaccine technology due to its accuracy and high efficiency has given promising results in various diseases. The objective of this research was to create a novel mRNA vaccine with multiple epitopes targeting Brucella melitensis. Seventeen antigenic proteins and their appropriate epitopes were selected with immunoinformatic tools and surveyed in terms of toxicity, allergenicity, and homology. Then, their presentation and identification by MHC cells and other immune cells were checked with valid tools such as molecular docking, and a multi-epitope protein was modeled, and after optimization, mRNA was analyzed in terms of structure and stability. Ultimately, the immune system’s reaction to this novel vaccine was evaluated and the results disclosed that the designed mRNA construct can be an effective and promising vaccine that requires laboratory and clinical trials.
Introduction
Brucella is an intracellular microorganism responsible for inducing a severe febrile ailment in humans, which is referred to as brucellosis. 1 Brucellosis is a substantial global health concern, exhibiting elevated incidence levels in specific geographical areas, where it can reach up to 200 cases per 100 000 individuals. 2 This disease has established endemicity in numerous nations, posing a significant risk to human well-being and inflicting considerable financial setbacks within the livestock sector. 2 Brucellosis is a resurging infectious disease passed from animals to humans, presenting a range of symptoms and an extended duration of illness, frequently resulting in misidentification, elevated treatment expenses, and the inefficient allocation of medical resources. 3 The genus Brucella comprises 12 species among which, B melitensis is the most pathogenic species, causing ovine and caprine brucellosis and resulting in the most extreme manifestation of the illness in humans. 4 It persists as a prevalent issue in diverse regions like Latin America, the Middle East, Africa, and Asia, affecting roughly 500 000 individuals globally each year. 5 Transmission to humans occurs through the ingestion of unpasteurized dairy products, aerosol inhalation, or occupational exposure. 6 Despite the presence of antibiotic therapy for the treatment of brucellosis, the disease significantly debilitates the human body, often necessitating long-term recovery. The intracellular inclination of Brucella limits the effectiveness of antibiotics, as only a limited set of options can adequately combat this infection. In addition, relapses occur in 5% to 40% of cases, requiring extended treatment with different combinations of antibiotics. 7 The lack of an approved vaccine for human brucellosis exacerbates the problem in regions where the disease is prevalent. 8 At present, there are no officially approved vaccines for human use against Brucella, and the existing vaccines developed for animals come with various adverse effects, including the potential for infection. 9 In addition, bacterial vaccines that are either inactivated or attenuated have their own set of constraints.8,9 While early-stage animal brucellosis prevention relied on M5, H38, and 45 of 20 vaccines, the current animal vaccines include S19, Rev.1, S2, RB51, and SR82.8,9 Nonetheless, these vaccines possess certain disadvantages, such as retaining some level of virulence and causing interference with standard serological examinations.8,9 Today, mRNA vaccines provide a compelling alternative to conventional vaccine strategies, thanks to their remarkable efficacy, swift development prospects, economical production, and safe delivery.10-14 Over the previous 10 years, substantial technological progress and substantial research investments have propelled mRNA into the forefront as a valuable tool in the realm of vaccine creation and protein replacement therapy.15,16 In the case of Brucella, numerous surface proteins play crucial roles in host cell binding and entry, making them suitable targets for the design of an effective mRNA vaccine. 16 Seventeen surface proteins have been selected for vaccine development: VirB5, VirB2, Cyclic β-1-2-glucans (CβG), BtpA, bvrS, bvrR, OM25, OM31, OMP10, outer membrane protein, immunogenic protein, FrpB, FlgE, flgK, BRUME 26-kDa periplasmic immunogenic protein, Dihydrolipoyllysine, and Ribosome-recycling factor. 17 The VirB operon encodes the Type IV secretion system (T4SS), which plays a critical role in conjugation, DNA uptake and release, as well as acting as a significant virulence factor for Brucella. 17 The T4SS facilitates substrate transport across the cell membrane and enables the secretion of proteins outside the cell. Comprising 12 genes (VirB1 to VirB12), the VirB operon, encompassing 12 genes (VirB1 to VirB12), encodes the T4SS. 18 This T4SS plays a crucial role in various Brucella functions, including conjugation, DNA uptake and release, and serves as a significant virulence factor. 18 The T4SS facilitates the transport of substrates across the cell membrane and enables the secretion of proteins outside the cell. 18
Cyclic β-1,2-glucan (CβG) is a key virulence factor in Brucella pathogenesis and its intracellular life cycle. CβG is composed of cyclic polymers with 17 to 25 D-glucose molecules linked by β-1,2 linkages. Its synthesis is catalyzed by a membrane-bound enzyme called Cgs (cyclic glucan synthase), which exploits UDP-glucose as the sugar donor. 19
BtpA, a 30-kDa cytoplasmic antigen in Brucella spp., hinders with the Toll-like receptor 2 (TLR-2) signaling pathway in the host, thereby reducing innate immunity. BtpA also modulates the function of dendritic cells (DCs), impairing their function and exerting a cytotoxic effect, collectively promoting the occurrence and persistence of chronic brucellosis. 20
Omp10 as a main membrane protein of B melitensis, has demonstrated significant immunogenicity in previous experiments, making it a suitable candidate for vaccine design. Similarly, Omp25, another outer membrane protein of B melitensis, exhibits strong immunogenicity. 21 Furthermore, Omp31 has revealed remarkable immunogenicity in animal infection models and is considered a promising choice for vaccine design. 21
Given the significance of cellular immunity in brucellosis and the absence of a viable vaccine for human use, this study sought to develop an mRNA-based vaccine that can elicit both cellular and humoral immune responses to combat brucellosis, which is primarily caused by B melitensis in humans.
