
Abstract
Environmental pollution has become a worldwide concern that requires rigorous efforts from all sectors of society to monitor, control, and remediate it. In environmental pollution control, Cupriavidus gilardii CR3 has become a model organism to study resistance to heavy metals as a means of bacterial bioremediation. This research aimed to single out regulatory element analysis and conduct a comparative genome study of the heavy metal resistance genes in the complete genome of C gilardii CR3 using bioinformatics and omics tools. Comparative genome analysis, promoter prediction, common motif identification, transcriptional start site identification, gene annotation, and transcription factor identification search are the major steps to understanding gene expression and regulation. MEME Suit, TOMTOM, Prokka, Rapid Annotation utilizing Subsystem Technology (RAST), Orthologous Average Nucleotide Identity Software Tool (OAT), and EziBio databases or programs were the major tools used in this study. Fourteen transcriptional factors were identified and predicted from the most credible and lowest candidate motifs with an e-value of 3.0e-009, which was statistically the utmost remarkable candidate motif. A detailed evaluation was further performed, and 14 transcriptional factors were identified as in activation, repression, and dual functions. The data revealed that most transcriptional factors identified were used for activation rather than repression. The C gilardii CR3 genome contains many genes responsible for resisting heavy metals such as mercury, cadmium, zinc, copper, and arsenate. As a result, regulatory elements will lay a solid basis for understanding genes responsible for heavy metal bioremediation. It was concluded that further studies with wet lab support could be conducted for confirmation. Moreover, other advanced bioinformatics and omics technologies are needed to strengthen the results.
Introduction
Environmental pollution is the current challenge to sustainable development in the world. The challenge arises from the natural and anthropogenic activities of humankind. Pollution has become a global concern requiring rigorous efforts from all sectors of society to monitor, control, and remediate. As industrialization and urbanization have accelerated over the past century, pollution has become a major problem for both developing and developed countries. The use of pesticides and herbicides in modern agricultural practices also contributes to environmental pollution. 1 Unmanaged industrial waste can leak into soil and groundwater, contaminating the environment with heavy metals. Pollution caused by mining is also one of the main causes of environmental degradation. 2 Heavy metal effluents from wastewater and agricultural inputs also contribute to major environmental pollution due to their extreme poisoning. Heavy metals are also dissolved in water bodies contaminating ground water, lakes, rivers, and oceans. This causes damage to the food chain and web systems. Environmental pollution threatens humans, animals, and marine life by affecting ecosystem services that support life. 1
Microorganisms have developed specific adaptation capabilities to thrive in harsh environmental conditions. 3 Bacteria have a variety of coping mechanisms despite challenging environmental conditions such as food scarcity, metabolic and biological alterations, and extreme temperatures. 3 The mechanisms include extracellular barrier, metal ions transporting actively, extracellular, bioaccumulation, intracellular sequestration, and reduction of heavy metals.3,4 C gilardii CR3 is a strain of bacteria belonging to the genus Cupriavidus, which is known for its ability to metabolize a variety of substances including heavy metals. It is often studied for its potential for bioremediation particularly in contaminated environments where heavy metals are present. This bacterium has been shown to utilize various carbon sources anaerobically and aerobically through pathways. Therefore, it is used in cleansing up contaminated sites and its role in nutrient cycling in various ecosystems. Cupriavidus spp., Pseudomonas putida, Pseudomonas aeruginosa, Pseudomonas nitroreducens, and Pseudomonas alcaligenes, 5 Klebsiella species, 6 and recombinant Escherichia coli are few bacteria that have mechanisms to resist cadmium heavy metal. 7 C gilardii CR3 (see Figures 1–4) encodes all heavy metal resistance genes such as arsenate, cadmium, chromate, copper, mercury, and nickel among Cupriavidus spp. On the contrary, C gilardii CR3 encodes for genes on conjugative elements, which are chromosomally integrated. For efficient uses of bacteria in HMR, temperature is one of the utmost essential factors for microbes to succumb to extreme environmental conditions. 8 It affects protein expression levels and gene regulation in microorganisms.

The sequence logos for HMR predicted common motifs. MEME suite version 5.5.2 was utilized for the prediction.

(A) Czc ABC family, (B) mercury operon families, and Cobalt-Resistant and Copper-Zinc- and Cadmium-Resistant gene families predicted by RAST. RAST indicates Rapid Annotation utilizing Subsystem Technology.

(A) Graphic circular genome mapping for C gilardii CR3 and a sequence obtained from the NCBI database with accession number CP010516.1. (B) Zoomed circular genome map for C gilardii CR3 obtained from NCBI. (C) Zoomed circular genome map with the location of sequence length and/or contig, stop, start, ORF, GC skew+ and GC skew–. ORF indicates open reading frame; RAST, Rapid Annotation utilizing Subsystem Technology.

(A) Sequence similarity of OrthoANI and the other closely related strains, (B) phylogenetic tree and evolutionary relationships of taxa for Cupriavidus gilardii CR3, and (C) a circular phylogenetic analysis of the complete genomes of C. gilardii CR3. The phylogenetic tree shows heavy metal resistance gene locations.
Bioinformatics focuses on manipulating biological and genomic data using computational tools. It provides an exploratory view of bioremediation in environmental conservation by addressing proteomic and genomic perspectives. 9 Biological data are important for information collected, stored, analyzed, and disseminated through computer technology, in the domain of omics and bioinformatics. Sequences of DNA and amino acids, or annotations of those sequences, are widely used in different areas of study such as health, agriculture, industry, and environment proposed for disease identification, gene prediction that increases productivity, strain improvement, and environmental bioremediation, respectively.
