
Abstract
Genomes may now be sequenced in a matter of weeks, leading to an influx of “hypothetical” proteins (HP) whose activities remain a mystery in GenBank. The information included inside these genes has quickly grown in prominence. Thus, we selected to look closely at the structure and function of an HP (AFF25514.1; 246 residues) from Pasteurella multocida (PM) subsp. multocida str. HN06. Possible insights into bacterial adaptation to new environments and metabolic changes might be gained by studying the functions of this protein. The PM HN06 2293 gene encodes an alkaline cytoplasmic protein with a molecular weight of 28352.60 Da, an isoelectric point (pI) of 9.18, and an overall average hydropathicity of around −0.565. One of its functional domains, tRNA (adenine (37)-N6)-methyltransferase TrmO, is a S-adenosylmethionine (SAM)-dependent methyltransferase (MTase), suggesting that it belongs to the Class VIII SAM-dependent MTase family. The tertiary structures represented by HHpred and I-TASSER models were found to be flawless. We predicted the model’s active site using the Computed Atlas of Surface Topography of Proteins (CASTp) and FTSite servers, and then displayed it in 3 dimensional (3D) using PyMOL and BIOVIA Discovery Studio. Based on molecular docking (MD) results, we know that HP interacts with SAM and S-adenosylhomocysteine (SAH), 2 crucial metabolites in the tRNA methylation process, with binding affinities of 7.4 and 7.5 kcal/mol, respectively. Molecular dynamic simulations (MDS) of the docked complex, which included only modest structural adjustments, corroborated the strong binding affinity of SAM and SAH to the HP. Evidence for HP’s possible role as an SAM-dependent MTase was therefore given by the findings of Multiple sequence alignment (MSA), MD, and molecular dynamic modeling. These in silico data suggest that the investigated HP might be used as a useful adjunct in the investigation of Pasteurella infections and the development of drugs to treat zoonotic pasteurellosis.
Keywords
Introduction
Next-generation sequencing (NGS) has shortened the time it takes researchers to collect massive volumes of data. 1 The difficulty of attributing functions to genes is growing as the genomes of more and more species are sequenced. Among the all sequenced data, more than 30% of proteins in various animals are called “Hypothetical Proteins” (HPs) because their molecular activities are unknown. 2 The increased quantity of raw HP is compelling researchers to find ways to use them. Characterizing hypothetical proteins in silico aids in the determination of their 3-dimensional (3D) structures, which may lead to the discovery of previously unknown domains, motifs, pathways, protein networks, etc.3-5 Potential biomarkers and pharmaceutical targets may potentially be uncovered by structural and functional annotation of HPs. 6 One such example is the newly discovered Shigella dysenteriae ATCC 12039 HP, which shows promise as a treatment against that particular bacteria. 1 Meanwhile, newly characterized M4, a bacterial metalloprotease, demonstrating their use in the development of antimicrobial vaccines and biotechnological enzymes. 7 In addition, an HP from Orientia tsutsugamushi str. Karp shows promise as a new antibacterial medication targeting the bacterium. The roles of putative proteins in several pathogenic bacteria have been effectively annotated using a number of bioinformatics databases and techniques.8-10 Pasteurella multocida (PM) is an example of a pathogenic bacterium; it is rod-shaped, gram-negative, facultative anaerobic, coagulase-negative, and causes several zoonotic diseases across a wide range of hosts and habitats.1-4 The bacterium is a common commensal or opportunistic pathogen that lives in the upper respiratory tracts of many different types of animals. 11 This includes dogs,12-15 cats,16-18 rabbits,19-21 cattle,22,23 goat,24-27 bison, 28 swine, 11 ,29-31 marine mammals, 32 chimpanzee, 33 and komodo dragons.34,35 This implies that the PM may infect a wide variety of species and is responsible for a number of economically significant illnesses such avian fowl cholera, bovine hemorrhagic septicemia, zoonotic pneumonia, and swine atrophic rhinitis.2,3,5,9 In humans, respiratory infections are uncommon, but those who suffer from chronic pulmonary sickness are particularly vulnerable.36,37 Severe consolidation pneumonia, epiglottitis, lymphadenopathy, and abscess formation are all possible symptoms of pasteurellosis in such situations.15,38 In the previous 30 years, the number of human cases of pasteurellosis has increased from 20 to 30, and this trend seems to be continuing. It has been estimated that more than 300 000 persons in the United States visit emergency rooms annually due to animal scratches or bites, with PM being the most often linked illness type.15,39 A total of 162 cases of Pasteurella infections were reported in Hungary during the years of 2002 and 2015. 