Materials and Methods
Compilation of proteomes
To gather data, 6 distinct Brucella melitensis strains with complete protein sequences were sourced from the GenBank database. Subsequently, the PSORTb v.3.0.2 online server 22 was employed for subcellular localization determination.
Extract of bacterial protein sequences
For the retrieval of the amino acid sequences of target proteins, the UniProt Knowledgebase was used.
Forecasting of immune cell epitopes
To predict B-cell epitopes, the ABCpred web server 23 was employed. This prediction employed an artificial machine learning approach with a 0.5 threshold for each protein sequence. The desired epitope length was set at 16 amino acids, and the overlap filter was activated.
For the anticipation of cytotoxic T-cell lymphocyte (CTL) epitopes, the Immune Epitope Database MHC 24 used the ANN 4.0 method. The predicted epitopes were organized based on their IC50 values.
In addition, for the forecasting of helper T-cell lymphocyte epitopes, the Immune Epitope Database MHC-II employed the NN-align method. 24
Evaluation of human homology
To ascertain the homology, all peptides in the vaccine construct were cross-referenced with the Homo sapiens (TaxID: 9606) protein database using the BLASTp tool Peptides with an E-value surpassing 0.05 were considered potential nonhomologous peptides.
Estimation of epitope antigenicity, allergenicity, and toxicity
All chosen epitopes underwent an assessment of their antigenicity, allergenicity, and toxicity. Antigenicity evaluation was carried out using the VaxiJen web server, 25 which relies on an alignment-independent method hinging on the physicochemical properties of the epitopes. A threshold of 0.4 was applied, with specific consideration of bacteria in the prediction process. The AllerTOP V.2.0 web server 26 was employed to determine the allergenicity of the epitopes, with all parameters maintained at their default settings. Furthermore, the ToxinPred server 27 was used to forecast and evaluate the toxicity of the epitopes, including the generation of potential mutants while keeping the default parameters. Only epitopes demonstrating antigenicity, nontoxicity, and nonallergenicity were retained for further research stages.
Molecular docking between T lymphocyte epitopes and MHC alleles
A subset of the extracted T lymphocyte epitopes (80 in total) underwent a molecular docking simulation to assess their binding affinity with their corresponding MHC alleles. The 3D structures of the MHC alleles were acquired from the RCSB PDB database. 28 Structural processing, which involved the removal of unnecessary ligands, was performed using PyMOL software. 29 Energy minimization of these structures was carried out via the Swiss-PDB Viewer. 30 Simultaneously, the selected epitopes for docking were transformed into three-dimensional structures using the PEP-FOLD 4 server 31 and subjected to energy minimization with the Swiss-PDB Viewer. The ClusPro 2.0 server 32 was employed for the docking procedure.
Design of vaccine construct
The proposed mRNA vaccine construct is characterized by a sequence that spans from the N-terminus to the C-terminus. It incorporates a sequence featuring a 5′ m7GCap, a 5′ UTR, a Kozak sequence, a tPA (Signal peptide), an EAAAK Linker, an RpfE (Adjuvant), a GPGPG linker, HTL Epitopes, KK, LBL Epitopes, an AAY Linker, CTL Epitopes, the MITD sequence, a Stop codon, a 3′ UTR, and a Poly (A) tail.
To ensure proper separation of the epitopes, three distinct linkers are employed: AAY, KK, and GPGPG. These linkers are intentionally designed to be cleavable, flexible, and rigid, facilitating the optimal functionality of the domains.33-35
Anticipating biological and physicochemical properties of the construct
The antigenicity of the vaccine construct was assessed using the VaxiJen 2.0 25 and ANTIGENpro. The prediction of antigenicity is a pivotal step as it determines the capacity of an antigen to induce an immune response and stimulate the formation of memory cells. VaxiJen 2.0 relies on the physicochemical properties of the vaccine for predictions, while the ANTIGENpro employs machine learning algorithms and microarray analysis data.
The input for the created mRNA vaccine exclusively encompasses the amino acid sequence of the translated ORF, excluding the tPA and MITD sequences. Allergenicity was evaluated via the AllerTOP 2.0, while vaccine toxicity was predicted using the ToxinPred. 27 Furthermore, the ProtParam web server was harnessed to predict various physicochemical properties of the vaccine.
In silico immune simulation
The C-ImmSim simulation server 36 was engaged to execute a dynamic imitation of the immune response for the studied vaccine. In the default setup, the C-ImmSim uses epitopes and lymphocyte receptors to simulate the immune response. In this particular study, the immune simulation involved the administration of 3 doses, each containing 1000 vaccine units (ng/mL), over a span of 4 weeks. This regimen aligns with the recommended practice for many contemporary vaccines, where 2 to 3 doses are administered within a 4-week window. The injections were given at time-step 1, 84, and 168, and all parameters were put to their default values.
Codon optimization of the vaccine construct
To ensure effectual expression within human cells, the codon sequences within the designed construct required optimization. The GenSmart Codon Optimization Tool, provided by GenScript (GS), was the tool of choice for this purpose.
