
Abstract
Soil contamination by hydrocarbons due to oil spills has become a global concern and it has more implications in oil producing regions. Biostimulation is considered as one of the promising remediation techniques that can be adopted to enhance the rate of degradation of crude oil. The soil microbial consortia play a critical role in governing the biodegradation of total petroleum hydrocarbons (TPHs), in particular polycyclic aromatic hydrocarbons (PAHs). In this study, the degradation pattern of TPHs and PAHs of Kuwait soil biopiles was measured at three-month intervals. Then, the microbial consortium associated with oil degradation at each interval was revealed through 16S rRNA based next generation sequencing. Rapid degradation of TPHs and most of the PAHs was noticed at the first 3 months of biostimulation with a degradation rate of pyrene significantly higher compared to other PAHs counterparts. The taxonomic profiling of individual stages of remediation revealed that, biostimulation of the investigated soil favored the growth of Proteobacteria, Alphaprotobacteria, Chloroflexi, Chlorobi, and Acidobacteria groups. These findings provide a key step towards the restoration of oil-contaminated lands in the arid environment.
Implications for Practice
This study has clear implications in developing strategies to enhance the efficiency of bioremediation of oil-contaminated soil in the arid regions using a climate-adapted bacterial consortium.
The identified microbial taxa represent a bacterial population which is tolerant to highly toxic PAHs and resilient to an environment prevailing in the weathered crude oil in an extremely arid environment.
This knowledge can be further utilized for applying microbial-based remediation strategy for the restoration of landscapes characterized by the presence of aged oil-contaminated soil in the Arabian Gulf region.
Introduction
Petroleum pollution causes serious threats to the ecological environment. Long-term soil pollution by hydrocarbons led to environmental deterioration through damaging the soil structure, altering plant growth and food chains, putting at risk animal, and human health. 1 Arabian Gulf region represents one of the biggest area engaged in oil production and refining. This region produces more than 50% of the marine transported oil in the world. 2 As a result, hydrocarbons are widespread in soil due to the accidental spills during the petroleum processing, transport, storage, and leakage of oil pipelines posing constant threat of contamination. This massive scale of crude oil contamination is responsible for causing imbalance in the fragile ecosystem and it is of great public concerns. 3 The desert areas of Kuwait contain high level of oil contamination up to 20% or more 4 resulting in reduced diversity of organisms in this ecosystem.
The crude oil mainly consists of aliphatic, alicyclic, as well as aromatic hydrocarbons (ie, polycyclic aromatic hydrocarbons (PAHs)). These hydrocarbon molecules exhibit great variability in terms of volatility, degradability, bioavailability, and toxicity to other organisms.5,6 PAHs are among the common petrochemical compounds containing 2 or more fused phenyl and/or pentacyclic rings. 7 Some of these PAHs are considered as possible cancer-inducing agents for human, and their distributions at low concentration in the environment causes hazard to human beings. 8 Apart from their toxicity and mutagenic properties, the PAHs are persistent in soils and sediments, and also bioaccumulative, and hence considered as priority pollutants by the US-EPA.9–11
Restoration of the oil-contaminated soil remains a challenge, as all the factors contributing to the degradation of the hydrocarbons need to be understood. The degradation of petrochemical compounds is affected by both biotic and abiotic processes, such as chemical oxidation, photolysis, adsorption, and volatilization. Although physical and chemical approaches are used for remediation, they lead to air pollution or ground water toxification. 12 Thus, bioremediation by indigenous microorganisms is considered the primary pathway for removal of hydrocarbons and PAHs from soils.13,14 Among such microorganisms, several bacterial strains colonizing polluted sites play a major role in hydrocarbon degradation. The analysis of this microbial community during biostimulation offers a good strategy for addressing the challenge and effectively improve the degradation efficiency. Compared to physicochemical methods, bioremediation techniques are cost-effective, versatile, environmentally sustainable, and can be highly effective to detoxify the environmental pollutants. 15 This eco-friendly technique enhances the biodegradation rates through settling its limiting factors.