40 Forty-four instances of Pasteurella infections were reported in the United States between 2000 and 2014, with 8 patients requiring intensive care unit (ICU) treatment. 41 Invasive pasteurellosis, however, was associated with a 27.1% mortality rate in Hungary and a 21% mortality rate in the United States.40,41 Because of this, there is now a pressing need to investigate and study zoonotic pasteurellosis extensively to contain its spread. In this light, PM subsp. multocida str. HN06, the whole genome of which was just released. The National Center for Biotechnology Information (NCBI) database states that it encoding 2117 proteins (AFF25514.1). Nevertheless, expression and function data are missing for approximately 2000 predicted protein-encoding coding sequences. The word “hypothetical” has been used to these chains. These HPs account for almost half of all proteins in the genome (47.6%). For these HPs to discover their potential roles in the cell and provide light on novel structures and functions in this bacterium’s participation in the illness process, functional annotation is essential. Because of the potential importance of this organism’s genome to the success of a medication or vaccine still in development in labs, in silico examination of these putative proteins is crucial. In this work, we use many different bioinformatics programs to investigate the structure and function of a putative protein (accession no. AFF25514.1; 246) from PM subsp. multocida str. HN06.
In light of these considerations, the purpose of this study is to define a PM subsp. multocida str. HN06 HP and investigate its potential as a therapeutic target of the bacterium, which may be helpful in combating zoonotic pasteurellosis as well. Thus, we predicted the 3D structure of this bacterium’s HP, annotated its function, and described it using a variety of computational methods. Researchers also identified its role as an altered form of a protein essential to their replication machinery called S-adenosylmethionine (AdoMet or SAM)-dependent methyltransferase (MTase). The potential of this HP in preventing pasteurellosis was effectively identified. Ultimately, this study may be used as a future hope for preventing and treating PM zoonosis.
Materials and Methods
Retrieval of protein sequence
By searching the NCBI Protein database (https://www.ncbi.nlm.nih.gov/protein/) for the phrase “Hypothetical proteins AND Pasteurella multocida” we were able to locate the 246-residue HP of PM subsp. multocida str. HN06. Among the hits found, we randomly selected an HP (accession no. AFF25514.1, GI| 380873147|), and its sequence was acquired in FASTA format for further examination. A sequence-based peptide search was also performed in the UniProt database (https://www.uniprot.org/peptidesearch/) to determine whether or not the protein is redundant. The whole research plan is shown in Figure 1.

The study’s overarching notion is shown in a flowchart. Cyan, light green, and blue boxes represent HP’s sequence analysis, structural evaluation, and molecular interaction tests, respectively.
Physicochemical properties analysis
The chemical and physical attributes of the favored HP were assessed using the ProtParam tool on the ExPASSy website (https://web.expasy.org/protam/). The analyzer provides theoretical metrics such as molecular mass, amino acid composition, totally positive and negative residue count, extinction coefficient, theoretical pH, aliphatic index (AI), instability index (II), and grand average of hydropathicity (GRAVY) score. 42
Annotation of functional domain
Functional annotation was applied to the HP to reveal its functions. Several publicly available tools and databases, including NCBI CDD (https://www.ncbi.nlm.nih.gov/cdd/), 43 InterProScan (https://www.ebi.ac.uk/interpro/search/sequence/), 44 and SUPERFAMILY (https://supfam.mrc-lmb.cam.ac.uk/SUPERFAMILY/) 45 were used to annotate precisely the conserver and functional domain within HP. The default settings were considered in each case. These databases and other bioinformatics tools aid in the identification of conserved domains, which are then used to classify the proteins.
