
Abstract
The mitigation of cadmium (Cd) pollution, a significant ecological threat, is of paramount importance. Pseudomonas aeruginosa harbors 2 Cd resistance genes, namely, cadR and cadA. Presently, our focus is on the identification and characterization of the cation-transporting P-type ATPase (cadA) in Pseudomonas aeruginosa BC15 through in silico methods. The CadA protein and its binding capacities remain poorly understood, with no available structural elucidation. The presence of the cadA gene in P aeruginosa was confirmed, showing a striking 99% sequence similarity with both P aeruginosa and P putida. Phylogenetic analysis unveiled the evolutionary relationship between CadA protein sequences from various Pseudomonas species. Physicochemical analysis demonstrated the stability of CadA, revealing a composition of 690 amino acids, a molecular weight of 73 352.85, and a predicted isoelectric point (PI) of 5.39. Swiss-Model homology modelling unveiled a 33.73% sequence homology with CopA (3J09), and the projected structure indicated that 89.3% of amino acid residues were situated favourably within the Ramachandran plot, signifying energetic stability. Notably, the study identified metal-binding sites in CadA, namely, H3, C30, C32, C35, H48, C89, and C106. Docking studies revealed a higher efficiency of Cd binding with CadA compared to other heavy metals. This underscores the crucial role of N-terminal cysteine residues in Cd removal. It is evident that CadA of P aeruginosa BC15 plays a crucial role in Cd tolerance, rendering it a potential microorganism for Cd toxicity bioremediation. The structural and functional elucidation of CadA, facilitated by this study, holds promise for advancing cost-effective strategies in the remediation of cadmium-contaminated environments.
Graphical abstract

Introduction
Heavy metals pose a serious threat to the environment, originating from diverse sources such as industrial discharge, refineries, and waste treatment facilities, as well as indirect pathways like soil, groundwater, and atmospheric deposition via rainfall.1-6 Notable among these pollutants are cadmium (Cd), lead (Pb), and mercury (Hg), which exhibit toxicity even at low concentrations.7,8 Among them, Cd stands out as a significant global hazard,9-11 presenting severe health risks to both human and wildlife populations. Its accumulation in plants leads to various physiological, biochemical, and structural alterations, affecting processes such as mineral nutrient absorption, photosynthesis, and carbohydrate metabolism, along with changes in antioxidant mechanisms.12,13 Therefore, effective environmental Cd removal methods are imperative, given the associated health risks and ecological implications. Current Cd remediation techniques include chemical precipitation, oxidation-reduction processes, ion-exchange, filtration, electrochemical treatments, reverse osmosis, evaporative recovery, and solvent extraction. However, these conventional methods face challenges such as unpredictable metal ion removal and the generation of potentially hazardous sludge. Therefore, there is a pressing need to develop more efficient, sustainable, and environmentally friendly Cd removal methods.14,15
Bioremediation offers an alternative approach utilizing natural and engineered microorganisms to reduce or eliminate hazardous contaminants, including Cd.3,16,17 This method is recognized for its cost-effectiveness and environmentally friendly nature. Bacteria play a crucial role in bioremediating toxic metal ions. Particularly, Pseudomonas aeruginosa demonstrates notable Cd tolerance and applications in removing Cd and other heavy metals from various environments such as water, soil, contaminated waste, and solutions. P aeruginosa, known for its resilience and adaptability, employs diverse mechanisms to resist Cd toxicity. 18 Several studies have highlighted its ability to withstand Cd in different environmental conditions.3,16,17,19 These mechanisms include efflux pumps that actively expel Cd ions from the cell, reducing their intracellular accumulation, and utilizing metallothioneins, cysteine-rich proteins, to bind and sequester Cd ions, thereby minimizing their harmful effects. For instance, strains like P aeruginosa KUCD1 and san ai 20 can tolerate high Cd concentrations, up to 8 mM,3,18 and 7.2 mM respectively, in nutrient agar medium at a pH of 7.2. Some P aeruginosa species produce pyoverdine (PVD) under Cd stress, potentially aiding in reducing Cd accumulation in plants. In addition, P aeruginosa PU21 biomass effectively removes Cd, Cu, and Pb from contaminated water. Similarly, the non-viable biomass of P aeruginosa and strains isolated from active sludge exhibit robust adsorption abilities for Cd and Pb in aqueous solutions. 3 Furthermore, adapted cells of P aeruginosa and genetically engineered strains have been found effective in Cd removal.
Notably, P aeruginosa BC15, an environmental isolate, displays resistance to Cd and can biosorb Cd as well as other metals like Pb, Ni, and Cr. Induction studies on P aeruginosa BC15 using sub-lethal Cd concentrations have revealed the development of adaptive resistance to lethal Cd doses, further highlighting its potential in bioremediation efforts.3,18 This adaptive capability highlights the resilience of P aeruginosa BC15 in confronting heavy metal stress. In addition, previous research underscores the efficacy of P aeruginosa BC15 in mitigating Cd and Zn contamination in industrial wastewater and soil.3,18 The strain demonstrated notable resistance to these metals and exhibited substantial biosorption capacity, with Cd absorption reaching 82% and Zn reaching 85% in binary solutions. Furthermore, analysis of the Cd resistance (cadR) gene in P aeruginosa BC15 revealed its similarity to other Pseudomonas strains. Cloning cadR into E coli BL21 facilitated robust growth in Cd-supplemented media, indicating its potential for heavy metal remediation. Moreover, live cells of Pseudomonas BC15 displayed the ability to biosorb Cd, as well as other metals like Pb, Ni, and Cr, showcasing its versatility in metal detoxification. Overall, these findings highlight the potential of P aeruginosa BC15 as a valuable tool in addressing heavy metal contamination in the environment.