To understand how the genome works, it is crucial to identify DNA sequences that have been preserved in a wide range of organisms over millions of years. It pinpoints genes essential to life and highlights genomic signals that control gene function across many species. It helps us to gain a deeper understanding of how genes relate to various biological systems. Comparative genomics is also a powerful tool for studying evolution. 10 The appearance, behavior, and biology of living things have changed over time. This can be done by using and analyzing evolutionary relationships between species in their DNA. 11 Comparative genomics identifies real genes based on nucleotide conservation patterns during evolutionary time. The discovery of regulatory elements help decide whether a gene turns on or off based on nucleotide conservation patterns. 12 It is widely used in agriculture, the environment and biotechnology as model organism identification for various functions. C gilardii CR3 genomes were compared with related species of Cupriavidus, which have various genes associated with heavy metal bioremediation.
Bioremediation is an attractive technology for cleaning polluted environments. Microorganisms, especially bacteria, have developed specific adaptation abilities to a wide range of extreme conditions to flourish in such habitats. These conditions include food deprivation, biochemical, biological changes, and extreme temperatures. It is common for bacteria to accumulate metal ions in their extracellular and intracellular cell walls. 13 The ability of bacteria to absorb and convert energy is essential for their survival and performance in harsh environments. A better understanding of the gene regulation of HMR bacteria could be powerful in biological waste management. 14 Numerous studies have been conducted on bacterial resistance to heavy metals in the last two decades. However, the mechanism by which a bacterium resists heavy metals, its gene regulation mechanisms, and gene function is not fully reported. This study was aimed at the identification of regulatory element analysis and comparative genome investigation of genes in the complete genomes of C gilardii CR3 through comprehensive understanding by using omics and bioinformatics tools. Therefore, bioinformatics and comparative genome analyses will lay a solid foundation for understanding gene regulatory mechanisms. This study identified genes associated with HMR as a means of heavy metal bioremediation approach.
Materials and Methods
Prediction for transcriptional start site and promoter regions
The exact sequences of HMR genes were obtained from the National Center for Biotechnology Institute genome search engine at https://www.ncbi.nlm.nih.gov/gene. For this study, 15 protein-coding gene sequences were retrieved after checking the search results in the sequence databases in Table 1. We predicted whether starting codons were found on positive and negative strands or on both strands for further analysis. The transcriptional start site (TSS) was studied by collecting 1 kb sequences from genome coordinates areas and acquiring them in a FASTA format. The search query sequences’ FASTA file format was used for an additional promoter prediction investigation via Neural Network Promoter Prediction (NNPP). For NNPP prediction, about 1 kb upstream sequences from the start codons were prepared in the FASTA file format and forwarded to NNPP version 2.2 (https://www.fruitfly.org/seq_tools/promoter.html) tools to obtain a possible TSS. 15 The NNPP version 2.2 program was used with an appropriate minimum predictive activator score with a default cut-off threshold of 0.8 for prokaryotic cells and projected to reduce zero counts by 80% from the query sequences before transformation. 15 The output of NNPP, activator prediction sequences region, for those having more than one TSS, the highest prediction score value was evaluated for consistency and accuracy cut-off values. The remaining TSS regions were utilized solely for simple comparative gene analysis.16 -18
TSS, its promoter predictive score values, and distance from 5′-UTR region of the corresponding gene predicted and function.

TSS, transcriptional start site; UTR, untranslated region.
The NNPP (Neural Network Promoter Prediction) tool prediction findings are considered reliable at 0.8 cut-off values for the prokaryotic organism.
Common motifs and transcriptional factor determination
The promoter sequence segments that met the required standards were imported and investigated with the MEME Suite, a motif-based sequence analysis tool (5.5.2 version) via the website provided by the National Biomedical Computational Resource (https://meme-suite.org/meme/tools/meme). 19 The common candidate motifs were identified as binding sites of transcriptional factors (TFs) to regulate HMR gene expressions. The MEME-Suite was utilized for motif identification and exploration, motif alignment investigation, motif scanning, and motif comparisons. 20 Statistically, significant candidate motifs predicted by MEME-Suit in the sequence were picked and used for further comparison. The MEME-suit predicted and discovered gene sequences that fit the best motifs (fixed-length repetitive patterns with the lowest e-values) were submitted to TOMTOM online database to obtain TFs. This technique was utilized to discover common motifs that served as binding spots for TFs believed to influence HMR gene expression stages. However, before beginning the search for the selected sequences, the motif distribution menu’s basic criteria for searching were defined. These included motif distribution, motif positions, zero or one incidence per sequence, or multiple incidences per sequence.