Although, several bacteria seem to have developed efficient metabolic system for the natural attenuation of these toxic components, the process of degradation is slow.16–21 Therefore, appropriate remediation techniques need to be applied to enhance the rate of degradation of crude oil to clean up the contaminated sites. High temperature and the availability of essential nutrients and water are the main limiting factors for the bioremediation of soils contaminated by hydrocarbons and PAHs in the gulf region. 22 Nevertheless, previous studies conducted in this region have shown that biostimulation of indigenous hydrocarbon-degrading microorganisms is an adequate technique in this arid environment to improve the degradability of TPHs and PAHs.23,24 Even if further enhancement of bioremediation techniques can be made with further research, the limiting factor has been the identification of the microorganisms present in the polluted site. 24 The knowledge of the bacterial community dynamics during bioremediation is essential for designing an adequate bioremediation strategy effective for removal of TPHs and PAHs from contaminated soils. Since recently bioaugmentation has become a promising approach, 25 the development of bioaugmentation technology consisting of the isolation and inoculation of the identified consortia of bacteria with inherent biodegradation potential could be efficient to tackle soil pollution by hydrocarbons in extreme environment such as deserts. Furthermore, the identification of the bacterial community associated with natural degradation of these pollutants could provide valuable information for monitoring biotreatment. 26 These bacteria could be used as oil contamination indicators.27,28
Although cultivation of these bacteria is the classic methodology for assessing microbial community changes during the bioremediation process, less than 1% of environmental bacteria are cultivable. Recently, culture-independent techniques have been successfully applied as new approach to investigate the diversity of soil microbial communities.1,29 These techniques enable the capture of a molecular image of the existing microorganisms at a given time.
For successful application of bioremediation strategies for restoration of oil-contaminated soil, the screening for hydrocarbon degrading bacteria using high-throughput methods are extremely needed. Thus, the main goal of the present study is to monitor the degradation of TPHs and PAHs in parallel with soil bacterial community analysis during 9 months pilot scale biostimulation process for biopiles contaminated with hydrocarbons. This can reveal the microorganisms linked to the high hydrocarbons and PAHs degradation rates observed in this soil undergoing treatment. An unbiased, culture-independent method that could provide an overview of the bacterial community present during bioremediation has been used. The microbiome of Kuwait soil biopiles was sequenced using 16S rRNA during the bioremediation process and compared results with clean soil and oil-contaminated soil.
Material and Methods
Soil bioremediation and sampling
Based on detailed soil investigation at petroleum-contaminated locations around a petroleum refinery at southern Kuwait, a site having a history of petroleum contamination, moderately contaminated soil by TPHs was excavated and transported to stock pile area after segregating to different categories depending on soil nature and TPHs concentrations. Soil samples were collected from a moderately contaminated stock pile (50 000 mg/kg) subjected to bioremediation experiment. This pile was aerated by mixing and fertilized with a combination of inorganic nutrients (urea; triple phosphate) and compost (50 kg/m3) corresponding to a total amount of C:N:P = 50:1:1 to enhance the growth of the indigenous microorganisms. 30 The average of the environmental temperature was 30°C. A control clean soil was taken from an area directly adjacent to the excavated contaminated soil at a depth of 20 to 30 cm. The compost was added to the clean soil at the same ratio as the contaminated soil. Three samples were collected from the contaminated stock pile immediately after treatment (T0: zero day after treatment); T3: 3 months after treatment; T6: 6 months after treatment and T9: 9 months after treatment. T0 was considered as baseline (starting point) to compare the effect of treatment in all experiments. Each solid sample was prepared by pooling and homogenizing 5 samples corresponding to 5 locations randomly chosen along the pile. The soil samples were sieved through a 2 mm pore size sieve and stored at 4°C for further physicochemical characterization and total DNA extraction.
Soil physicochemical characteristics
The physicochemical characteristics and concentration of TPHs and PAHs in the oil-contaminated soil measured in triplicates are summarized in Table S1. Moisture was determined by placing pre-weighed soil samples in an oven at 105°C for 24 h. pH of the soil was determined according to USEPA method 9045D. Organic matter content was calculated by combustion at 440°C. Total organic carbon (TOC) was determined according to Walkley and Black. 31 Total phosphorus (TP) was analyzed by Inductively-coupled-plasma spectrometry (ICP) and total nitrogen (TN) was determined by the Kjeldahl method (ISO 11261 32 ).
TPHs analysis was performed using method based on a modified version of EPA method 418.1. The concentration of the light and heavy (C8-C40) fractions of TPHs was determined using the preparation method EPA 3546 and the analysis method FL-PRO at the ALS Environmental facility in Jacksonville laboratory, Florida.