Following optimization, a quality valuation of the optimized sequence was executed using the Rare Codon Analysis tools, also provided by GS. In this analysis, the efficiency of mRNA translation is gauged by the Codon Adaptation Index (CAI), which measures the alignment of codon usage in a given sequence with the codon usage bias of the target organism—in this case, human cells. In addition, the Codon Frequency Distribution (CFD) analysis was used to identify the incidence of any tandem unusual codons, indicating regions with atypical codon usage patterns.
Secondary structure prediction of the designed mRNA vaccine
The RNA secondary structure of mRNA is important in mRNA vaccines because it plays a crucial role in determining the stability, efficiency, and immunogenicity of the vaccine. The secondary structure of mRNA refers to how the nucleotides in the RNA molecule interact with each other to form stable base pairings and secondary structures such as hairpins and loops.
In this study, Structure of the mRNA vaccine was anticipated by means of the RNAfold tool from the ViennaRNA Package 2.0. 37 This tool applies McCaskill’s algorithm to calculate the minimal free energy (MFE) of the projected secondary structure. Both the MFE structure and the centroid secondary structure were reported, along with their respective minimum free energy values.
Anticipating and validating secondary and 3D structures of the designed vaccine
For predicting the secondary structure of the peptide, the PSIPRED 38 was applied. This prediction relied on position-specific scoring matrices and achieved an accuracy of 84.2%.
In the quest for predicting the 3D structures of the peptide, the Robetta server 39 was called into action, yielding five potential structures. To validate and confirm the most optimal structure, the ProSA-web, 40 PROCHECK, 41 and ERRAT were employed.
Foreseeing conformational B-cell epitopes
The tertiary structure of a protein can give rise to the formation of new conformational B-cell epitopes. To predict these discontinuous B-cell epitopes within the protein structure, the ElliPro online server was enlisted. ElliPro leverages the geometrical properties of the 3D model to make these predictions. Among the various prediction tools for discontinuous B-cell epitopes, ElliPro stands out with the highest AUC (area under the curve) value of 0.732 for any protein model, signifying its accuracy and reliability in predicting these specific epitopes.
Molecular docking study of the designed vaccine
The PIPER docking algorithm, available through the ClusPro, was used to dock the 3D structure of the desired vaccine with TLR-4 or TLR-3.
Molecular dynamics simulation
To verify solidity and scrutinize the physical movements of atoms and molecules within the TLR4-vaccine and TLR3-vaccine complex structures with the lowest binding energy, dynamic simulation analysis was done by means of the iMODS server. 42
Results
Retrieval of bacterial protein sequences
To obtain the amino acid sequences of specific proteins, we used the UniProt Knowledgebase. The following proteins and their respective accession numbers were retrieved: (1) virB5 (accession number—A0A144KBH0), (2) virB2 (accession number—A0A1S6ZGJ3), (3) cyclic β-1,2-glucans (CβG) (accession number—O85166), (4) BtpA (accession number—A0A1S6ZGK7), (5) bvrS (accession number—O68164), bvrR (accession number—O67996), (7) Omp25 (accession number—Q45321), (8) Omp31 (accession number—P0A3U4), (9) Omp10 (accession number—P0A3N8), (10) outer membrane protein (accession number—P0A3U4), (11) immunogenic protein (accession number—P0A3U4), (12) FrpB (accession number—Q8YDS0), (13) FlgE (accession number—Q8YDL6), (14) flgK (accession number—C0RMR9), (15) BRUME 26-kDa periplasmic immunogenic protein (accession number—P0A3U8), (16) dihydrolipoyllysine (accession number—Q9L6H8), and (17) Ribosome-recycling factor (accession number—P94340).
Prediction and assessment of B-cell epitopes
In the pursuit of epitopes for inclusion in the vaccine construct, we employed a rigorous selection process. The ABCpred web server was employed to predict epitopes, and the top five epitopes for each protein were chosen. Subsequently, we retained only those epitopes that met specific criteria: they had to be antigenic, nonallergenic, and nontoxic. To make these determinations, we made use of the VaxiJen, AllerTOP, and ToxinPred web servers, respectively. Furthermore, we scrutinized the predicted epitopes for any homologs in Homo sapiens (TaxID: 9606) to mitigate the risk of autoimmunity induction, and peptides with an E-value exceeding 0.05 were excluded from consideration. We also ensured that all desired epitopes were located within the conserved regions of the proteins under investigation. Variants of the selected proteins were obtained from the NCBI database and aligned using the Bioedit 7.2 program. In the end, we extracted a total of 12 B-cell epitopes from the 17 proteins studied, and these were incorporated into the vaccine construct (Table 1).
A compilation of 72 potential epitopes derived from 17 Brucella melitensis proteins, along with the induced cell types for each epitope.

Assessment of CTL epitopes
To predict and evaluate CTL epitopes, we relied on the immune epitope database (IEDB), focusing on the 9 proteins included in this study. Only epitopes with an IC50 value greater than 500 were selected for further analysis. We applied the same criteria as with B-cell epitopes, ensuring that the chosen epitopes were antigenic, nonallergenic, nontoxic, and nonhomologous. A total of 32 epitopes, situated within the conserved regions of the proteins, were chosen for inclusion in the vaccine construct (Table 1).