Furthermore, the number of motifs and persistent motif width (6–50 bps) were set to default. Following the completion of the MEME searches, a search results page was linked to the MEME output in HTML. This stage is an important preliminary point of view for the expected value (e-value). The smaller the e-value, the better result was expected as reported in earlier studies. 20 At the bottom of the MEME HTML output, one or more potential motifs were provided for additional investigation. TOMTOM Web server database tool was used to identify common TFs. The sequences matching with the best candidate motifs in their respective TF were identified for the imported gene sequences. As part of TOMTOM output, LOGOSS represents the alignment of the candidate motif and TF with the P-value and q-value (a measure of false discovery rate) and links back to the parent transcription database for more detailed information on the matches in TOMTOM output.20,21
Comparative genome analysis of C gilardii CR3 genome by Prokka and Rapid Annotation utilizing Subsystem Technology
The sequences responsible for the HMR were obtained from the NCBI database, which is freely available at https://www.ncbi.nlm.nih.gov/. Heavy metals such as arsenate, cadmium, mercury, chromium, zinc, and cobalt resistance of DNA sequences were used in the study. Comparative genome analysis of C gilardii CR3 (CP010516.1), Cupriavidus metallidurans CH34 (NC_007973.1), Cupriavidus nantongensis X1 (CP014844.1), Cupriavidus ISTL7 (CP066227.1), Cupriavidus MP-37 (NZ_CP085344.1), and Cupriavidus gilardii WM02 (CP104386.1) for the HMR was performed by using Rapid Annotation utilizing Subsystem Technology (RAST) online engine accessed through (https://rast.nmpdr.org/rast.cgi). The RAST is a prokaryotic genome annotation service used to re-annotate for gene prediction. 18 Similarly, a C gilardii CR3 circular genome analysis was performed using CGView (Circular Genome Viewer) a Java application and library for generating high-quality and zoomable maps of circular genomes. 17 Finally, gene expression, gene sequences, location, and function are used to identify genes. Similarly, SEED Subsystem category distribution approach technology was used to characterize functional genes from the C gilardii CR3 genome and other related strains. 19
Comparative genome analysis of C gilardii CR3 by using OAT software
In this study, we used Orthologous Average Nucleotide Identity Software Tool (OAT) online to figure out the Orthologous average nucleotide identity (OrthoANI) of 16S rRNA from closely related species acquired from EziBio Cloud (www.ezi.biocloud.net/sw/oat).22,23 Genomic sequences of C gilardii CR3 CP010516.1 and related strains such as Cupriavidus gilardii WM02, C metallidurans CH34, C nantongensis X1, Cupriavidus ISTL7, and Cupriavidus MP-37 were investigated for comparative genomic analysis of HMR genes for heavy metal bioremediation. P putida PD584 (CP115665.1) was exploited as an out-group for the comparisons. 22
Results
TSS and promoter determination
Identifying the transcription start point is critical for understanding gene expression. In a conceptual sense, where an RNA polymerase enzyme interacts before the initiation spot. With primer extensions, TSSs can be pinpointed to the exact nucleotide. This approach involves binding an oligonucleotide primer to mRNA to start initiations. To accurately predict promoter regions and interpret patterns of gene expression, as well as build and understand genetic regulatory networks, a correct prediction of promoter sites has become a necessary element. 24 Nowadays, bioinformatics tools can also be used to determine the exact sites of TSS. The NNPP version 2.20 databases were utilized to work out the TSS, which is often used in HMR. The promoter region located upstream of 1 kb of the TSS was characterized with the assumption that the promoter’s functional gene elements could be discovered within the region. Table 1 summarizes and provides the TSS predicted values for each of the HMR gene’s coding sequences. Consequently, the gene variety has several TSS values ranging from 1 to 38. Interestingly, the minimum and maximum of the TSS values identified correspond to genes CzcA/CzcB and CadD, respectively, as shown in Table 1. Similarity genes symbolized by CzcD and CadA had the same TSS values on the different strands with −323 and 303 far away from codons, respectively.
A start codon’s location has a great deal to do with transcriptional initiation and gene regulation. There can be an enhancement or hindrance to gene regulation based on the location of the start codon. Accordingly, about 36.67% and 73.33% of TSS was found on the negative strands and the positive strands, respectively, as summarized in Table 1. The minimum and maximum TSS values were 38 and 550 for genes found on negative strands. These values were linked to the CutC and ChrB genes. In contrast, the minimum and maximum TSS values were 30 and 2994 for those located on the positive strand that corresponds to CadC and CzcA, respectively, as found in Table 1. The CzcA and CzcB genes showed 2994 and 1415 the highest location of the best TSS predicted by NNPP at the fourth and second rounds, respectively. The CadC gene had the lowest location of the best TSS values of 30 values.
Prediction of common motif and TFs
There is evidence that a wide range of factors affect gene expression. One of the most significant things was the motif. Thus, the five common candidate motifs were predicted and searched by the MEME suite algorithm version 5.5.2 available https://meme-suite.org/meme/. For this study, data show that the CMFs_3 by e-values of 1.3e_009 having seven numbers of promoters with motive width of 50 was the best candidate motif as mentioned in Table 2 compared to 84 in the databases of MEME. Thus, CMFs_3 had the lowest e-values having 63.64% binding sites. Table 2 also showed that the highest and lowest numbers of promoter binding sites had been eight and six, respectively with 3.0e_010 and 3.8e_010/3.0e_009 e-values. As presented in Table 2, the CMFs_4 candidate motif shares with that of the best candidate motifs of the number of binding sites.
List of predicted motifs and the number and proportion of promoter-containing motifs.