Multiple sequence alignment and phylogenetic analysis
Sequence similarities with the studied HP were searched using NCBI’s Basic Local Alignment Search Tool (BLAST) (https://blast.ncbi.nlm.nih.gov/Blast.cgi). We used NCBI’s BLASTp method 46 to search for matches in a unique protein database. Multiple protein sequences were initially retrieved from the NCBI protein database, all of which were assumed to have the same purpose. The Molecular Evolutionary Genetics Analysis X (MEGA X) program was then used to conduct the multiple sequence alignment (MSA) and phylogenetic analysis between the HP and recovered protein sequences. 47 The ClustalW method, which works in steps, was employed for the MSA analysis. 48 To further illustrate the evolutionary separation of the linked proteins, a phylogenetic tree was built by homologous sequence alignment. We used the standard settings (maximum likelihood, or ML, techniques) with 1000 replicates of the bootstrap. 49
Secondary structure prediction of selected hypothetical protein
The PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred)50,51 and SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/npsaautomat.pl?page=/NPSA/npsasopma.html) servers were used to make predictions for the HP’s secondary structure (2D). Comparatively, SOPMA predicts a protein’s secondary structure by consulting the “DATABASE.DSSP,” whereas the PSIPRED service employs feed-forward neural networks and the PSI-BLAST algorithm.50,51 Secondary structure prediction was performed in both instances using the HP’s FASTA sequence. 52
Tertiary structure prediction of protein
The tertiary (3D) structure of the HP was predicted by the HHpred (https://toolkit.tuebingen.mpg.de/tools/hhpred)53-56 and I-TASSER (https://zhanggroup.org/I-TASSER/) servers.57-59 Using the MODELLER software developed at the Max Planck Institute for Developmental Biology,53-56 the HHpred predicts the 3D structure of a hitherto uncharacterized protein. In addition, beginning with an amino acid sequence, I-TASSER generates 3D atomic models using multiple threading alignments and iterative structure assembly simulations. 60 For homology modeling, both HHpred and I-TASSER used their respective default values for all parameters. The 3D structures predicted by the HP were refined, and their energy was minimized using the YASARA energy minimization server 61 (http://www.yasara.org/minimizationserver.htm). GalaxyRefine (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) 62 was then used to further enhance the refined 3D structures. GalaxyRefine generates several possible structures; the best quality and performance ones are hand-picked. PyMOL and BIOVIA Discovery Studio were then used to create 3D images of the HP’s structures.
Model quality assessment of studied hypothetical protein
The energy-minimized and fine-tuned 3D structure of the HP was evaluated using the PROCHECK 63 and ERRAT 64 modules of the SAVES server (https://saves.mbi.ucla.edu/). The Z-score of the projected models was also forecasted using the ProSA server (https://prosa.services.came.sbg.ac.at/prosa.php).65,66 The HP 3D model was further validated using the SWISS-MODEL Structural Evaluation tool67,68 developed by the Swiss Institute of Bioinformatics (SIB). In the end, the highest-quality model was selected for future study.