Assessment of HTL epitopes
Numerous potential HTL epitopes were identified through examination of the 17 proteins associated with Brucella melitensis, as previously mentioned. We exclusively focused on epitopes that were antigenic, nonallergenic, nontoxic, and nonhomologous. These epitopes were assessed for their potential to induce IL-4, IL-10, and IFN-γ. In the end, A total of 72 epitopes were identified (Table 1), with 28 epitopes specifically selected for vaccine design based on the analyses conducted, all of which are located within the conserved regions of the proteins and demonstrated the ability to induce the mentioned cytokines (Table 1).
Docking among MHC alleles and studied T lymphocyte epitopes
Our investigation successfully identified a total of 60 lymphocyte epitopes, all of which demonstrated recognition by a diverse array of 90 MHC alleles. Intriguingly, some of these epitopes displayed a predilection for binding to a single MHC allele, while others exhibited the versatility to interact with multiple alleles. These findings are comprehensively detailed in Table 2.
Selected T lymphocyte epitopes (CTL + HTL epitopes) alongside their respective MHC alleles.

In our quest to delve deeper into the nature of these interactions, we executed a series of molecular docking experiments involving 12 selected epitopes and their respective MHC alleles, with comprehensive information provided in Table 3. The crystallographic structures of the MHC alleles employed in this analysis were thoughtfully obtained from the RCSB PDB, and their specific PDB IDs are thoughtfully presented in Table 3.
Docking analysis of some CTL epitopes alongside their respective MHC alleles.

The outcomes of these meticulously conducted molecular docking experiments, shedding light on binding affinities, are meticulously documented in Table 3. It is worth highlighting that the epitope IAAPFVVIW emerged as a standout performer, displaying the highest binding affinity with a remarkable score of −935.9 kcal/mol when paired with its corresponding MHC allele, HLA-B5801. On closer examination of this particular interaction, we observed the epitope fitting precisely into the binding cleft of the MHC allele. Figure 1 illustrated the cartoon representation of docking results of some epitopes and corresponding MHC alleles (Figure 1). Overall, the findings of this section indicate that the epitopes forecasted by the examined servers are situated within the antigen-binding groove of the corresponding MHC alleles during the molecular docking process.
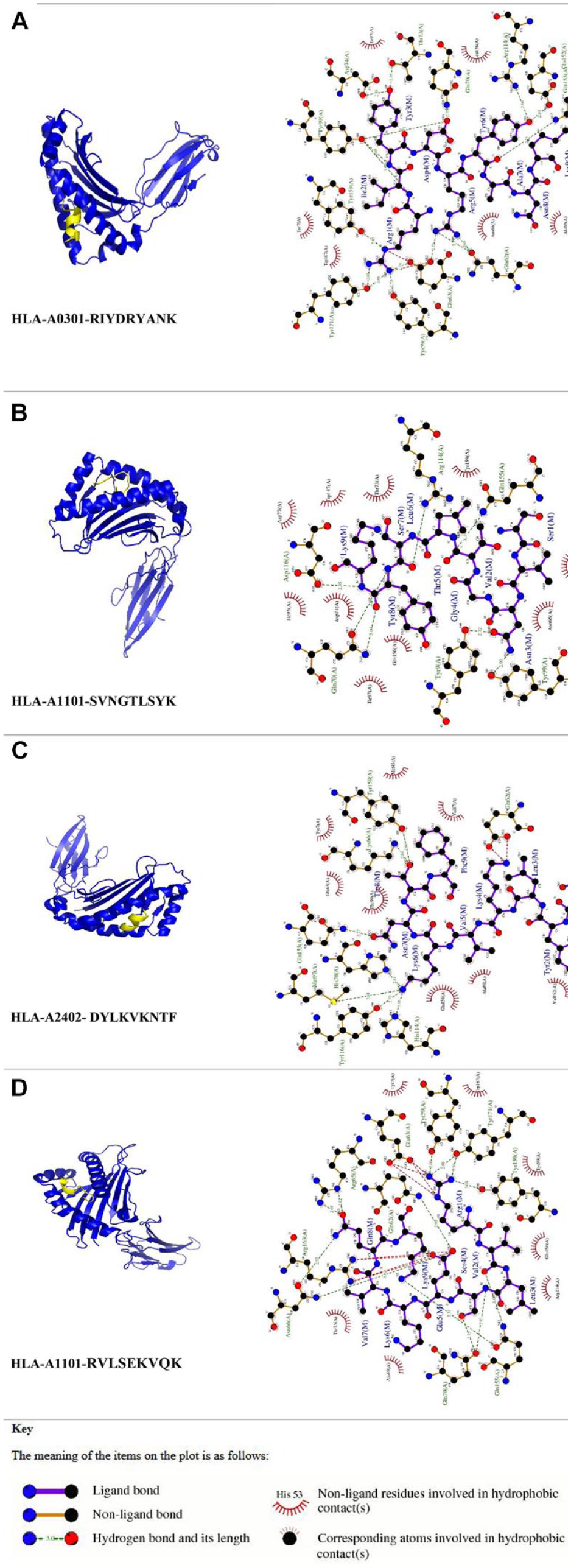
Docking among the epitopes and their corresponding MHC alleles.