PAHs extraction and analysis were performed according to Song et al, 33 in triplicates. A 5 g air-dried soil sample was extracted using 20 ml of dichloromethane after spiking with deuterated internal standard (1 µg/ml) listed in Table S2. Samples were concentrated to 1 ml using rotary evaporator and loaded into a clean-up column with silica gel and anhydrous sodium sulfate. Extracts were dissolved in 1 ml hexane after condensation by evaporation of the dichloromethane under a stream of nitrogen. The concentration of PAHs and their identification was determined by GC-MS. The GC oven temperature program for PAH analysis: initial temperature: 45°C, initial time: 2.00 min, rate of temperature increase (10°C/min), final temperature (290°C: final hold time 8 min), total run time: 33.70 min. The mean recovery of the PAHs was 80% to 110% depending upon individual PAHs. The biodegradation rates of PAHs were calculated using this formula:

Where, Ci is the concentration of 16 PAHs during the treatment and C0 is the initial concentration of 16 PAHs at T0
DNA extraction, library preparation and 16S rRNA sequencing
Total microbial DNA was extracted from 5 g soil sample at different stages of bioremediation (T0, T3, T6, and T9) and from control clean soil using the MoBio Power Max Soil DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA). The amplification and sequencing of 16S rRNA was performed according to Dowd et al, 34 Briefly, the 16s universal Eubacterial primers 530F (5ʹ-GTGCCAGCMGCNGCGG) and 1100R (5ʹ-GGGTTNCGNTCGTTG) were used for amplifying the 600 bp region of 16S rRNA genes. A single step 30 cycle PCR using HotStarTaq Plus Master Mix Kit (Qiagen, Valencia, CA) was used under the following conditions: 94°C for 3 min, followed by 28 cycles of 94°C for 30 seconds, 53°C for 40 seconds and 72°C for 1 minute and a final elongation step at 72°C for 5 minute. A secondary PCR was performed to incorporate tags and linkers into the primary amplicon. All amplicon products from different samples were mixed in equal concentrations and purified using Agencourt Ampure beads (Agencourt Bioscience Corporation, MA, USA). The samples were sequenced using the Roche 454 FLX titanium instruments and reagents following the manufacturer’s guidelines.
Analysis of 16S rRNA sequencing data and taxonomic assignment
The sequenced data was checked for quality before and after trimming, using FastQC v0.10.1.
35
The raw data was trimmed for barcodes and primer sequences, any ambiguous bases. Sequences containing homopolymers of
The trimmed sequences were checked for chimeras against RDP Gold database v9 reference sequences, and chimeric sequences were removed using UCHIME v7.0. 37 Chimera filtered sequences were used for identification of Operational Taxonomic Units (OTU) clusters with a minimum similarity of 97% using UCLUST, 38 and representative sequences were selected. The representative sequences were filtered further to remove any singletons. The filtered representative sequences were aligned pair-wise using PyNAST. 39 The alignment file was further filtered for positions with gaps, and outliers (sequences dissimilar to the alignment consensus). The filtered representative sequences were mapped against green genes database 40 with a similarity of 80% using Ribosomal Database Project (RDP) classifier. 41 All the data analysis steps were performed using the QIIME pipeline. 36
Statistical analysis and visualizations
Filtered alignments were considered for various statistical analysis and phylogenetic tree construction. Alpha diversity representing the diversity and richness for each sample was performed by rarifying a small percentage of sequences randomly picked, and considering 10 iterations each time. The lowest sample count was considered as rarefaction depth. The Shannon index indicating the diversity and Chao1 index indicating the richness of microbial population were calculated using the taxonomic classifications and phylogenetic tree. Beta diversity indicating the diversity across samples was calculated using weighted and unweighted UniFrac.
42
Principal Coordinate Analysis (PCoA) was also performed using the UniFrac results. All the statistical analyses were performed using QIIME.
36
Significant differential abundances of taxonomic assignments across samples were identified using Metastats.
43
Phylum or genus having a q-value
Results
TPHs biodegradation
The concentrations of TPHs and the light and heavy (C8-C40) fractions of TPHs in aged oil-contaminated soil are presented in Figure 1. Data shown are average values of 3 replicate measurements. The initial concentration of TPHs in this soil (T0) was 54 223 mg/kg. After treatment, there was a rapid 67% reduction in TPHs in the first 3 months, then followed by a slower decrease of 15% and 20% of the remaining concentrations after 6 and 9 months respectively (Figure 1A). The concentration of TPHs in the control clean soil was under the detection limit.

Concentration of TPHs (A), C8-C40 (B) during the bioremediation process of oil-contaminated soil. Error bars represent ± SD.
The initial concentration of the light and heavy (C8-C40) fractions of TPHs in this soil (T0) was 38 100 mg/kg. After treatment, the degradation rate was 84% in the first 3 months, then 25% and 31% of the remaining concentrations were degraded after 6 and 9 months respectively (Figure 1B). The concentration of the light and heavy (C8-C40) fractions of TPHs in the control clean soil was under the detection limit.
PAHs biodegradation
Changes in the concentration of 16 PAHs during the bioremediation process are presented in Figure 2. Data shown are average values of 3 replicate measurements. The initial PAHs concentration at T0 was 3.1 mg/kg. The concentration of 6 PAHs (naphthalene, benzo[a]anthracene, benzo[b]fluoranthene, benzo[k]fluoranthene, indeno[1,2,3-c,d]pyrene, and dibenz[a,h]anthracene) was under the detection limit. After 3 months of bioremediation, the percentage reduction in total PAHs was 58%. The rate of degradation of pyrene was significantly rapid compared to other PAH counterparts. Moreover, a significant reduction of chrysene was observed after 6 and 9 months of bioremediation while no significant degradation was observed for the other PAHs. The concentration of PAHs in the control clean soil was under the detection limit.