In the intricate network of Cd resistance mechanisms in bacteria like P aeruginosa, CadA stands as a frontline defender, orchestrating a timely response to Cd exposure. 21 CadA is a membrane-bound efflux pump that actively expels Cd ions from the bacterial cell, reducing intracellular accumulation and mitigating toxicity.3,18,22 Its role extends beyond direct Cd efflux; it also triggers the induction of the CzcCBA efflux system, crucial for extruding multiple heavy metal ions from the bacterial cytoplasm, including Zn, Cu, and Cd. This coordinated response ensures the swift removal of Cd ions from the cellular milieu, safeguarding cellular integrity and functionality. P aeruginosa possesses chromosomally encoded Cd resistance facilitated by 2 inversely transcribed genes, cadR and cadA. CadR, sourced from P aeruginosa, encodes a transcriptional regulatory protein that exhibits a robust response to Cd(II), followed by Zn(II) and Hg(II), at its cognate promoter P cadA . CadA shares structural similarities with Cd-transporting ATPases identified in Gram-positive bacteria. 3 Functionality studies of CadA have referenced E coli ZntA sequences.18-20 Recent work by Kumari et al 23 unveiled the role of metal bioremediation mediated by CadC and CadA in the B megaterium strain YC4-R4.
Furthermore, CadA plays a vital role in protecting bacterial cells from sudden surges in Zn levels, acting as a primary defense mechanism against Zn toxicity. The regulation of CadA expression is tightly controlled by the CadR transcriptional regulator, which orchestrates the cellular response to heavy metal exposure.18,24 In P aeruginosa, the Cation-transporting P-type ATPases (CadA) protein is encoded by the cadA gene, often located on genetic elements associated with heavy metal resistance, such as plasmids or genomic islands. 18 This CadA protein serve as key players in cellular ion homeostasis and heavy metal resistance mechanisms. Understanding their function is essential for elucidating bacterial adaptation to metal-contaminated environments and developing strategies for environmental remediation and antimicrobial therapy. Research on CadA in P aeruginosa, a Cd resistance determinant, is essential due to its significance in understanding bacterial adaptation to heavy metal stress. Understanding the genomic context of the cadA gene is crucial for elucidating the regulatory networks and genetic pathways involved in Cd detoxification. Genomic analyses have identified neighbouring genes associated with Cd resistance or related cellular processes, providing insights into the genetic architecture of Cd tolerance in P aeruginosa. 25
Research on cation-transporting P-type ATPases sheds light on fundamental cellular processes and microbial adaptation to environmental challenges. These ATPases, crucial for mediating resistance to heavy metal ions like Cd, Cu, and Zn, offer avenues for combating antimicrobial resistance, enhancing metal bioremediation, and engineering microorganisms with improved metal tolerance. Understanding the structural basis and mechanisms governing CadA’s cadmium binding affinity is vital for these advancements, holding promise for innovation in medicine, biotechnology, and environmental remediation.23,26 Recent research efforts have increasingly focused on computationally characterizing the active sites of proteins.27,28 This has led to the development of online resources dedicated to locating, delineating, and quantifying concave surface regions within 3D protein structures, enabling differentiation of metal resistance proteins. Physico-chemical attributes, including atom count, molecular weight, isoelectric point, and conserved domains, can be readily assessed.28,29 In addition, homology modelling and comparative proteomics approaches have been extensively employed to forecast the 3D structure, binding sites, and potential functionalities of metal regulatory proteins.30,31 However, the CadA protein and its binding capacity remain inadequately understood, with no prior reports elucidating its functional and structural attributes, including interactions with metals and metal complexes. Hence, this study endeavours to employ in silico methodologies for the identification and characterization of the 3D structures, metal binding proficiency, and removal mechanism of the cation-transporting P-type ATPase (CadA) in Pseudomonas aeruginosa BC15.
Experimental Section
Target sequence retrieval and phylogenetic analysis
The sequences for both nucleotide and protein of the CadA protein were obtained from the National Center for Biotechnology Information (NCBI) databases. 32 It was observed that the cadA gene was positioned upstream of the cadR promoter. To confirm this arrangement, the cadR gene (KU302252) was submitted to NCBI for further analysis, leading to the successful identification of the cadA gene. Subsequently, the specific nucleotide sequence of the cadA gene was retrieved from the NCBI database (http://www.ncbi.nlm.nih.gov/) by the accession number KU302252. Furthermore, the 2-dimensional structure of the heavy metals was acquired from the PUBCHEM database in SDF file format.