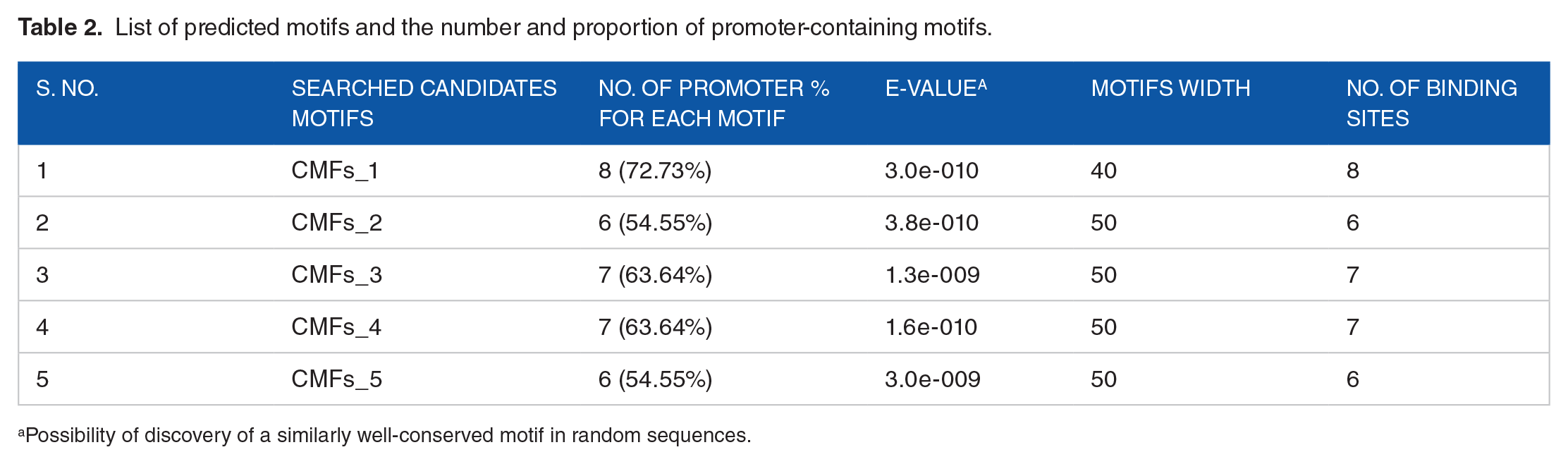
Possibility of discovery of a similarly well-conserved motif in random sequences.
The prospective common motif with the lowest e values (1.3e-009) indicates a significantly different and functionally important motif, which was successfully imported into TOMTOM a motif comparison tool version 5.5.2 for further comparison, a database that is freely available for transcription element predictions that may be matching to known regulatory elements.25,26 TOMTOM includes LOGOSS, which stands for the matching of observed motifs with probable TFs. The database’s TOMTOM output includes interactions with a parent TFs database for more information. This includes the matched motif’s activation, repression, and dual regulatory functions. Furthermore, conformational information connected to the TF databases of monomers, dimers, tetramers, unpredicted, and other factors is anticipated (see Tables 3 and 4). The data showed that CMFs_3 had the lowest e-value (1.3e-009) and was statistically significant with nine matching TFs from the databases obtained with matched e-value criteria of 14 or below, as tested and seen in the TOMTOM database. Figure 1 describes a forward and reverse strand of statistically significant strands.
Lists of matching candidates from EXPREG transcription factor (TF).
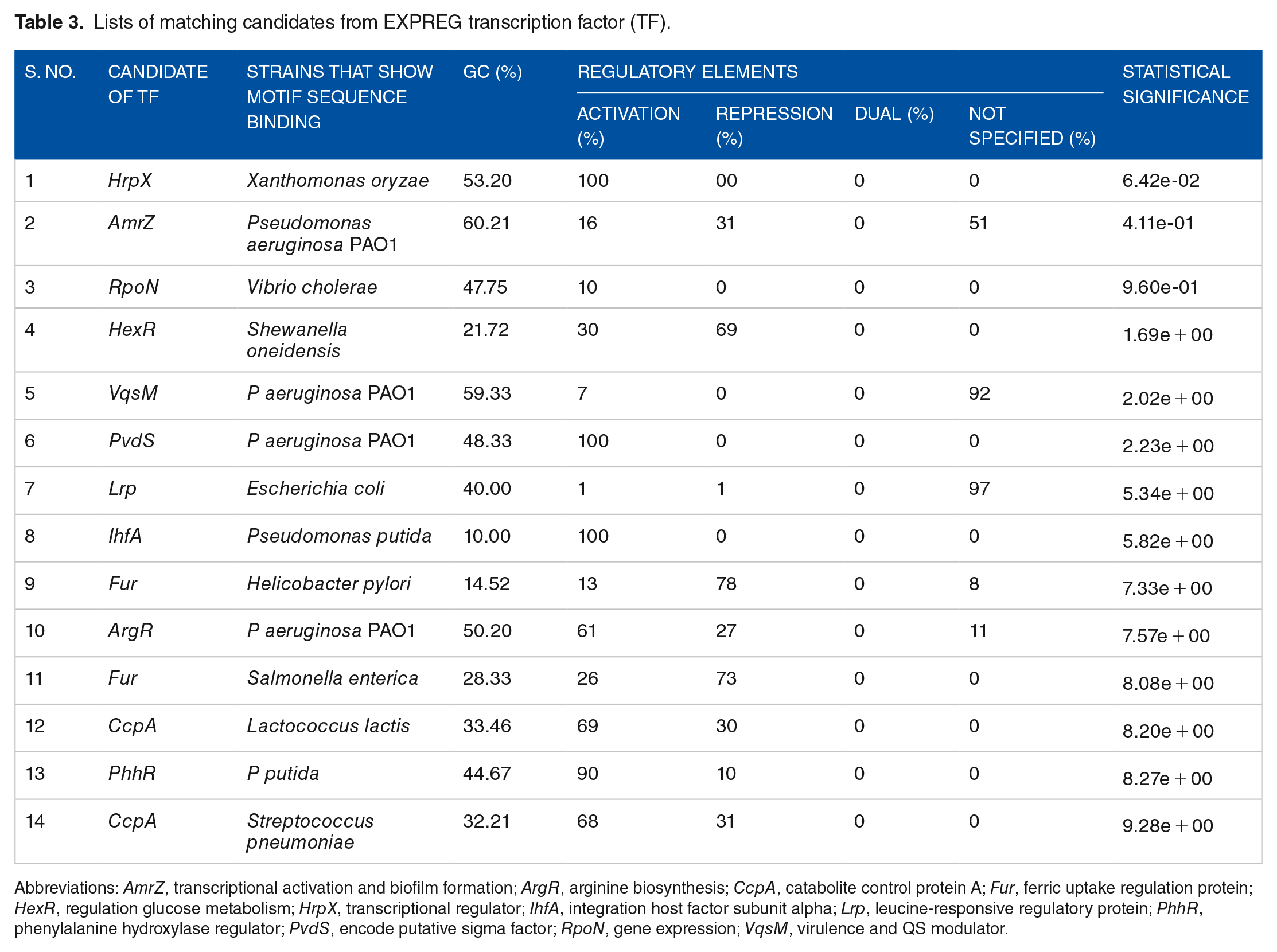
Abbreviations: AmrZ, transcriptional activation and biofilm formation; ArgR, arginine biosynthesis; CcpA, catabolite control protein A; Fur, ferric uptake regulation protein; HexR, regulation glucose metabolism; HrpX, transcriptional regulator; IhfA, integration host factor subunit alpha; Lrp, leucine-responsive regulatory protein; PhhR, phenylalanine hydroxylase regulator; PvdS, encode putative sigma factor; RpoN, gene expression; VqsM, virulence and QS modulator.