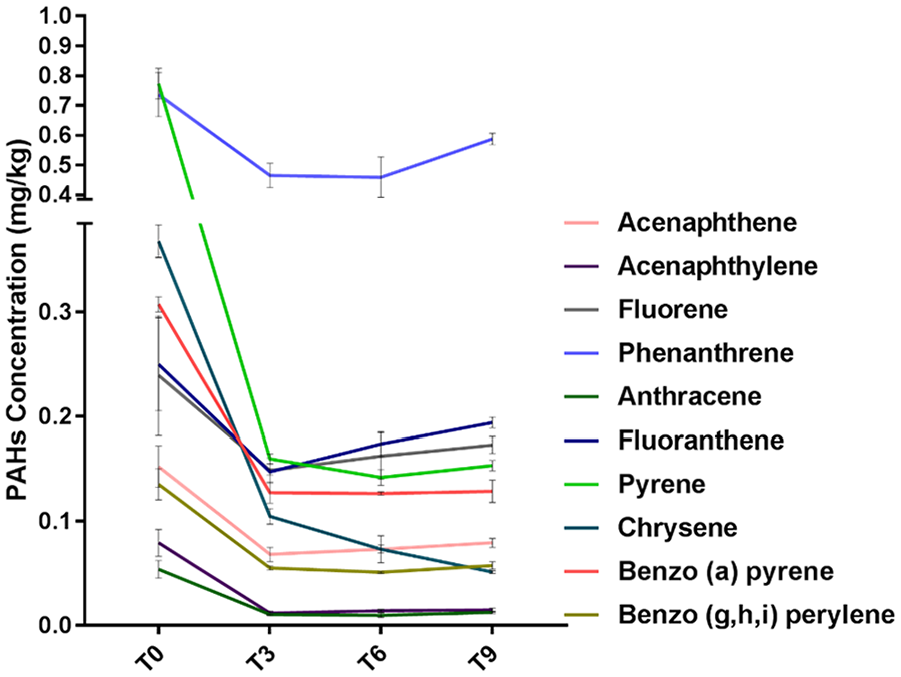
Concentration of PAHs during the bioremediation process of oil-contaminated soil. Error bars represent ± SD.
Analyses of bacterial community using 16S rRNA sequence data
The 16S rRNA gene sequencing of 15 samples (5 groups with 3 replicates) provided a total of 3 025 440 sequences. Around 84% of data (2 533 751 sequences) was retained after stringent quality filtering (Tables 1 and 2). The quality score based filtering removed 7% of the reads. A total of 141 726 chimeric sequences corresponding to 4.7% of the raw reads were detected and removed (Table 1). We identified 60 308 OTUs across all the samples, of which 19 870 were retained after filtering singletons (OTUs containing only one sequence). The filtered OTUs represent 2 493 313 sequences, corresponding to an average number of 166 220 reads per sample. The representative set of sequences obtained from filtered OTUs produced alignment hits for all except for 47 sequences. After filtering the alignments with gaps, and removing outliers, 19 770 aligned sequences were obtained.
Summary of read data after various quality filtering steps.

Summary of raw and filtered reads per sample.

Control: clean soil with compost; T0: zero day after treatment; T3: 3 months after treatment; T6: 6 months after treatment; T9: 9 months after treatment.
Around 2.4 million sequences were assigned to various taxa with at least 80% similarity. Only 54 446 sequences were not assigned to any phylum. A total of 31 taxa at phylum level were identified across samples, of which 9 are represented by at least 0.5% of total assigned sequences. Phylum level distribution across samples for these 9 taxa is represented in Figure 3A and B. Major percentage of sequences were assigned to the phylum Proteobacteria (54.4%), followed by Bacteroidetes (19.4%) and Actinobacteria (9.1%).

Phylum level distribution of microbial population across samples. Phyla with at least 0.5% abundance (9 of total 31 phyla) are represented using (A) bar plot and (B) heat map.
The results showed that the predominant phyla were Proteobacteria, Bacteroidetes, and Actinobacteria in clean soil and oil-contaminated soil at the beginning of the treatment (T0). The phylum Actinobacteria was present in the clean soil (8%) but more distinctly represented in T0 (25%) whereas the phylum Bacteroidetes was found more abundant in the clean soil (28%) versus 23% in T0. The abundance of phylum Proteobacteria was 35% in the clean soil and 31% in T0 (Figure 3).