Investigation of template
The CadA protein sequences obtained were analyzed using the BLASTp algorithm within the standard protein BLAST suite. To conduct structural and functional analysis, both the CadA protein and gene sequences were sourced from the SWISS-PROT database. Sequences exhibiting noteworthy similarity were aligned through the ClustalW algorithm, integrated into the Molecular Evolutionary Genetic Analysis (MEGA 6) software, accessible at http://www.megasoftware.net. The alignment was further refined to generate a consensus sequence based on distance matrix analysis.
Examination of physicochemical characterization
Using the ProtParam tool, which is available at http://web.expasy.org/protparam/, the CadA protein in P aeruginosa BC15 underwent physicochemical characterization, including analysis parameters such as isoelectric point (pI), molecular weight, atom count, instability index, aliphatic index, and grand average of hydropathicity (GRAVY). Using CELLOv.2.5, subcellular localization was predicted. The PSIPRED server was used for secondary structure analysis, and the PRINTS server was used for fingerprint analysis.
Evaluation of CadA protein CDD and motif
The motif search tool (http://www.genome.jp/tools/motif) was employed to identify the conserved region within the CadA protein. In addition, the functional unit of the protein was annotated by the conserved domain database (http://www.ncbi.nlm.nih.gov). The secondary structure predictions were conducted by PDBsum (www.ebi.ac.uk/pdbsum), SOPMA server (http://npsa-prabi-ibcp.fr/sopma.html), and Phyre2 (www.sbg.bio.ic.ac.uk/phyre2). Protein-protein interactions were assessed using the STRING server.
Validation of CadA protein 3D structure modelling
The cadA gene encoded CadA protein sequences were retrieved from P aeruginosa (Uniprot ID: A0A0H2ZF06) in FASTA format and subjected to BLASTp evaluation against the PDB database for the identification of the suitable template for modelling. The SWISS MODEL tool, Phyre2, and RaptorX servers were used to predict the 3D (3-dimensional) structure of CadA protein. The obtained model was visualized by SWISS MODEL (http://www.expasy.org/spdbv). The CadA protein 3D structure model quality was assessed using PyMOL, further validation was conducted by the Ramachandran lot analysis. QMEANZ score was calculated to evaluate and refine the predicted CadA protein model.
Analysis of the metal protein interactions by docking using PatchDock
In order to investigate interactions between metal-protein complexes, 2-dimensional structures of heavy metals (arsenate, cadmium, chromium, cobalt, copper, zinc, lead, silver, and nickel ions) were obtained from the PUBCHEM database in SDF file format. These structures were then converted to PDB file format using an online molecular converter tool (http://cactus.nci.nih.gov/translate). Docking analyses of the 3D structures were performed using the PatchDock tool, a molecular modelling simulation software known for its effectiveness in protein-ligand docking studies.
Results
Retrieval of target sequences (cadA gene) from Pseudomonas aeruginosa
The cadA gene sequence of P. aeruginosa BC15 was identified using cadR gene sequences. The cadA gene sequence was obtained in FASTA format with an accession number of NC_002516 (Figure S1A). Subsequently, the cadA gene sequence was subjected to the ExPASy Translate tool (https://web.expasy.org/translate/), resulting in the protein sequence AVZ34575.1, as shown in Figure S1B & C.
BLAST analysis of CadA protein from P aeruginosa BC15
The results of the BLAST analysis of the cadA gene from P aeruginosa BC15 revealed a high degree of similarity with other Pseudomonas species. The analysis indicated that the cadA gene from P aeruginosa BC15 had a 100% score and 98% identity with the CadR and CadA genes from P putida HB3267, a 100% score and 96% identity with the genes from P monteilii SB3101 and SB3078, a 100% score and 96% identity with the genes from Pseudomonas sp. DRA525, and a 100% score and 96% identity with the genes from P putida and P plecoglossicida (Figure S2). The BLAST analysis of the CadA protein from P. aeruginosa BC15 also revealed a high degree of similarity with other Pseudomonas species. The analysis indicated that the CadA protein from P. aeruginosa BC15 had a 100% score and 99.86% identity while the CadA protein from Pseudomonas sp., and a 100% score and 99% identity (Table 1).
BLAST analysis of cadA gene from Pseudomonas aeruginosa BC15.

Phylogenetic distribution and evolutionary relationship of cadA gene and CadA protein
The phylogenetic trees were constructed by Neighbour-joining method using cadA gene and its protein sequences of Pseudomonas aeruginosa. The phylogenetic tree of cadA gene and its protein sequences showed the evolutionary relationship between strains Pseudomonas genera (Figure 1).

Phylogenetic analysis of the cadA gene and its protein across Pseudomonas species and gram-positive bacteria. (A) Evolutionary relatedness of the cadA gene sequences from Pseudomonas aeruginosa (highlighted) compared to homologous sequences from other Pseudomonas species and gram-positive bacteria. The phylogenetic tree demonstrates the genetic divergence and common ancestry among these cadA gene sequences. (B) Phylogenetic tree depicting the evolutionary relationship of the CadA protein sequences from P aeruginosa (highlighted) in comparison to related proteins from other Pseudomonas species and gram-positive bacteria. The analysis highlights the conservation and divergence of CadA protein sequences across these bacterial taxa.