Active site prediction of hypothetical protein of Pasteurella multocida strain HN06
The HP’s active site and residues were determined with the use of the Computed Atlas of Surface Topography of Proteins (CASTp) (http://sts.bioe.uic.edu/castp/calculation.html) 69 and FTSite (https://ftsite.bu.edu/) 70 servers. Protein Data Bank (PDB), 71 UniProt, 72 and Structure Integration with Function, Taxonomy, and Sequence (SIFTS) 73 databases were also used. When a protein’s structure and its sequence are correlated, as they are in the CASTp server, rapid residue-level annotations become possible. 69 The predicted active site and residues were further validated by molecular docking (MD) analysis. In addition, the docking investigation verified the anticipated active site and residues. Small organic compounds of varying sizes and polarities may bind to ligand-binding sites, as shown by the FTSite server’s implementation of an algorithm verified by experimental data. Without employing evolutionary or statistical data, the program achieves near experimental accuracy. 70
Subcellular localization and function prediction of hypothetical protein
The spatial environment that governs a protein’s interaction patterns and biological networks influences a protein’s ability to function at its best. 74 For this context, the subcellular localization of the HP was predicted by multiple servers including PSLpred (https://webs.iiitd.edu.in/raghava/pslpred/), 75 SOSUIGramN (https://harrier.nagahama-i-bio.ac.jp/sosui//sosuigramn/sosuigramn_submit.html), 42 Gneg-PLoc (http://www.csbio.sjtu.edu.cn/bioinf/Gneg-multi/),76,77 DeepTMHMM 2.0 (https://dtu.biolib.com/DeepTMHMM), 78 and PSORTb (https://www.psort.org/psortb/) servers. 79
Molecular docking of hypothetical protein with S-adenosylmethionine and S-adenosylhomocysteine
Molecular docking is frequently employed to investigate and evaluate the intermolecular interactions between ligands and macromolecules. 80 Hence, docking experiments were performed on the HP using both SAM and S-adenosylhomocysteine (SAH) as the ligands. The Structured Data File (SDF) formatted data files and the structures of both ligands were downloaded from the PubChem (https://pubchem.ncbi.nlm.nih.gov/) 81 database and then converted to the PDB format using the PyMOL program. AutoDock Vina82,83 software and the SeamDock (https://seamless.rpbs.univ-paris-diderot.fr/cloudless/instance/5208806/ctx/index.html)84-86 server were then used to conduct a docking study between the HP and ligands. AutoDock Vina was used for both site-specific and blind docking, with the program being run with exhaustiveness = 24 and energy range = 4. Except for adjusting the exhaustiveness number to 24, the SeamDock server’s default settings were used for the MD analysis. Even yet, this server merely underwent the blind docking method.
Molecular dynamic simulation
The stability and function of every protein complex depend on the atoms’ mobility, which may be analyzed computationally using molecular dynamic simulation (MDS).86-88 For this reason, MDS was performed on the HP-ligand complexes, such as HP-SAM and HP-SAH, predicted by the AutoDock Vina, using the Internet server “WebGRO for Macromolecular Simulations” (https://simlab.uams.edu/). 89 The ligand topology files, which are required for the simulation run, were generated using the GlycoBioChem PRODRG2 Server (http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg). 90 Selecting “neutralize” and “add 0.15 M salt” and using the SPC 91 box type of triclinic water model were other necessary parameters in addition to using the Gromos96 43a192 force field on the Webgrow server. Moreover, the energy minimization settings 93 include a steepest descent integrator and 5000 steps. NVT/NPT (here, N-Constant number, V-Constant volume, T-Constant temperature, P-Constant pressure) equilibration, 300 K temperature, 1 bar pressure, 50 ns simulation period, and 1000 estimated frames per simulation are also recommended for MDS runs. 94 Finally, the results of the MDS analysis have been interpreted, and the stability and flexibility of the docked complexes have been assessed using metrics such as the root mean square deviation (RMSD) of the given structure over time, the root mean square fluctuation (RMSF) of each residue in the given structure, the average number of H-bonds in each frame over time, the radius of gyration (Rg) or structural compactness, and the solvent-accessible surface area (SASA). 89
Result and Discussion
Retrieval of protein sequence
The NCBI Protein database was queried at random, yielding the HP PMCN06 2293, which is the PM strain HN06 HP. The acquired sequence was then used to search UniProt, a public, free database of protein sequences and their functional annotations. For the sake of analysis, the HP’s attributes have been saved. This includes the HP’s locus, definition, accession, version, and version as well as the HP’s total number of amino acids and FASTA sequence. There are a total of 246 amino acids in the HP, which has been labeled as PMCN06 2293 and assigned the locus, accession, and version numbers of AFF25514, AFF25514, and AFF25514.1 (Table 1).