During the bioremediation treatment, bacteria of a few phyla were found to be varying significantly. An average of ~30% of phylum Proteobacteria identified in the clean soil and oil-contaminated soil (T0) groups drastically increased to an average of 65% after treatment (T3, T6, and T9) (Figure 3). The high abundance of phylum Proteobacteria in the treated group was mainly due to class Gammaproteobacteria. Moreover, an average of 2% Chloroflexi and 0.06% Chlorobi phyla in the clean soil and oil-contaminated soil (T0) groups increased to 10% and 1%, respectively in treatment groups. On the contrary, an average of approximately 28% of phyla Bacteroidetes, 10% of Firmicutes and 5% of Thermi bacteria in the clean soil and oil-contaminated soil (T0) groups reduced to 13%, 1% and 0.1%, respectively in treated groups. BRC1 phylum, which had a low abundance found to be increased by 40 fold in treatment compared to clean soil and oil-contaminated soil (T0) groups. Some bacteria from phyla Gemmatimonadetes and TM7, though having less abundance (<0.5%), decreased with a minor fold differences (<1.2) in treatment compared to clean soil and oil-contaminated soil (T0) groups. A complete list of assigned taxa at phylum level is presented in Table 3.
Summary of abundance at phylum level in control and treatment groups.

A complete list of assigned taxa at phylum level and summary of their abundance in control (clean soil and T0) and treated groups (T3, T6 and T9).
Microbial population corresponding to 15 genera indicated at least 10 fold differences between clean soil and oil-contaminated soil (T0) groups and the treated groups. The genus Alcanivorax was the most abundant in the treated group. Similarly, microbial species of genera Pseudidiomarina, HTCC, Luteibacter, Parvibaculum, and Zhouia showed an increase in the treatment groups with a fold ranging from 20 to 25 compared to the clean soil and oil-contaminated soil (T0) groups. However, the genera B-42, KSA1, Jiangella, Lentibacillus and Actinomadura showed a 30-50 fold higher abundance in clean soil and oil-contaminated soil (T0) compared to treated groups. Species from other genera, such as Lutibacterium and Dietzia, though having less abundance across all samples showed minor differences of <3 fold. Species from genera Sphingomonas, Geotgenia, and Balneola also having less abundance across samples showed >10 fold increase in the treated groups compared to clean soil and oil-contaminated soil (T0). In the treated groups, Salinimicrobium, Zhouia, and Pseudidiomarina genera showed a significant increase at the end of the treatment (T9) simultaneously with reduced concentrations of TPHs and PAHs. Genus level distribution across samples with >1% and at least 0.5% abundance is represented in Figure 4 and Supplementary Figure 1, respectively.
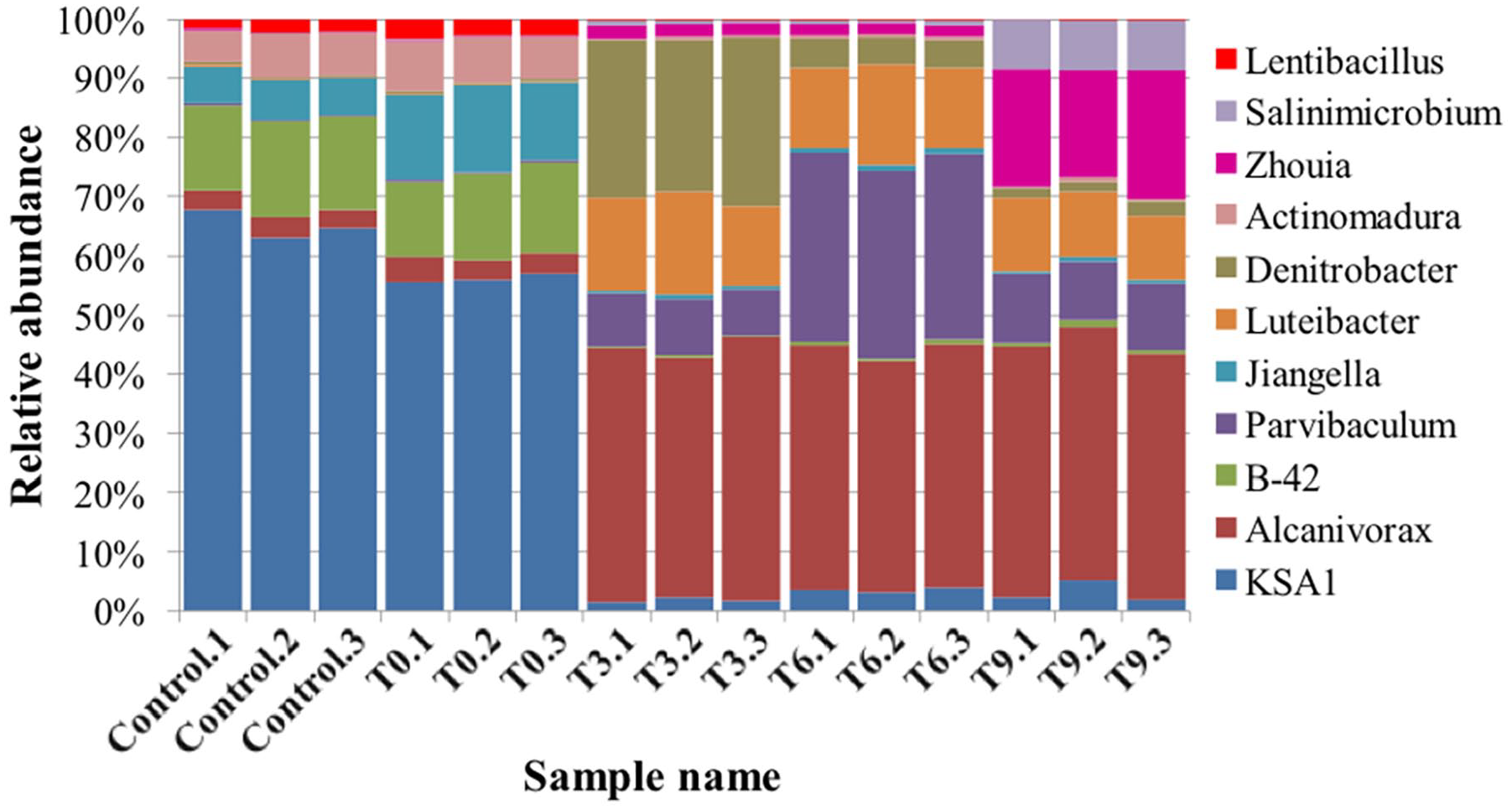
Genus level distribution of microbial population across samples. Genera with >1% abundance (11 of total 216 genera) are represented.
Significance enrichment analysis indicated Bacteroidetes and Actinobacteria to be as the most significant phyla in control clean soil and T0 groups respectively compared to all the other groups, with a P-value of < .001 (Table 4). BRC1, and Acidobacteria were found to be the most significant enriched phyla in T3 and T9 treatment groups, respectively. With the considered threshold, no phylum was found to be significant in T6 treatment group. At the genus level, KSA1, and Desulfotomaculum were found to be the most significant enriched genera in control and T0 groups, respectively. Whereas, Denitrobacter, Parvibaculum and Salinimicrobium genera showed significant enrichment in T3, T6, and T9 treatment groups respectively.