Physicochemical parameters
The physicochemical parameters were analysed using the ProtParam tool. The CadA protein was predicted to consist of 690 amino acids with a molecular weight of 73 352.85 Daltons and a theoretical isoelectric point (pI) of 5.39. The amino acid composition revealed that alanine comprised the highest percentage at 14.8%, followed by leucine at 13.8%, arginine at 6.7%, and other amino acids occurring in less than 9% abundance. A total of 80 residues were negatively charged, while 66 residues were positively charged. The instability index (II) was calculated to be 31.60, and the aliphatic index was determined to be 113.20. The grand average of hydropathicity (GRAVY) was found to be 0.241 (Table 2). Subcellular localization analysis predicted the target CadA protein to be present in the cytoplasm (value 2.493) as determined by CELLOv2.5. Secondary structure analysis of the CadA protein identified parameters such as alpha helix and random coil using PSIPRED (Figure S3B). The fingerprint predictor from the PRINTS server revealed that the CadA protein contains 2 fingerprints: CATATPASE, which consists of 5 out of 6 motif numbers, and CDATPASE, which contains 3 out of 9 motifs.
The physicochemical properties of the CadA protein.

Conserved domain and motif analysis
The 5 conserved domains/functional motifs in CadA were analysed using Motif Finder (Figure S3A). The aligned sequences of a consanguineous protein family were examined using the SOPMA algorithm. Each sequence line was visually labelled with colour-coded predictions for helix, sheet, turns, and coils. The proportions of alpha helix and beta strand were determined by SOPMA using a homology-based technique. The identification of beta turns, important secondary structure components, greatly aided in the formation of an interim stage. These beta turns were also observed in the secondary structure, contributing to the formation of an intermediate structure that eventually folds into a 3-dimensional structure (Figure S4). Protein-protein interactions of the CadA protein were evaluated using the STRING database. The resulting coloured lines indicate different sources of evidence for each interaction within the proteins (Figure S5).
Conserved domain analysis by Clustal Omega
The other MerR cation transport ATPase proteins of Pseudomonas sp. were analysed through multiple sequence alignment. Clustal Omega was utilized for aligning the selected protein sequences. The multiple sequence alignment results revealed the conservation of amino acid residues from the Pseudomonas sp. cadA gene (Figure 2). Hydrophobic amino acids are represented in green, polar ones in blue, aliphatic residues in black, and positively charged amino acids in red.

Comparative analysis and quality assessment of CadA protein: insights from sequence alignment and QMEAN evaluation. (A) Consensus of cadA gene from Pseudomonas species. (B) The figure displays the sequence alignment results between the template protein E coli CopA (PDB code 3J09) and the target protein CadA. The alignment was performed using standard bioinformatics tools and algorithms, highlighting regions of similarity (identical or similar amino acids) and gaps (insertions or deletions). The percentage of sequence identity, indicating the proportion of identical amino acids between the 2 sequences, is provided. Higher sequence identity suggests a closer evolutionary relationship and potentially similar functions between the proteins. The alignment results provide insights into the structural and functional similarities between E coli CopA and CadA, aiding in understanding their roles in cellular processes. (C) Quality Model Energy ANalysis (QMEAN) Quality assessment of CadA protein. The QMEAN assessment of the CadA protein yielded a score of -4.91, falling within the standard range of 0 to 1. The evaluation indicates that the predicted model generally exhibits satisfactory quality. While certain residue clusters scored as low as 0.1, implying lower quality, the majority of amino acid patches displayed higher values exceeding 0.6, suggesting the accurate and stable behavior of the CadA protein. Notably, the blue peak in the assessment chart represents the highest prediction value, indicating superior quality results (> 0.85) for the predicted model. In addition, the purple regions also demonstrated good accuracy, with scores ranging from 0.6 to 0.85. Conversely, the orange areas indicated less stable behavior, with scores below 0.6. Overall, the evaluation suggests that the predicted CadA model exhibits satisfactory stability and warrants further analysis.
Homology modelling was employed using the SWISS-MODEL software to construct the 3D structure of CadA. The primary sequence of CadA was obtained from UniProt, and template searching was conducted using SWISS-MODEL software. Figure 2B illustrates the selection process, where the crystallographic structure of E coli CopA (PDB code 3J09), resolved via X-ray method at a resolution of 1.30 Å, was chosen as the template due to its highest sequence identity of 33.73% with the target sequence among 50 given templates. The final 3D structure was obtained from the SWISS-MODEL software and utilized for subsequent docking studies. Overall, the evaluation suggests that the predicted CadA model exhibits satisfactory stability and warrants further analysis (Figure 2C).