Lists of matching candidates from EXPREG transcription confirmation factor (TCF).

Abbreviations: AmrZ, Transcriptional activation and biofilm formation; ArgR, arginine biosynthesis; CcpA, Catabolite control protein A; Fur, Ferric uptake regulation protein; HexR, regulation glucose metabolism; HrpX, transcriptional regulator; IhfA, Integration host factor subunit alpha; Lrp, Leucine-responsive regulatory protein; PhhR, phenylalanine hydroxylase regulator and VqsM is virulence and QS modulator; PvdS, encode putative sigma factor; RpoN, gene expression.
Transcription factors regulate gene transcription, that is, their copying into RNA, on the way to protein making. A TF is a sequence-specific DNA-binding protein that controls the transcription of a set of genes, and thus regulates gene expression in a cell. 27 Thus, for this study, about nine TFs were identified from TOMTOM databases that handle the regulation of gene expression in the bacterium of HMR. AmrZ, ArgR, CcpA, Fur, HexR, HrpX, IhfA, Lrp, PhhR, PvdS, RpoN, and VqsM are TFs with diverse functions in gene regulation and expression. The primary functions of these TFs were activation and repression as seen in Table 3. P aeruginosa PAO1, Streptococcus pneumoniae, P putida KT2440, Xanthomonas oryzae, Vibrio cholerae, Shewanella oneidensis, Salmonella enterica, Helicobacter pylori, Lactococcus lactis, and E coli as revealed in Table 3. The sequences of all motif-binding sites were obtained from prokaryotic organisms.
Table 3 shows that HrpX, AmrZ, RpoN, HexR, PvdS, Lrp, CcpA, ArgR, PhhR, and IhfA were predicted TFs that activate transcription by 100%. The rest of the TFs that act as activators, repressors, and others have yet to be identified. As shown in Table 3 above, none of the identified TFs was used in dual. It was found that several TFs exist, but their functions have not yet been revealed, as in the case of Lrp and VqsM. Additional research using various tools and wet labs may be needed for further investigation. Only Fur and HexR TFs had the highest functions of repression on 78% and 69% transcriptional gene expressions, in Table 3. This shows that these TFs take part in greater transcription repression than activation. In general, the data show that most identified TFs are used in activation rather than repression. From an environmental perspective, the PhhR TF has been exploited in a variety of biotechnological applications. It was employed in different technological areas in addition to agriculture, biocatalysts, bioremediation potential applications, and bioplastic production. 10
Table 4 also shows the TF conformational mode of application of the identified TFs as monomers, dimers, tetramers. For instance, about 35.71% of the identified TFs’ conformational mode were not known while 7.14% and 21.43% were identified as monomer and dimer TF conformation mode applications. As shown in Table 4, no TFs were associated with the tetramer TF conformational mode.
Figure 2 revealed the open reading frame (ORF) in which the predicted genes were located. It is usually located at the 5′-UTR region of the upstream of the transcripts. It was known that six ORFs are found in molecular biology, three on the positive, and three on the negative strands. This refers to reading frames in the RNA code used by ribosomes in protein synthesis. As a result, it may serve as a repository for genes with functional elements that could be targeted by heavy metal bioremediation in current studies. In Figure 2, the red color indicates that genes with similar sequences are grouped together in the same number and color. Genes whose relative position is conserved in other species are functionally coupled and share gray background boxes. The focus gene always points to the right, even if it is located on the minus strand. For instance, the czcA and merR genes were pinpointed out of the reading frame in red color in the same direction. The same reading frame varied in size, sequence, and location within the genome.