The properties of HP protein retrieved from NCBI protein database.

Abbreviations: HP, hypothetical protein; NCBI, National Center for Biotechnology Information.
Physicochemical properties analysis
Several physicochemical parameters of the HP PMCN06 2293 were analyzed using the ProtParam tool of the ExPASSy service, and the findings are shown in Table 2. The server predicted that the HP has a 246 amino acid sequence and a molecular weight of 28 352.60 Da. A theoretical pI value of −9.18 was calculated as well for the HP by the server, indicating that it is an alkaline protein with a high negative charge. Protein stability is a crucial factor in various biological processes. One way to determine the stability of a protein is by calculating its II. If the II of a protein is less than 40, it is anticipated to be stable. However, if the II is more than 40, the protein is expected to be unstable. 95 This predicts that HP is an unstable protein with a stability score of 56.57. The AI of a protein is the ratio of the volume occupied by its aliphatic side chains (alanine [Ala], valine [Val], isoleucine [Ile], and leucine [Leu]) to the overall volume of the protein. 96 Therefore, an AI of 84 is predicted for HP, indicating the protein’s widened temperature stability. For each amino acid in the query sequence, its hydropathy value is computed and then divided by the total number of residues to get the GRAVY score for the peptide or protein. The computed value for HP is −0.565, proving that it is a hydrophilic protein. According to the Beer-Lambert law, the extinction coefficient serves as a proportionality constant and measures the intensity of a certain wavelength of light absorbed by a protein. 97 Therefore, the extinction coefficient of the HP was calculated to be 25 565. There are plenty of tyrosine, tryptophan, and cysteine around because of the high extinction coefficient. 95 However, Table 2 provides a comprehensive overview of the physicochemical properties of HP. These features will be helpful when working with the protein in future studies.
The physicochemical properties of HP protein predicted by ExPASSy server.

Abbreviations: HP, hypothetical protein; pI, isoelectric point.
Annotation of functional domain
Predicted by the servers to be present in the HP is the well-known conserved domain of tRNA (adenine(37)-N6)-methyltransferase TrmO (Supplementary Table 1). Escherichia coli yaeB or tRNA (adenine(37)-N6)-methyltransferase TrmO is an SAM-dependent MTase variant that has also been identified.98,99 Moreover, this variant of SAM-dependent MTase has been classified as a unique AdoMet-dependent methyltransferase Class VIII.98,99 This organism’s version of SAM-dependent MTase is responsible for the formation of N6-methyl-threonylcarbamoyl adenosine (m6t6A) by methylating t6A at position 37 of tRNA-Thr.98,99 It has been shown that the attenuation activity of the operon is considerably improved after N6 methylation of t6A to m6t6A, which is consistent with the effective decoding of ACC codon.98,99 In addition, most known MTases use SAM as a co-factor of methylation or a donor of the methyl group, which is thereafter converted into SAH by cleavage of the CH3 group. 100 Many computational methods have speculated that the HP’s conserved domain functions like E coli’s tRNA (adenine(37)-N6)-methyltransferase TrmO or a modified AdoMet-dependent methyltransferase of Class VIII.