Designing the vaccine construct
The design of the proposed vaccine construct followed a sequence from the N-terminus to the C-terminus, structured as follows: 5′ m7GCap–5′ UTR–Kozak sequence–Signal peptide(tPA)–EAAAK-Adjuvant(RpfE)ASATMEAEEEYDFFI
The vaccine contains the tissue plasminogen activator (tPA) secretory signal sequence in its 5′ region. This sequence acts as a signal to aid in the excretion of epitopes after translation outside the cell, if necessary. 43
Assessing the biological and physicochemical characteristics of the studied vaccine
To ensure the suitability and safety of the vaccine construct, we conducted a comprehensive evaluation encompassing various aspects. This included the assessment of antigenicity, allergenicity, and toxicity through using esteemed servers such as VaxiJen, ANTIGENpro, AllerTOP, and ToxinPred. In addition, we delved into the physicochemical properties of the vaccine construct, with the results thoughtfully compiled in Table 4. Our thorough evaluation revealed that the vaccine not only displayed antigenicity but also exhibited characteristics of nonallergenicity and nontoxicity.
Physicochemical properties of studied mRNA vaccine.

Furthermore, our investigation confirmed the thermal stability of the vaccine construct, as supported by its physicochemical properties. We also identified the vaccine’s hydrophilic nature, evident through a measured Grand Average of Hydropathicity (GRAVY) of −0.496. The results of this server show that the designed protein vaccine has a suitable weight and would be stable, and its half-life in the mammalian body is about 4.4 hours. In general, these findings imply that the designed multi-epitope mRNA vaccine construct has the potential to be an assertive candidate for use as a vaccine.
Predicting population coverage
To gauge the potential impact of the vaccine, we sought to assess its coverage across the global population. The combined analysis of the 60 epitopes and their respective 90 alleles was executed using the IEDB Population Coverage tool. The outcome of this evaluation indicated that the vaccine holds the promise of providing coverage to an estimated about 98% of the global population (Supplementary 1).
Simulating the immune response against the vaccine
To gain insights into the immune response triggered by the vaccine, a series of in silico simulations were conducted, as visually depicted in Figure 3. The observations unveiled that both secondary and tertiary responses surpassed the primary response. Notably, the production of immunoglobulin (IgM) outpaced that of IgG. Following antigen reduction, the levels of immunoglobulins remained notably elevated, signifying the potential emergence of immune memory resulting from antigen exposure and the development of acquired immunity.
The enduring presence of B-cell isotypes for an extended duration suggests isotype switching and memory formation within the B-cell population. In addition, an upsurge in the generation of memory CTL and HTL cells was observed. Macrophage activity exhibited enhancement, while dendritic cell activity remained relatively stable. Furthermore, the levels of IFN-γ and IL-2 experienced an increase, with heightened activity of epithelial cells contributing to innate immunity. Ultimately, the low Simpson index (D) suggests a discernible difference in the immune response.
The combined global population coverage of the 60 epitopes and their corresponding 90 alleles was determined using the IEDB Population Coverage tool. It was discovered that the vaccine has the potential to cover approximately 98.04% of the global population.
In silico simulation of the immune response to the vaccine
In the realm of in silico simulations, we administered 3 doses of the vaccine to mimic the immune response, as visually represented in Figure 2. Our observations shed light on several key aspects of the simulated response:

An immune simulation using computational methods was performed to assess the effectiveness of the mRNA vaccine obtained from the C-ImmSim server. (A) the production of immunoglobulins following antigen injection (B) the B-cell population post 3 injections, (C) the distribution of B-Cell Population in various states, (D) the Helper T-Cell Population, (E) the Helper T-Cell Population distribution by state, (F) the Cytotoxic T-Cell Population distribution by state, (G) the Macrophage Population in different states, (H) the Dendritic Cell Population in different states, and (I) the production of cytokines and interleukins alongside the Simpson Index, which provides insights into the immune response’s distinctiveness and specificity.
Secondary and tertiary responses: notably, the secondary and tertiary responses surpassed the primary response. This suggests an increasingly robust and effective immune reaction with subsequent exposures to the vaccine.
IgM dominance
The production of immunoglobulin (IgM) exceeded that of IgG, indicating the initial activation of the immune system and the body’s rapid response to the antigen (Figure 2A).
An intriguing observation regarding the antibody response following booster doses is the unexpected decrease in their production levels. Following the second and third administrations, a decline in antibody titers was noted, possibly attributed to the short intervals between the doses, resulting in a phenomenon suggestive of immune tolerance.
Isotype switching and memory formation
The presence of B-cell isotypes for an extended duration provides evidence of isotype switching and the formation of immune memory within the B-cell population, further underlining the vaccine’s potential to establish lasting protection (Figure 2B and C).
Augmented generation of memory CTL and HTL cells
An increase in the generation of memory cytotoxic T lymphocytes (CTL) and helper T lymphocytes (HTL) cells was observed, contributing to a more robust and specific immune response on reexposure to the antigen (Figure 2D to F).
Stable dendritic cell activity
Dendritic cell activity remained stable, ensuring the continued surveillance and communication between innate and adaptive immunity (Figure 2G).
Enhanced macrophage activity
Macrophage activity was notably enhanced, indicating an active role in phagocytosis and antigen presentation, which are crucial components of the immune response (Figure 2H).
Elevated IFN-γ and IL-2 levels
The levels of important immune signaling molecules, such as interferon-gamma (IFN-γ) and interleukin-2 (IL-2), experienced an increase, signifying a heightened and coordinated immune response (Figure 2I).
Increased epithelial cells
An increase in the population of epithelial cells, which play a role in innate immunity, was also observed, further reinforcing the body’s defense mechanisms (Supplementary 2).
The low Simpson index (D) indicated a discernible difference in the immune response, reflecting the vaccine’s capacity to induce a distinct and specific defense against the targeted pathogen.