Phyre2
In the Phyre results, matching residues between the query and template are shaded with a gray background. Predicted and confirmed secondary structures are delineated as α-helices. Conservation levels for both the query (CadA) and template sequences are represented by horizontal bars, with thicker bars denoting greater conservation. The ‘Conservation’ rows classify residue conservation into 3 states. No symbol indicates lack of conservation, a thin gray bar suggests moderate conservation, and a large block signifies a high degree of conservation. Details regarding confidence lines can be found in the Secondary Structure and Disorder section. Alpha helices are depicted in green, beta strands in blue, and random coil with faint lines. The ‘SS confidence’ line indicates the level of confidence in the prediction, with red signifying high confidence and blue indicating lower confidence. Phyre generates a series of 3D protein models through structural alignment. In addition, it identifies sequence homologues using PSI-BLAST (Figure 3).
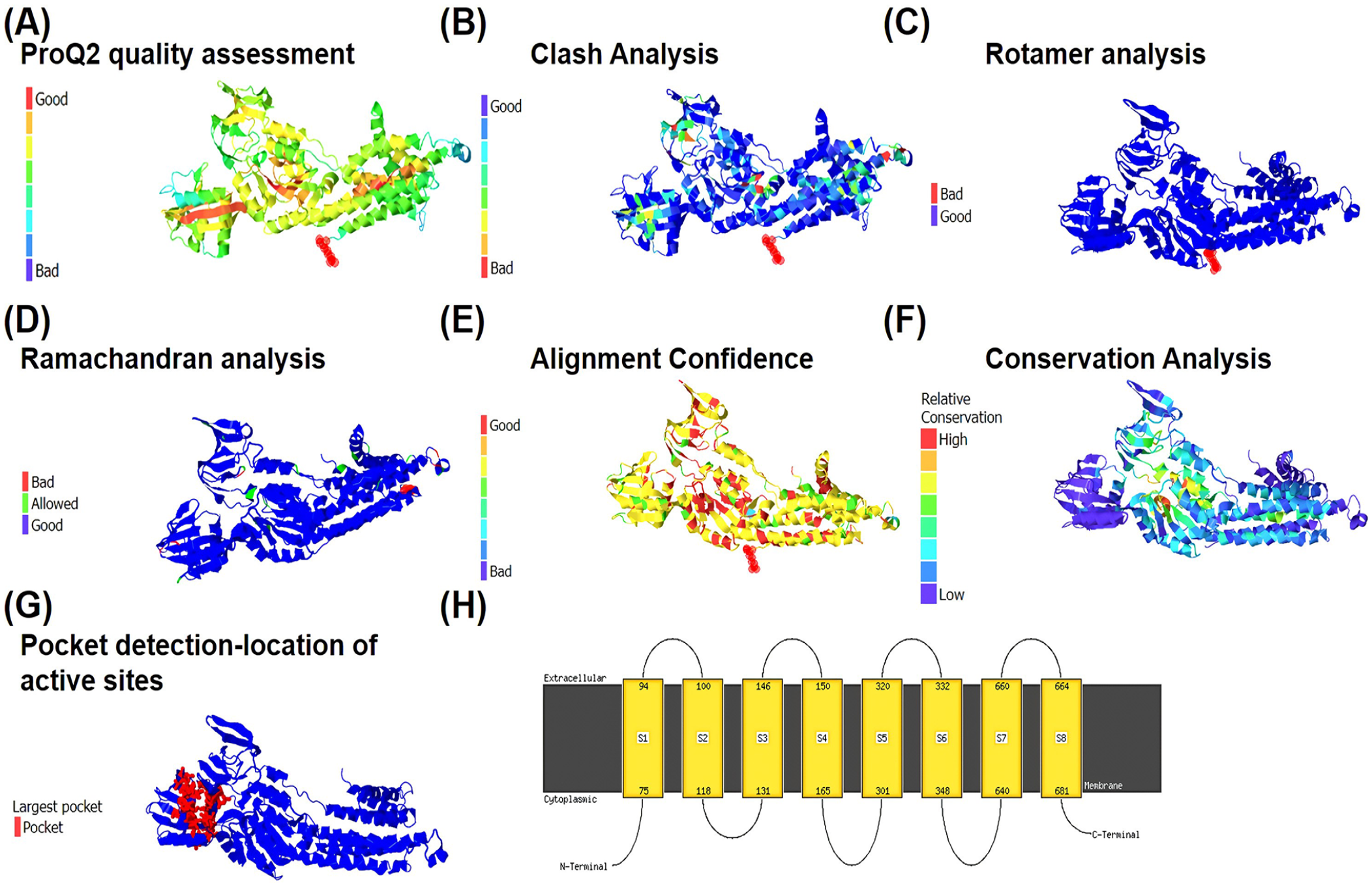
Validation of CadA protein 3D structure by Phyre2. (A) Model quality assessment by ProQ2. The colour gradient indicates the reliability of the model, with red regions representing areas of low confidence and blue regions indicating higher confidence. (B) Clash analysis of CadA protein. The colour gradient represents clash severity, where blue regions denote minimal clashes and red regions signify significant clashes. (C) Analysis of rotamers. The colour gradient reflects the quality of rotamer conformations, with blue regions indicating well-packed and favourable conformations, and red regions representing unfavourable conformations. (D) Ramachandran analysis for CadA protein. The colour gradient represents the distribution of phi-psi angles, with blue regions indicating favourable backbone conformations and red regions representing outliers. (E) Alignment confidence from HHsearch. The colour gradient is reversed, with red regions indicating higher confidence in the alignment and blue regions representing lower confidence. (F) Detection of sequence features using the Conserved Domain Database (CDD). The colour gradient signifies conservation levels, with red regions indicating highly conserved regions and blue regions representing less conserved areas. (G) Pocket detection by fpocket2. The largest pocket of the active site is highlighted in red. (H) Transmembrane helices of CadA protein. 615 residues (89% of CadA sequence) have been modelled with 100.0% confidence by the single highest-scoring template.