Significantly enriched phyla in control and treated sample groups.

A phylum was considered enriched in a group if it had the least P-value in that group compared to the samples from all other groups combined. Control: clean soil with compost; T0: zero day after treatment; T3: 3 months after treatment; T6: 6 months after treatment; T9: 9 months after treatment.
The species richness and diversity was calculated using Chao1 and Shannon diversity indices. Chao1 index indicated a slight increase in the richness of species in treatment compared to clean soil and oil-contaminated soil (T0) groups (Figure 5 and Table 5). Shannon index representing the sample diversity indicated enough sampling depth at 30 000 sequences (Figure 6). Beta diversity PCoA plot using weighted and unweighted UniFrac analysis indicated that the samples from same group clustered together. PCoA plot from unweighted UniFrac analysis is shown in Figure 7. This result was further corroborated by the UPGMA clustering (Figure 8). A phylogenetic tree was constructed using Mega 7 showing the closely related species implicated in oil degradation (Supplementary Figure 2).

Species richness indicated by Chao1 rarefaction measure. Control: clean soil with compost; T0: zero day after treatment; T3: 3 months after treatment; T6: 6 months after treatment and T9: 9 months after treatment.
Shannon and Chao1 index for the control and treated samples.

Control: clean soil with compost; T0: zero day after treatment; T3: 3 months after treatment; T6: 6 months after treatment; T9: 9 months after treatment.

Species diversity estimated by Shannon index. Control: clean soil with compost; T0: zero day after treatment; T3: 3 months after treatment; T6: 6 months after treatment and T9: 9 months after treatment.

PCoA plot of samples using unweighted UniFrac analysis. Control: clean soil with compost; T0: zero day after treatment; T3: 3 months after treatment; T6: 6 months after treatment and T9: 9 months after treatment.

UPGMA clustering of samples based on phylogenetic distribution. Control: clean soil with compost; T0: zero day after treatment; T3: 3 months after treatment; T6: 6 months after treatment and T9: 9 months after treatment.
Discussion
Soil contamination by hydrocarbons due to oil spills has become a major global concern and it has more implications in the oil producing regions. A complete degradation of hydrocarbons could be achieved by bioremediation treatment, a cost-effective approach compared to physicochemical treatments. The microbial dynamics in a given ecosystem is highly influenced by the type of contamination, the surrounding environment, and the remediation method used for clean-up. Thus, it is hard to obtain general conclusions about the microbial population responsible for oil-degradation applicable to all regions. The present study adds valuable information about hydrocarbon degraders in the hot desert gulf region. A culture-independent method, 16S rRNA amplicon sequencing, was used to identify these degraders in parallel with analytical analysis of TPHs and 16 priority PAHs degradation during a biostimulation treatment.