Genome re-annotation and genome mapping of C gilardii CR3 genome
Circular genome mapping for C gilardii CR3 genomic DNA was performed using a Prokka online server that can be accessed at https://proksee.ca/, which replaced CGView. The GC content, ORF, GC skew (+/-), rRNA, starting codon, stop codon, regulatory elements and CDS were predicted on the genome mapping (see Figure 3A to C). The sequence of C gilardii CR3 was obtained via the NCBI database and re-annotated utilizing subsystems category distribution technology. An analysis was performed on the circular genome for C gilardii CR3, which showed the position of GC content, sequence length and/or contigs, stop, start, ORF, GC skew+ and GC skew– (see Figure 3A to C). Several genes have been thought to be associated with HMR mechanisms in environments with contamination, following this investigation. Genes such as chrA, chrB, chrR, arsA, arsB, copC/D, czcA, czcB, czcD, czc ABC, cadiC, and cadiC play a significant role in the polluted environment to clean or decrease pollutant levels after it had been expressed (see Figure 3A to C). Similar investigation of circular genome analysis for the trehalase gene from the complete genome of Shigella sp. PAMC28760 from Antartica were predicted with CDS, GC contents, rRNA, tRNA, GC skew+ and GC skew– and ORFs have been predicted. 8 The RAST server identified genes with their functions, locations, and levels of protein expression for C gilardii CR3 (CP010516.1) and other related associated strains were shown in Figure 3A. For this study, the number of coding sequences (3399), GC contents (67.7), contigs (1), shortest contig size (3539530), media sequence sizes (3539530), mean sequence sizes (3539530.0), longest contig sizes (3539530), the sequence length in base pairs (N50 values; not predicted) and the smallest number of contigs (L50) (1) whose length sum makes up half of genome size were predicted.
The prediction revealed that the genome contains multiple genes involved in co-factor production, chromosomal replication initiator protein DnaA, vitamin biosynthesis, protein metabolism, nitrogen metabolism, RNA metabolism, nucleosides and nucleotide biosynthesis, stress response and aromatic compound metabolism, copper-sensing two-component system response regulator, mercuric resistance operon regulatory protein, lead, cadmium, zinc and mercury transporting ATPase, and chromate transport protein in Figure 3A.
Comparative genome analysis by using OAT software tools
The 16S rRNA sequences were obtained from WGS C gilardii CR3 (CP010516.1) and related other strains such as C metallidurans CH34 (NC_007973.1), C nantongensis X1 (CP014844.1), Cupriavidus sp. ISTL7 (CP066227.10156), Cupriavidus sp. MP-37 (NZ_CP085344. The phylogenetic tree was performed using the OAT offline server. The sequences were aligned with ClustalW. P putida PD584 (CP115665.1) was used as an out-group in Figure 4A and B.
The neighbor-joining method was used to infer evolutionary history. 28 The best tree was shown with branch lengths. Based on 1000 bootstrap replications through confidence points, evolutionary distances were calculated using neighbor-joining. A bar, 0.01 substitutions per nucleotide location. 29 The evolutionary distance was calculated by p-distance techniques. 30 The investigation connected eight nucleotide sequences as one nucleotide sequence was used as an out-group. All locations having gaps and missing data were eliminated. Finally, the evolutionary relationship investigation was performed with OAT offline server. 22 P putida PD584 (CP115665.1) was selected and used as an out-group in the evaluation; other related sequences were obtained from the EzTaxon-e server and annotated with the RAST server before tree construction (b) UPGMA dendrogram Heatmap for OrthoANI. 22 The OrthoANI values of ⩾ 96% show grouped genomes originating from strains of the same species together (see Figure 4A and B).
A circular phylogenetic analysis of the complete genome of C gilardii CR3(CP010516.1) was studied. The phylogenetic tree shows heavy metal resistance genes location in the complete genome of the organism in Figure 4C. The data also show that C gilardii CR3 is 99.41% related to Cupriavidus sp. ISTL7. Figure 4A and B shows C gilardii CR3 (CP010516.1) has a close relationship to Cupriavidus sp. ISTL7 (CP066227.10156) and C gilardii MW02 (CP104386.1) and less related to that of C nantongensis X1 (CP014844.1) and C metallidurans CH34 (NC_007973.1). The 16S rRNA gene sequence results of C gilardii CR3 (CP010516.1) were highly evolutionary related to Cupriavidus sp. ISTL7 (CP066227.10156), C gilardii, C gilardii WM02 (CP104386.1), and Cupriavidus sp MP-37 (NZ_CP085344.1) as seen in Table 5. The data also revealed that it was less evolutionary related to that of C metallidurans CH34 (NC_007973.1). Thus, the evolutionary relationship between the organisms was 99.41%, 98.85%, 98.41%, and 94.39%, respectively (see Table 5). The OrthoANI again revealed a weak correlation among C metallidurans CH34 (NC_007973.1) compared with C gilardii CR3 (see Figure 4A and B). As shown in Figure 4A and B, the sequence similarity of OrthoANI between C gilardii CR3 (CP010516.1) and Cupriavidus sp. ISTL7 (CP066227.10156), C gilardii CR3 (CP010516.1) C gilardii, C gilardii CR3 (CP010516.1) and C gilardii WM02 (CP104386.1), C gilardii CR3 (CP010516.1) and C nantongensis X1 (CP014844.1), C gilardii CR3 (CP010516.1) and Cupriavidus sp. MP-37 (NZ_CP0 85344.1) C gilardii CR3 (CP010516.1) and C metallidurans CH34 (NC_007973.1) were 99.41%, 98.85%, 98.41%, 84.10%, 84.21%, and 80.85% (see Figure 4A and B). The larger sequence similarities among the current genome annotated sequences of our strains were calculated between C gilardii CR3 (CP010516.1) and Cupriavidus sp. ISTL7 (CP066227.10156), OrthoANI is a genomic similarity measuring technique. It was utilized to evaluate the same species of organism with a demarcation cut-off value of 95% to 96%, and massive comparison tests showed that both algorithms produced identical reciprocal similarities (see Figure 4A and B) which confirms our phylogenic tree. Accordingly, the OrthoANI obtained between C metallidurans CH34 (NC_007973.1) and P putida PD584 (CP115665.1) as well as other related genera in the present investigation were lower of the demarcation cut-off values.
Features of re-annotated genomic DNA for Cupriavidus gilardii CR3 genome, its allied reference strains, and out-group.