Multiple sequence alignment and phylogenetic analysis
The NCBI protein database served as a BLASTp server, which returned HP values for the proteins that were found. In this instance, the software was run against a nonredundant protein database to return the microorganisms with the largest percentage of identical protein sequences, the lowest e-value, and the highest query coverage. These results suggest that the HP and tRNA (N6-threonylcarbamoyladenosine(37)-N6)-methyltransferase TrmO may have comparable purposes (Table 3). After that, the MEGA X program was used to do sequence alignment and phylogenetic tree building. For MSA and tree building, we used the MEGA X software’s ClustalW algorithm and ML technique, respectively, for their iterative processes. The HP and Pasteurella tRNA (N6-threonylcarbamoyladenosine(37)-N6)-methyltransferase are 100% identical in sequence, placing them in the same clade of the evolutionary tree (Figure 2). Nevertheless, HP has also revealed that Pasteurella oralist tRNA (N6-threonylcarbamoyladenosine(37)-N6)-methyltransferase has 81% sequence similarity with that of Actinobacillus rossii, the closest relative outside of the subtree. As a tRNA (N6-threonylcarbamoyladenosine(37)-N6)-methyltransferase or Class VIII SAM-dependent MTase, HP was also shown to have sequence similarity with an unidentified ancestor’s tRNA (N6-threonylcarbamoyladenosine(37)-N6)-methyltransferase (Figure 2).
The identical proteins with the HP, aligned by BLASTp algorithm, the NCBI.

Abbreviations: HP, hypothetical protein; NCBI, National Center for Biotechnology Information.

The evolution and ancestral relationship of the HP with the top aligned sequences. The red marked sequence represents the HP, whereas the tree nodes represent the ancestral relationship.
Secondary structure prediction
The HP’s secondary structure has been predicted using tools like PSIPRED and the SOPMA servers. As a quick summary, the PSIPRED server projected that the HP structure will include the most random coils, followed by prolonged strands, and finally an alpha-helix area (Figure 3). The SOPMA server agreed with the PSIPRED’s assessment that the HP would have a greater proportion of random coil than extended stand or alpha helix (Table 4 and Supplementary Figure 1).

The secondary structure of the HP predicted by PSIPRED server. The strand, helix, and coil structures are depicted by the yellow, pink, and ash colors.
The predicted secondary structure of the HP by SOPMA server.

Abbreviation: HP, hypothetical protein.
Tertiary structure prediction
For accurate HP model prediction, we used the HHpred and I-TASSER servers. The HHpred server determined an optimal 3D model of HP by comparing it to a database of known protein structures and picking a template that best fit the protein’s structure. Using the criteria of a 100% success rate, an E-value of 6.7e−67, and a secondary structure score of 28.4, the template 7BTU_B was selected as the template to aim toward (Figure 4A). In addition, I-TASSER predicted a total number of 29 models for the intended HP, and the model with a C-score of 0.00, an estimated TM-score of 0.710.11, and an estimated RMSD of 5.83.6 was selected among all models predicted by I-TASSER (Figure 4B). Subsequently, the YASARA and GalaxyRefine servers have reduced and refined the anticipated models. PyMOL and BIOVIA Discovery Studio were used to examine and display the tertiary structures of the predicted and revised models.

The tertiary structure of the HP predicted by HHpred (A) and I-TASSER (B) servers. The spiral and arrow ribbon represent alpha-helix and beta-sheet structures, whereas the line ribbon represents coil structure of the HP, respectively.
Model quality assessment
The SAVES PROCHECK found that 89.9% of the amino acid residues in the HHpred-predicted model of the HP were located in the Ramachandran preferred area, but only 84.5% of the residues in the I-TASSER referenced model were located there (Figure 5). The ERRAT score is likewise greater in the HHpred-predicted model (87.5) compared with the I-TASSER-predicted model (85.3211) (Table 5 and Supplementary Figure 2). Both HHpred and I-TASSER provide a negative value for the HP model’s projected Z-score: −4.76 and −4.71 (Table 5 and Supplementary Figure 3). The SWISS-MODEL predicts that the HP created by HHpred has a MolProbity score of 3.64, a Ramachandran preferred area of 90.76%, a QMEAN of −3.64, and a QMEANDisCo Global of 0.61 0.05. The server also came up with a MolProbity score of 3.64, a Ramachandran preferred area of 90.57%, a Qualitative model energy analysis (QMEAN) of −3.64, and a Qualitative model energy analysis-distance constrainst (QMEANDisCo) Global value of 0.62 0.05 for the I-TASSER projected model (Table 5). We analyzed each anticipated model’s structure and settled on the HHpred model for further study.