Codon adaptability of the mRNA construct
The mRNA coding sequence (CDS) spanned 1896 nucleotides, and we evaluated the superiority of the optimized construct. The codon adaptation index (CAI) value, crucial for efficient expression, was determined to be 0.95. This CAI value surpasses the accepted threshold of 0.8, indicating the construct’s suitability for proficient expression within the human host.
Efficient expression requires the GC content of the construct to fall within the range of 30% to 70%. The optimized construct demonstrated an average GC of 66.78%, falling comfortably within this desired range. In addition, the Codon Frequency Distribution (CFD) analysis revealed a value of 0%, signifying that no codons hinder translation efficiency or functionality. It is worth noting that codons with values below 30 can disrupt the translational process (Supplementary 3).
Forecasting the secondary structure of the studied mRNA vaccine
The mRNA vaccine’s structural configuration was projected using the RNAfold server, and the associated free energy values were assessed (Figure 3). In this assessment, the optimized codons from the construct were employed as the input. The outcomes reveal that the mRNA vaccine is anticipated to possess stability, featuring a minimum free energy (MFE) for the structure at −1399.90 kcal/mol. Furthermore, the secondary centroid structure indicates a free energy of −1213.12 kcal/mol. These data offer valuable insights into the thermodynamic resilience of the mRNA vaccine structure.
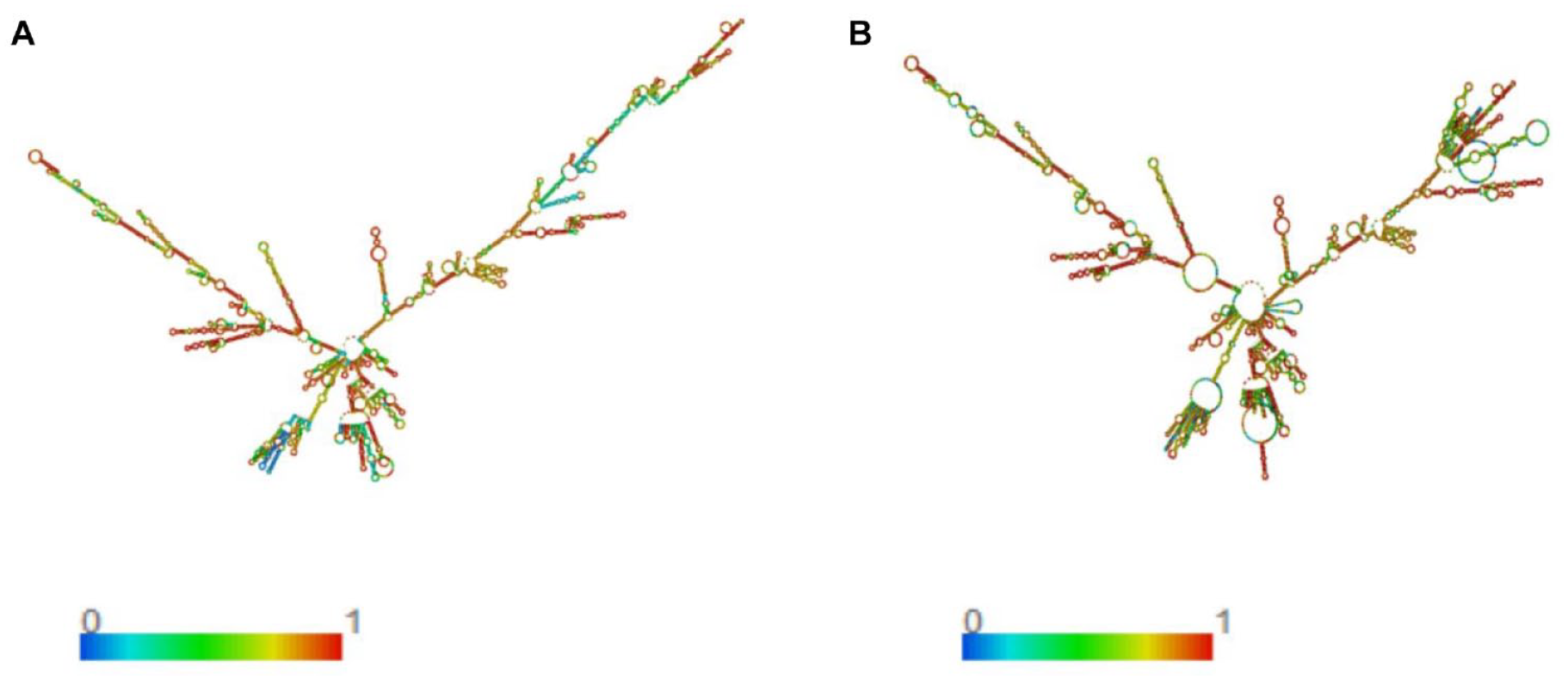
(A) Ideal secondary configuration, (B) Central secondary arrangement of the mRNA vaccine obtained through the RNAfold web server.
Estimation and verification of secondary and tertiary structures of the studied vaccine
The PSIPRED service was used to forecast the secondary structure of the vaccine. This analysis indicated a prevalence of alpha helices in the structure, as depicted in Figure 4A. Following that, we predicted the peptide’s tertiary structure using the Robetta, as displayed in Figure 4B. To evaluate the stereochemical accuracy of the structure, we used the PROCHECK, which considers the geometry of residues and the general structure for prediction. The Ramachandran plot, presented in Figure 4C, demonstrates that roughly 79.8% of residues occupy the most favored regions, 17.5% are within the additionally allowed zone, and 1.5% fall in the allowed regions. The total quality factor acquired was 94.6168, indicating a high level of structural quality. Furthermore, the ProSA projected a negative Z-score (−10.72), signifying substantial consistency in the 3D protein model, as illustrated in Figure 4D.