A number of studies recently have been focused on application of next generation sequencing to discover microbial populations in environmental samples.45-51 The work presented here focused on understanding the bacterial community structure associated with TPHs and PAHs degradation in desert soil at various stages of bioremediation process.
Oil-contaminated soil in this arid environment seems to be an appropriate lodging for hydrocarbon degrading bacteria as Proteobacteria, Actinobacteria, Chloroflexi, Chlorobi and Acidobacteria phyla. Proteobacteria which was the predominant phylum in the clean soil and the oil-contaminated soil (T0), was greatly stimulated during the treatment. This phylum is prevalent in soil libraries52,53 and it is known as an essential element in the carbon, nitrogen and sulfur cycles. 54 This finding is in accordance with several previous studies describing Proteobacteria as the most dominant phylum in oil-contaminated soil.1,29 In addition, a study in Nigeria revealed a correlation between the increase in the abundance of the phylum Proteobacteria and hydrocarbon removal from oil-contaminated site. 55 The phylum Actinobacteria was more abundant in the oil-contaminated soil compared to the clean soil in agreement with a recent study on the microbiome of crude petroleum oil-contaminated soil from agricultural biome of Gujarat, India showing Proteobacteria as the most abundant phylum followed by Actinobacteria. 1 Proteobacteria and Actinobacteria are known as main biocatalyst for hydrocarbon bioremediation harboring key enzymes involved in alkane degradation pathway (i.e. several families of alkane monooxygenases, ring-hydroxylation dioxygenase, and cytochromes P450).56,57 Recently, Jiang et al, 58 reported the phyla Proteobacteria and Acidobacteria to be directly involved in the uptake and degradation of phenanthrene. Our results are in accordance with this study since a significant reduction of phenanthrene was observed after 3 months of biostimulation. This reduction could be associated with the stimulation of the phyla Proteobacteria and Acidobacteria by the treatment.
The phyla Chloroflexi and Chlorobi which are less dominant in oil-contaminated soil, were stimulated by the biotreatment process. The increase in the Chloroflexi phylum was observed in a recent study conducted on crude and refined petroleum oil-contaminated soil in India showing that Chloroflexi was among the phyla majorly contributing to aromatic and aliphatic hydrocarbon degradation. Further, Peng et al, 59 found that Chloroflexi was among the dominant phyla in crude oil-contaminated soils in China exposed to different periods of oil pollution.
The phylum Bacteroidetes was reduced after treatment, which is in agreement with a study conducted by Yergeau et al, 60 showing a decrease in the abundance of this phylum during the bioremediation of diesel-contaminated Canadian high arctic soils. However, Balneola and Salinimocrobium genera increased after treatment. Balneola was among the dominant genus groups in aged soil that enhanced remediation of petroleum-contaminated soil collected from a crude oil spill site in Shengli Oilfield in China. 61
The high abundance of Proteobacteria phylum was mainly due to the class Gammaproteobacteria. Previous studies conducted on estuarine sediments contaminated with PAHs showed the predominance of this class and its association with pyrene degradation. 62 In the present study, the rate of degradation of pyrene is significantly rapid compared to other PAH counterparts. Pyrene reduction could be associated with the stimulation of the class Gammaproteobacteria by the treatment. These results show that the microbes show selective degradation of certain type of PAH under given conditions. 63 Most bacterial species under defined controlled conditions often degrade only a narrow range of PAHs and they show preferential degradation of selected PAHs. 64 The nature of the PAHs mixture in the environment, as well as the metabolic competition among degraders, influence the degradation of specific PAHs while inhibiting the degradation of other PAHs. Additionally, the catabolic genes from fluorene-utilizing Sphingomonas were unlike the genes involved in the degradation of other PAHs components.65,66
The detected class Gammaproteobacteria were mostly compound by Alcanivorax genus. Our results are in accordance with a metagenomic analysis of crude oil contaminated soil from Nigeria showing that the dominant Gammaproteobacteria were the genera Alcanivorax and Marinobacter. 67 Alcanivorax genus is known for its hydrocarbon degrading capability as well as glycolipid production. Currently, Alcanivorax species were reported as important oil and PAHs-degrader in marine environment. 68 They have been indicated as major degraders of petroleum-aliphatic-hydrocarbon in tropical marine environments. 69 Moreover, this genus was identified among the hydrocarbonoclastic bacteria isolated from Jakarta Bay and Seribu Islands that are known as the most polluted marine environment in Indonesia. 70 In addition, a recent study showed the increase of Alcanivorax genus during the remediation of crude oil-polluted soil by bacterial rhizosphere community. 71 Moreover, Alcanivorax borkumensis which is a marine alkane-degrading bacterium harbors enzymes involved in alkane oxidation (cytochromes CYP153 and AlkB homologs which are well-characterized particulate membrane-bound hydroxylase). 57 Further, this strain which was isolated from eastern Mediterranean sea in presence of crude oil as carbon source produced biosurfactant, 72 an extracellular polymeric substance that contributes to enhancing the solubility of PAHs and thereby facilitates improved degradation.73,74
Among Gammaproteobacteria class, the genera Luteibacter, and Pseudidiomarina were stimulated by the bioremediation treatment with a fold ranging from 20 to 25. Cui et al, 75 reported Luteibacter genus as main contributor to petroleum hydrocarbons degradation in an activated sludge from Oilfield Sewage Treatment Plant in China. Pseudidiomarina species were reported as potential petroleum-hydrocarbon degraders in tropical seas in Singapore. 69 Furthermore, this genus was isolated from oil polluted marine sediments in Indonesia. 70 Moreover, indigenous Pseudidiomarina from mangrove surface sediments of Nay band Bay in Iran showed high potential to degrade fluorene and phenanthrene. 76 In our study, Pseudidiomarina genus showed abundance at T9 simultaneously with an enduring high level of phenanthrene.