The data were predicted and generated by RAST (Rapid Annotation utilizing Subsystem Technology) and SEED (The database and infrastructure for comparative genomics) technologies.
Discussions
The objective of this work was regulatory elements analysis and comparative genome analysis of HMR bacteria extracted from C gilardii CR3 as a means of bioremediation. Sequence-specific DNA-binding TFs are usually revealed as “main regulators” because they bind to DNA and either activate or suppress gene expression. The identification of a gene’s TSS precedes the pointing out of the upstream promoter region, making gene expression analysis easier. Most of the sequences in this study showed multiple TSSs, which accounted for about 86.67% in Table 1. As a result, the gene sequence in consideration may have alternate transcription possibilities. However, TSS with a higher threshold value were evaluated for better prediction that related to earlier studies. 31 Genes that have multiple TSS enhance transcription initiation and are more likely to respond to environmental changes, which agreed with the findings of Dai et al. 32
Common motif identification was a core element of TF identification processes. Motifs recruit TFs, or regulatory elements to properly target genes in gene expression processes. 33 The MEME tools were utilized to identify a conserved candidate motif where these 14 TFs bind and motif sites were discovered (see Figure 5). In this study, common candidate motifs with different information content were identified. The densely populated motifs with their locations were found upstream between −100 and −600 (see Figure 5). These motifs might take part in the regulation of recognized promoters with contributors in gene expression. In this investigation, several binding sites were discovered in the promoter region of candidate motifs. These locations might enhance binding interactions and produce different regulatory effects.34,35
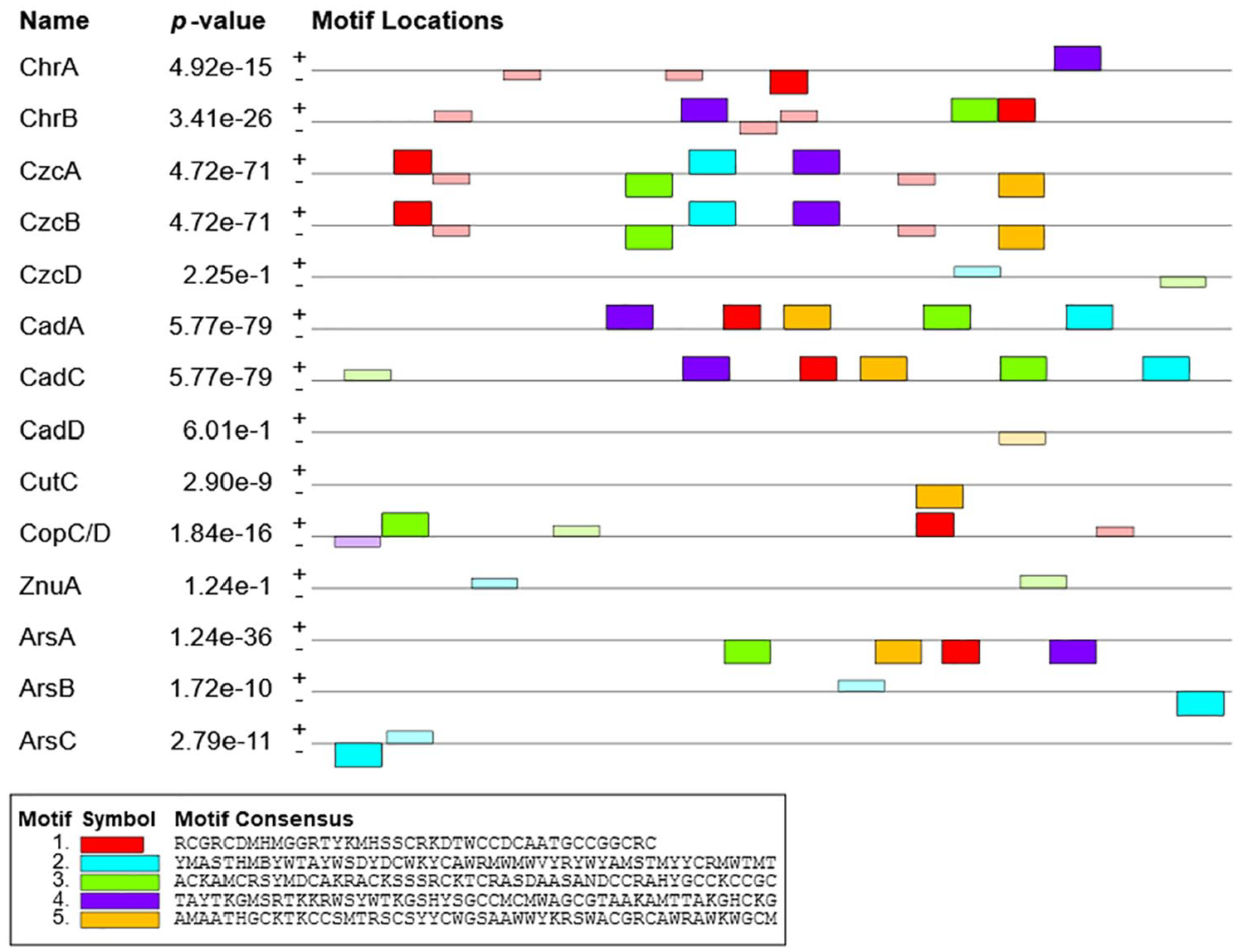
Block diagrams illustrate the relative location of potential motif + scanned sites in the promoter region relative to TSSs (transcription start sites). The bottom of the graph indicates the locations of nucleotides in the promoter region of heavy metal resistance genes. The locations range from the beginning of the TSS (+1) to upward of 1 kb (–1 kb) of the MEME suite outcome.