Model validation, energy minimization, and 3-dimensional structure modelling
Homology modelling was conducted using SWISS-MODEL. The sequence of the CadA protein underwent a BLASTp search against the PDB database to identify the closest matching crystal structure, which was then utilized as a template for modelling the target protein. The SWISS-MODEL server identified the PDB ID 3J09 (CopA protein) as a suitable template with 33.73% identity (Figure S6). The examination of the Ramachandran plot revealed that 89.3% of the residues occupied the favoured (red) region, with 7.3% falling within the allowed (blue) region, while the remaining 3.4% of residues were situated in either the generously allowed (yellow) or outlier region. The presence of over 89.3% of residues in the favoured region suggests the acceptability of the model (Figure 4A). The QMEAN value was determined to be -4.91 (Figure 4B and C). The structure was visualized, and docked complexes were analysed using PyMOL (Figure 4D).
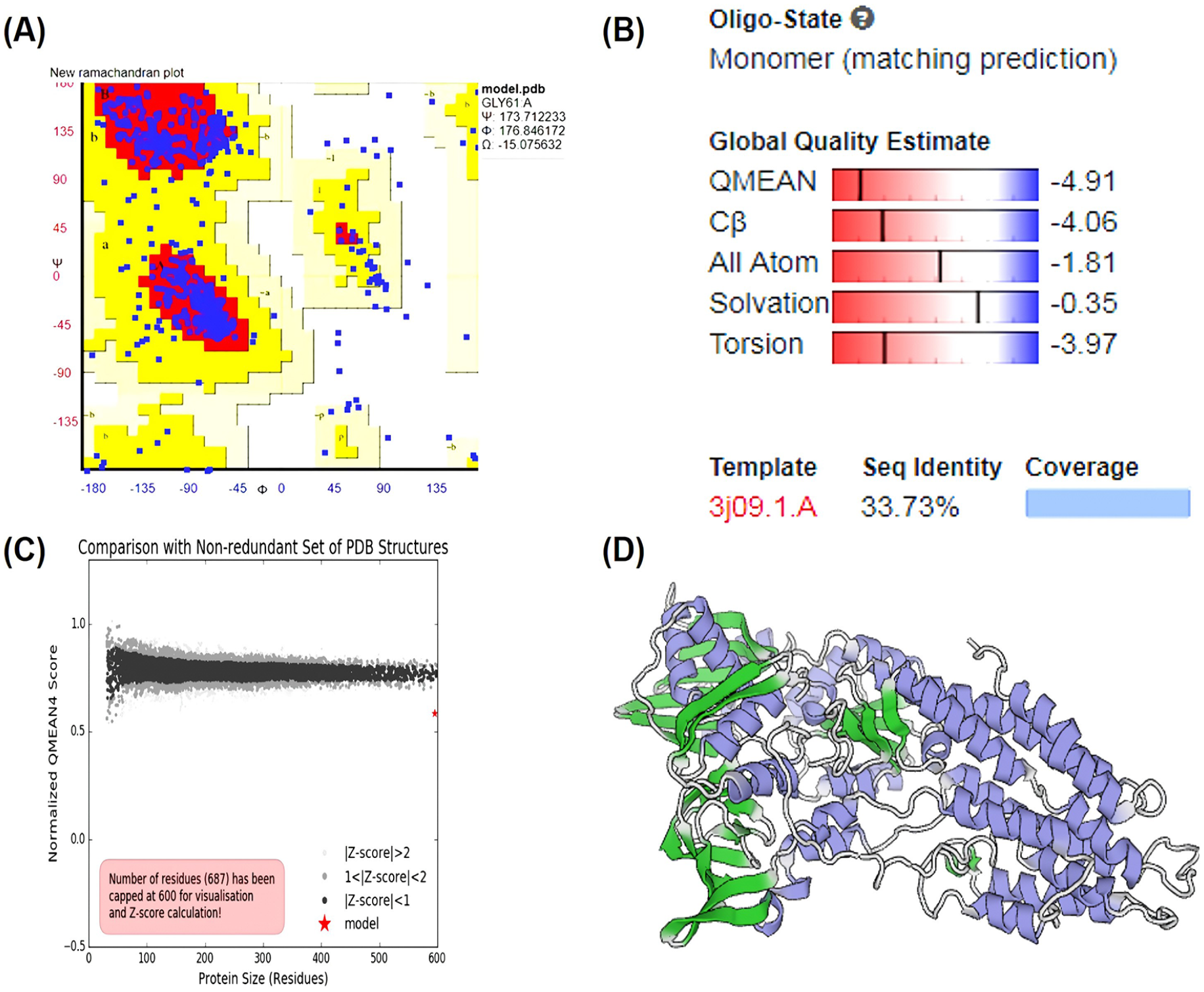
Energy minimization and validation of the model. (A) The Ramachandran plot illustrates the distribution of psi/phi values for the CadA protein. The colour legend denotes regions of varying favourability: red for the most favoured, yellow for allowed, pale yellow for generously allowed, and white for disallowed regions. (B) A graph compares the protein size with a non-redundant set of PDB structures, normalized for analysis. (C) The QMEAN4 score provides an assessment of the model’s quality, indicated by a red star. Models with Z-scores exceeding 2 are shaded in light gray. The red star represents the model. (D) The dimeric model of the CadA protein from Pseudomonas aeruginosa is depicted in cartoon form. The DNA and metal binding domains are also annotated. The 3D structure of the CadA protein model was visualized using PYMOL.