Forecasting and verification of the peptide vaccine construct’s structure: (A) Secondary structure prognosis of the vaccine, (B) Prediction of the tertiary structure. (C) Ramachandran plot, (D) Z-scores.
Foreseeing conformational B-cell epitopes
The folding of the model protein resulted in the creation of conformational B-cell epitopes. To pinpoint these epitopes, we engaged the ElliPro, which foretold 6 discontinuous B-cell epitopes. These epitopes encompass a total of 374 residues, with prediction scores ranging from 0.621 to 0.946, suggesting a high likelihood of their antigenic potential. The 2D and 3D models of these conformational B-cell epitopes are depicted in Figure 5, specifically in panels I and II, respectively.

Some anticipated conformational B-cell epitopes identified with the ElliPro: (I) Schematic Presentation of the Epitope Locations, (II) Three-Dimensional Models. Conformational Epitopes are Represented by Yellow Spheres.
Molecular dynamics simulation
To further explore the interactions within the vaccine-TLR3 and vaccine-TLR4 complexes, we conducted molecular dynamics simulations using the iMODS server. These simulations allowed us to evaluate how the receptor and ligand interacted over time and identify regions of the construct that displayed flexibility, visualized in the deformability graph (see Figures 6B and 7B).
We also employed normal mode analysis (NMA) to assess the flexibility of the proteins. The B-factor graph, which depicts the relationship between NMA and PDB areas in the complex, is presented in Figures 6C and 7C. Eigenvalues for the docked complexes were calculated to provide insights into their overall dynamics, as depicted in Figures 6D and 7D. These results demonstrated that the vaccine-receptor complex had a low deformation index, indicating high rigidity and enhanced steadiness.

Exploration of molecular dynamics, normal mode analysis, and interactions between receptor and ligand. (A) Complex formed by docking the vaccine with TLR4 using the ClusPro server (B) Graph illustrating deformability (C) Chart depicting B-factor analysis (D) Eigenvalue assessment of the vaccine-TLR4 and TLR3 (E) Visualization of the covariance matrix (F) Employment of an elastic network model via the iMODS server (G) Assessment of receptor-ligand interactions with the PDBsum web server (H) Examination of receptor-ligand interactions using the LIGPLOT web server.

Exploration of molecular dynamics, normal mode analysis, and interactions between receptor and ligand. (A) Complex formed by docking the vaccine with TLR3 using the ClusPro server (B) Graph illustrating deformability (C) Chart depicting B-factor analysis (D) Eigenvalue assessment of the vaccine-TLR4 and TLR3 (E) Visualization of the covariance matrix (F) Employment of an elastic network model via the iMODS server (G) Assessment of receptor-ligand interactions with the PDBsum web server (H) Examination of receptor-ligand interactions using the LIGPLOT web server.
Moreover, the covariance matrix (see Figures 6E and 7E) illustrated the connections between amino acid pairs located in dynamic regions. A connecting matrix based on an elastic network model, as revealed in Figures 6F and 7F, was used to recognize pairs of atoms connected by springs. Darker gray regions in this matrix represented higher stiffness within each chain of the complex.
Discussion
This study aimed to harness immunoinformatics methodologies to create a secure, engineered mRNA vaccine that is highly effective. Today, mRNA vaccines have garnered significant attention as promising alternatives to traditional vaccines due to their ease of production, cost-effectiveness, safety profile, and robust potency, particularly during pandemics.16,44 These vaccines have the ability to elicit a well-rounded immune response, encompassing both cellular and humoral immunity, and are not limited by specific MHC haplotypes. Moreover, mRNA serves as a safe vector as it carries transient information without interacting with the genome.16,44
To combat B melitensis infection, we developed an innovative in silico multi-epitope mRNA vaccine based on the bacterium’s major surface proteins. We thoroughly examined target proteins responsible for cell binding and attachment, seeking potential epitopes capable of triggering either humoral or cellular immune responses. The IEDB database was employed for predicting epitopes for HTL and CTL, while B-cell epitopes were determined using the ABCpred tool, which relies on machine learning methods. 24
One of the intriguing findings from our study was the identification of novel targets for the development of a vaccine. Using the complete bacterial proteome, we focused on surface-exposed proteins identified through domain signal analysis. The selection of appropriate servers for this purpose was a key strength of our study. The limited success in developing an effective brucellosis vaccine9,45 or addressing diagnostic challenges in serology-based kits46,47 may be attributed to the reliance on a small number of known bacterial proteins, highlighting the significance of the new targets identified in our investigation.
The predicted epitopes underwent scrutiny to evaluate their antigenicity, allergenicity, and toxicity through dedicated web servers. Specific linkers were employed to combine these epitopes, and immune simulations were carried out to validate the vaccine’s ability to provoke both humoral and cellular responses. The unexpected finding in our study was the decrease in antibody production following the administration of booster doses at a 4-week interval. This shortened interval may appear to induce a phenomenon of immune tolerance, suggesting the need to extend the interval between booster dose injections.