The data also show that the gene arsB at locus_tag = CR3_1877 with CDS located at complement (2282267. . .2283709) was a membrane product protein extracted from C gilardii CR3. In addition, the arsC and arsH genes at locus_tag = CR3_2746 and locus_tag = CR3_1262 were used as arsenate reductase and arsenical resistance functions, respectively. Moreover, the arsR gene with CDS complement (1487862. . .1488218) is located in the genome of C gilardii CR3, a family transcriptional regulator protein widely used for arsenate bioremediation capacities. Similar studies were performed in the complete genome of C metallidurans CH34 as a model bacterium of HMR. Resisting heavy metals by tri-component efflux systems were identified in pMOL30, with czcCBA present with resistance to Cd2+, Zn2+, and Co2+. 36 Environmental components such as soil and water contaminated areas with heavy metals are potential sources of HMR bacteria. These microbes could reduce, oxidize bio-accumulate, and efflux heavy metals through cell membranes and are widely used in bioremediation processes. 37 According to the comparative analysis, the genome of Cupriavidus sp. ISTL37 is 99.41% identical to the genomes of C gilardii CR3 as shown in Figure 5. A similar finding was observed on the C metallidurans MSR33; 38 however, it was less related to C gilardii CR3 as seen in Figure 5.
The MerR families include merP, MerT protein that is widely distributed in the bacterium of C gilardii CR3 and mercury resistance has also been investigated in this study (see Figure 3A). The finding is similar to the studied identified genes from Eubacteria. 39 The C gilardii CR3 regulates heavy metals through TFs controlled by the MerR, CopR, CadR, and ChrR protein families. We also studied the cadmium, copper, arsenate, zinc, and chromium resistance genes of CopB, CopC, CopCD, CopR, CadA, CadB, CadR, ArsA, ArsB, ArsC, ArsH, ArsR, CzcA, CzcB, ChrA, ChrB (see Figure 4C) resistant systems and demonstrated that the combined protein sequence analysis and analysis of DNA regulatory elements was possible to differentiate the genes involved in the metal resistant, in particular, mercury, copper, cadmium, zinc, and arsenate.40 -43 Several enzymes contribute to the efficient functions of the genes, including a multicopper oxidase, a cadmium transporter, a zinc transporter, and a copper/zinc/cadmium efflux system. 44
The RAST server identified gene calls based on their function, location, and degree of protein expression. In addition, sequence size, number of coding sequences, number of contings, GC content percentage, shortest contig sequence, mean sequence size, longest contig size, No50 values, L50 values, number of RNA, and a number of subsystems were predicted by the RAST server. The GC content percentage of C gilardii CR3 is similar to that of C gilardii WM02, which is 67.4%. However, the highest GC% is documented for Cupriavidus sp. ISTL7 (67.7%) while the lowest GC% is observed for P putida PD584 (61.6%). Similar findings were found in the study on Bacillus sereus. 45 Following C gilardii CR3 genomic DNA re-annotation with the RAST online server, subsystem category distribution and subsystem feature counts were anticipated. These subsystems are supposed to support cellular activity and metabolism. Strengthening our suggestion, it was projected that these subsystems would be used to provide broad metabolic information, increase annotation quality, and provide a framework for determining the statistical qualities required to properly exploit these tendencies. Our results anticipate carbohydrates and other chemical molecules. The OrthoANI, a tool that predicts sequence similarity between C gilardii CR3 and related strains, is an example of an OAT tool offline server. 22 When tested against OrthoANI, C gilardii CR3 (CP010516.1) displayed a genome sequence similarity of 99.16%. The C gilardii CR3 genomic DNA was found to be most similar to Cupriavidus ISTL7 with 98.85% OrthoANI (see Figure 4A), using the OAT tool. 22 Followed by C gilardii MW5 (98.80% OrthoANI), C gilardii strain WM02 (98.32% OrthoANI), Cupriavidus sp. MP,37 (94.39% OrthoANI), C nantongensis X1 (CP014844.1; 83.51% OrthoANI) and C metallidurans CH4.
Conclusions
In this study, 15 genes from C gilardii CR3 genomes were examined for HMR applications. To determine the functional conformation of the predicted genes, the promoter region, transcriptional regulatory elements, phylogenetic tree analysis and comparative genomic by OAT analysis, and RAST and SEED tools were utilized. Five common candidate motifs for binding sites related to regulatory elements were investigated. UniProtKB data showed additional research for a better understanding of the function of the TFs. The overall results revealed that identification of the common candidate motifs and their regulatory elements such as TFs, comparative genome analysis by OAT, and genome re-annotation by RAST with SEED technologies were employed for the investigation. The data show that 14 TFs were identified. In both positive and negative regulations,’ these TFs were widely used to regulate enzymatic biosynthesis and metabolism applications. In addition, comparative genome analysis, annotation, bioinformatics, and omics tools were expected to contribute significantly to our understanding of gene expression in prokaryotic. Therefore, more studies could be done to identify the screened TFs and their common binding sites in gene expression mechanisms in microorganisms with the help of a wet lab.
Footnotes
Acknowledgements
The authors are grateful to Adama Science and Technology University for the opportunities.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
DD, TD, and SM conceptualized and designed the study, analyzed, and interpreted the data. TD and SM analyzed, revised, and edited the article. All authors read and approved the final article to the published.
Ethical Approval and Consent of Participation
Not applicable.
Consent of Publication
Not applicable.
Availability of Data and Materials
The datasets used in this study were extracted from the NCBI database. Where necessary, it can be obtained from the corresponding author.