Docking analysis
The docking analysis, 3D structure of CadA protein was carried out by PatchDock. The 2D structures of heavy metal ions including Cadmium, Arsenic, Chromium, Cobalt, Copper, Nickel, Lead, Silver, Zinc, and Cadmium chloride were acquired from PUBCHEM in SDF format. Subsequently, they were transformed into PDB file format using an online molecular conversion tool at http://cactus.nci.nih.gov/translate/. (Figure S8). Target protein 3D structure of CadA bound Cd complex was visualized in PyMOL (Figure S7). Figure 5 show the protein-ligand interaction of CadA and heavy metals complexes. The binding of Cd with the E101 and H96 residues in the catalytic site indicated that the CadA protein is responsible for Cd detoxification in P aeruginosa. The result of the PatchDock analysis also supported that CadA has higher affinity to CdSO4 than other heavy metals based on the Atomic contact energy (ACE), Score and area values such as, -58.03 Kcal/Mol, 1702, 172.70 respectively (Table 3). PatchDock values of other heavy metals binding with CadA are shown in Figure 5.

Illustrates the results of PatchDock analysis conducted to examine the interaction between the CadA protein and various heavy metals, including cadmium, cadmium sulfate, arsenate, copper, chromium, cobalt, lead, nickel, silver, and zinc. (A) displays the predicted binding areas of the CadA protein with each heavy metal. (B) presents the binding efficiency scores of the CadA protein with the aforementioned heavy metals. (C) These analyses provide insights into the potential affinity and interaction strength between the CadA protein and different heavy metal ions, which are crucial for understanding the protein’s role in metal binding and detoxification processes.
CadA binding efficiency with heavy metals.

Discussion
Heavy metal contamination is a severe problem that results in the deterioration of human health. Specifically, cadmium (Cd) is an extremely toxic heavy metal for the environment, causing diseases such as itai-itai disease, cancers, osteoporosis, lung disease, and atherosclerosis in human.1,29,30 Studies have reported that Cd contamination in soil and drinking water has increased several times in many parts of the world. To control and remediate Cd contamination, a number of chemical and traditional methods are used. However, these methods have major disadvantages; they are more expensive and produce secondary compounds that are even more toxic.31,32
Therefore, an alternative biological method of Cd bioremediation is needed. The expression of Cd tolerance genes, under the regulation of CadA proteins, confers tolerance to Cd. In particular, Pseudomonas aeruginosa, a Cd-resistant bacterium isolated from industrial effluent wastewater, plays a major role in Cd removal using cadA and cadR genes, which encode P-type ATPase (CadA) and transcript regulatory protein (CadR). Moreover, P aeruginosa exhibits elevated Cd resistance capabilities, nearly 7.2mM
The identified and retrieved cadA gene sequence was used for NCBI BLAST analysis. Local alignment (BLASTp) and global alignment (Clustal Omega) were utilized to determine both near and distant relationships of the CadA protein with other species. BLASTp identified segments of the sequence that are conserved among other organisms, while Clustal Omega provided a more detailed analysis, displaying similarities among analogous sequences of amino acids of each compared protein. The sequence alignment of CadA from P aeruginosa BC15 clearly depicted conserved residues in other organisms. Conservation results obtained from Clustal Omega revealed highly conserved amino acid sequences with gap-free regions. In addition, a phylogenetic tree was constructed for the cadA gene-encoded CadA protein from P aeruginosa BC15, based on amino acid sequences, revealing the evolutionary relationship of Pseudomonas sp., which is consistent with earlier reports documented in the NCBI database.