The selected epitopes demonstrated compatibility with 90 distinct MHC alleles. Molecular docking analysis was performed to assess the interaction between the chosen epitopes and their corresponding MHC alleles, as this ligand-epitope interaction plays a pivotal role in vaccine design. The analysis included the examination of binding affinity and bond formation through ClusPro, as well as an analysis of the interactions between the epitopes and the MHC pockets.
Furthermore, as there is a vast diversity of human MHC alleles, the vaccine’s effectiveness hinges on individuals possessing specific MHC alleles capable of binding to the epitopes. According to predictions from the IEDB tool, one notable outcome of the vaccine formulated in this investigation was the broad coverage of epitopes obtained, encompassing over 98% of the alleles across the global population. This finding underscores the potential novelty and significance of the targets employed in this study as promising candidates for brucellosis prevention. In addition, the vaccine construct was subjected to docking with immune receptors TLR-3 and TLR-4 to assess its potential interaction with these receptors, which could trigger the development of innate and adaptive immunity. Molecular dynamic simulations were employed to evaluate the permanency of the vaccine complex.
Overall, the validity of in silico studies can be established through laboratory experimentation.
In a study conducted by Tarrahimofrad et al in 2022, a multi-epitope vaccine was developed using BtpA, Omp16, Omp28, virB10, Omp25, and Omp31 antigens targeting B melitensis, Brucella abortus, and Brucella suis. Through molecular docking analyses, molecular dynamics simulations including gyration, Root Mean Square Fluctuation (RMSF), and Root Mean Square Deviation (RMSD) assessments, and validation of tertiary protein structures, the constructed protein exhibited a stable conformation capable of interacting with TLR2/4 receptors. The researchers posited that this peptide-based vaccine could elicit immune responses in both B and T cells, thereby potentially preventing infections caused by B suis, B abortus, and B melitensis. 48
Notably, the divergence between the present investigation and the aforementioned study lies in the identification of novel targets sourced from a comprehensive proteomic analysis of the melitensis species. These selected targets represent novel candidates, and the vaccine under investigation is based on an mRNA platform. In contrast to the peptide vaccine delineated in the prior study, the current research elucidated that it could also trigger the immune response in celery, a crucial factor in the pathogenesis of the aforementioned bacterium.
Nguyen et al conducted a study in 2023 where they used the reverse vaccinology approach to design DNA vaccines with potential bacterial antigens against Nocardia seriolae. These potential vaccine candidates were identified through in silico analysis. In vitro assays confirmed the immunogenicity of these proteins, and DNA-based vaccines resulted in a significant gene expression and serum antibody production in the orange-spotted grouper Epinephelus coioides. The study concludes that the reverse vaccinology is a promising plan vaccine candidates against N seriolae infection. 49
In summary, this study introduces an innovative multi-epitope mRNA vaccine for B melitensis, developed through immunoinformatic approaches. While this research lays the foundation for future studies and vaccine development, further in vitro and in vivo investigations are imperative to confirm these findings and ensure the vaccine’s efficacy.
Supplemental Material
sj-pdf-1-bbi-10.1177_11779322241272404 – Supplemental material for Proteomics Exploration of Brucella melitensis to Design an Innovative Multi-Epitope mRNA Vaccine
Supplemental material, sj-pdf-1-bbi-10.1177_11779322241272404 for Proteomics Exploration of Brucella melitensis to Design an Innovative Multi-Epitope mRNA Vaccine by Maryam Asadinezhad, Iraj Pakzad, Parisa Asadollahi, Sobhan Ghafourian and Behrooz Sadeghi Kalani in Bioinformatics and Biology Insights
Supplemental Material
sj-pdf-2-bbi-10.1177_11779322241272404 – Supplemental material for Proteomics Exploration of Brucella melitensis to Design an Innovative Multi-Epitope mRNA Vaccine
Supplemental material, sj-pdf-2-bbi-10.1177_11779322241272404 for Proteomics Exploration of Brucella melitensis to Design an Innovative Multi-Epitope mRNA Vaccine by Maryam Asadinezhad, Iraj Pakzad, Parisa Asadollahi, Sobhan Ghafourian and Behrooz Sadeghi Kalani in Bioinformatics and Biology Insights
Supplemental Material
sj-pdf-3-bbi-10.1177_11779322241272404 – Supplemental material for Proteomics Exploration of Brucella melitensis to Design an Innovative Multi-Epitope mRNA Vaccine
Supplemental material, sj-pdf-3-bbi-10.1177_11779322241272404 for Proteomics Exploration of Brucella melitensis to Design an Innovative Multi-Epitope mRNA Vaccine by Maryam Asadinezhad, Iraj Pakzad, Parisa Asadollahi, Sobhan Ghafourian and Behrooz Sadeghi Kalani in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
We would like to thank the Students Research Committee (IR.MEDILAM.REC.1402.139), Ilam University of Medical Sciences, for their cooperation. Our appreciation goes to the Vice Chancellor for Research Affairs, Ilam University of Medical Sciences, Ilam, Iran, for their executive and financial support. Also, we appreciate Nazanin Omidi who assisted us in revising the revised version.
Author Contributions
The methodology was carried out by Maryam Asadinezhad. The results were analyzed and confirmed by Iraj Pakzad. Parisa Asadollahi revised the article draft. The analysis of the docking and visualization results was conducted by Sobhan Ghafourian. This study was designed, drafted, and supervised by Behrooz Sadeghi Kalani.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Ilam University of Medical Sciences, Ilam, Iran.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.