The class Alphaproteobacteria were mostly Parvibaculum and Sphingomonas genera. The genus Parvibaculum was detected in several hydrocarbon-contaminated environments.27,77 This genus was enriched in microcosms from Louisiana salt marsh sediment treated with crude oil. 78 It was also identified among a PAHs-degrading consortium isolated from deep-sea water and marine sediments of the Indian Ocean. 79 Members of the Sphingomonas genus are often isolated from petroleum-contaminated soils. 66 They possess a unique group of genes for aromatic compound degradation. 80 A study isolating Sphingomonas strains from petroleum-contaminated soils in the China’s biggest petroleum wastewater irrigation zone revealed that this genus plays a key role in the degradation of the PAHs fraction. 66 Since Sphingomonas were associated to pyrene degradation, 60 this genus might be a key element in the degradation of this compound in the investigated soil.
Salinimicrobium and Zhoui genera were abundant at the end of the biostimulation treatment meanwhile the TPHs concentration reached 1%. This could be due to the fact that genus Salinimicrobium could barely survive in the highly contaminated soil because of its sensitivity to petroleum hydrocarbons. As a result, this genus can be used to predict the end of contamination in the oil-contaminated soils in the hot desert gulf region.27,28 A recent study described Zhoui species isolated from mangrove sediment from East Asia as underexplored bacteria with lignocellulose degrading abilities. 81 Thus, the abundance of Zhoui at the end of treatment could be associated to the derivate with less complex ring structures resulting from the degradation of aromatic compounds with complex structure.
Conclusion
In this study, a diversity analysis of microbial communities in crude oil- contaminated soil suggests that biostimulation in hot arid environment favored the growth of Proteobacteria, Chloroflexi, Chlorobi, and Acidobacteria phyla. These groups have adapted catabolic machinery for hydrocarbons degradation. These findings can provide a platform for further utilization of a microbial consortia belonging to Alcanivorax, parvibaculum, and Sphingomonas genera for restoration of petroleum contaminated areas with hot arid conditions. Future work may focus on the isolation of these bacteria and the development of bioaugmentation strategy tailored to arid environment.
Supplemental Material
sj-pdf-1-evb-10.1177_11769343211016887 – Supplemental material for Insights into Bacterial Community Involved in Bioremediation of Aged Oil-Contaminated Soil in Arid Environment
Supplemental material, sj-pdf-1-evb-10.1177_11769343211016887 for Insights into Bacterial Community Involved in Bioremediation of Aged Oil-Contaminated Soil in Arid Environment by Rita Rahmeh, Abrar Akbar, Vinod Kumar, Hamad Al-Mansour, Mohamed Kishk, Nisar Ahmed, Mustafa Al-Shamali, Anwar Boota, Zainab Al-Ballam, Anisha Shajan and Naser Al-Okla in Evolutionary Bioinformatics
Footnotes
Acknowledgements
The authors thank the Kuwait Institute for Scientific Research for financial support. We thank Dr. Kshitish Acharya and his team from Shodhaka for their contribution in the analysis of the next generation sequencing data. The technical assistance of Dr. MT Balba is gratefully acknowledged.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by Kuwait Institute for Scientific Research.
Declaration of conflicting interests:
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author contributions
RR designed the research, wrote and edit the manuscript; VK contributing to the manuscript writing and data analysis; HA, NA, MA, AB, ZB, AS, NO performed the analytical analysis, AA, MK performed the NGS related experiments and data analysis
Data availability
The sequence data reported in this paper are available in the NCBI database (BioProject PRJNA699560)
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.