Physicochemical properties were analysed using the ProtParam tool. The analysis revealed that the CadA protein contains 14% alanine, 6.7% arginine, and other amino acids occurring in minimal percentages. Conserved amino acid regions were identified through motif analysis, employing the Conserved Domain Database search targeting sequences, superfamilies, and functional sites. Results from the PRINTS server indicated that CadA contains 2 fingerprints: CATATPASE, which has 5 out of 6 motifs, and CDATPASE, which has 3 out of 9 motifs. Secondary structure analysis performed by the SOPMA server showed 51.52% (355/689) alpha helix, 7.11% beta turn, 27.43% random coil, and 13.93% extended strand. These findings are consistent with the results reported by Rajkumar et al. 18
The research underscores the importance of understanding the 3-dimensional structure of proteins, particularly in fields like enzyme kinetics, ligand-protein interactions, and structure-based molecular compound design.18,20 By employing homology modelling techniques,33-37 the study successfully constructed the 3D structure model of the CadA protein using 3J09 as a template structure. Validation of the CadA protein structure through RAMPAGE revealed that 98% of its amino acid residues were located in the most favoured region, indicating the reliability of the model. This aligns with findings from Rajkumar et al, 18 which reported similar validation results for protein models.
In the context of bioremediation, metal sulfide precipitation emerges as a superior technique due to several advantages over conventional methods such as hydroxide, carbonate, or phosphate precipitation. It not only reduces sludge volume but also forms compounds with lower solubility products, enabling the recovery of valuable metals. Among the heavy metals targeted for remediation, cadmium sulfate (CdSO4) is particularly noteworthy for its stability and insolubility, making it an ideal candidate for remediation efforts. CdSO4 finds extensive use in various industries, including electroplating, pigments, batteries, and catalysis, underscoring its significance. However, the potential hazards associated with CdSO4 exposure, as evidenced by studies by Yorulmazlar et al 37 and Yin et al, 38 have highlighted the toxic effects of CdSO4 exposure, particularly when combined with other substances like α-naphthoflavone (ANF), leading to increased oxidative stress and down-regulation of protective genes in zebrafish embryos. These findings underscore the urgent need for effective bioremediation strategies targeting cadmium sulfate contamination to mitigate its adverse effects on the environment and human health. To address this need, PatchDock analysis was employed to predict the binding ability of the target CadA protein with different heavy metals, including arsenite, arsenate, arsenic, zinc, cadmium, copper, lead, cobalt, and silver. The analysis revealed that the CadA protein exhibits a higher binding affinity to Cd and CdSO4 compared to other heavy metals, with a binding score of -58.03 kcal/mol (ACE). Correspondingly, Bai et al 39 found out that most of the cadmium was removed by making cadmium sulfide through bioprecipitation, and only a little was taken out by biosorption.
In our study, we investigated the amino acid composition of the CadA protein, revealing notable proportions of alanine (A) and arginine (R), among others. These amino acids likely play critical roles in CadA’s interaction with cadmium sulfate. Specifically, alanine’s aliphatic nature facilitates hydrophobic interactions with metal ions, while arginine’s positively charged side chain enables electrostatic interactions with negatively charged groups in cadmium sulfate. Moreover, the physicochemical properties of CadA provide insights into its structural stability, which is essential for metal binding processes. As a P-type ATPase, CadA is implicated in active transport across membranes, potentially including the transport of Cd ions. This suggests its involvement in cadmium sulfate removal or detoxification within the cell. Furthermore, the identification of conserved regions and domains within CadA, along with its evolutionary conservation, underscores its significance in metal tolerance mechanisms across species. Thus, CadA likely plays a pivotal role in binding and removing cadmium ions, including those from cadmium sulfate, thereby aiding in heavy metal stress mitigation. The observed higher affinity of CadA from Pseudomonas for CdSO4 compared to Cd ions holds implications for cellular function and bioremediation strategies. Given CdSO4’s stability and insolubility, its efficient removal poses a significant challenge. However, CadA’s preferential binding to CdSO4 suggests a specialized mechanism for Cd sequestration and detoxification within the cell, potentially offering a targeted approach to remediation. Leveraging CadA’s cadmium-binding capabilities could lead to innovative bioremediation technologies tailored for CdSO4 removal, thereby advancing environmental cleanup efforts and mitigating contamination risks. Overall, CadA’s affinity for CdSO4 represents an intriguing adaptation with promising implications for both cellular function and environmental remediation, warranting further exploration of its molecular mechanisms and biotechnological applications.
Conclusions
In conclusion, this study identifies the CadA gene from Pseudomonas aeruginosa BC15, characterizing its high homology with related proteins and evaluating its physicochemical parameters and structural stability. Molecular docking studies reveal CadA’s preference for binding cadmium, highlighting its potential role in Cd tolerance mechanisms and bioremediation. This work contributes to understanding CadA’s 3D structure and its affinity for metal cations, paving the way for future research on Cd monitoring and environmental remediation strategies.
Supplemental Material
sj-doc-1-bbi-10.1177_11779322241266701 – Supplemental material for Elucidation of the CadA Protein 3D Structure and Affinity for Metals
Supplemental material, sj-doc-1-bbi-10.1177_11779322241266701 for Elucidation of the CadA Protein 3D Structure and Affinity for Metals by Rajkumar Prabhakaran and Rajkumar Thamarai in Bioinformatics and Biology Insights
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
T.R.K. conducted the experimental work, while P.R.K., as the corresponding author and research team supervisor, oversaw the experimental design, compilation, and revision of the full-length article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.