The Ramachandran plot of the predicted models by HHpred (A) and I-TASSER (B) server. The first represents the tertiary structure of the HP such as the beta-sheet region, where second and third quadrants represent the right-handed and the left-handed alpha-helix region, respectively. In addition, the red, yellow, gray, and white color regions depict the residues in most favored, additional allowed, generously allowed, and disallowed region, respectively.
The model quality assessment of the HP by SAVES, ProSA, and SWISS-MODEL structural assessment server.
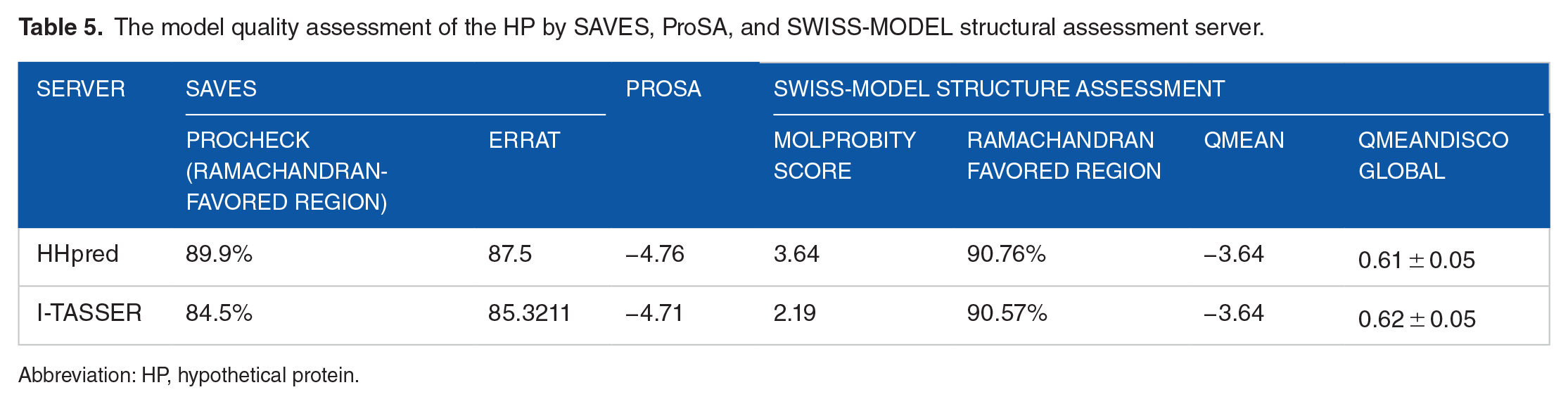
Abbreviation: HP, hypothetical protein.
Active site prediction
The CASTp server predicted a total number of 75 amino acid residues within the active site of HP. However, the active site has been predicted to be covered a total surface area and surface volume of 1811.175 and 2510.612 Å2, respectively (Figure 6A). In the meantime, the FTSite predicted 37 active amino acid residues within the active site of HP (Figure 6B). However, there are 27 common active amino acid residues reported from the servers including Lys-21, Phe-22, Ser-23, Val-24, Pro-25, Arg-26, Pro-28, Phe-63, Gln-64, Phe-65, Asp-66, Arg-94, Thr-96, Gly-103, Leu-104, Ser-105, Asp-127, Leu-128, Val-129, Thr-132, Gln-195, Asp-196, Pro-197, Arg-198, Pro-199, Ala-200, and Tyr-201 (Figure 6C).

The predicted active sites and active amino acid residues by CASTp (A) and FTSite (B) server and common active residues (C) from these servers. The cyan color denotes the protein, whereas the purple color indicates the active amino acid residues.
Prediction of subcellular localization
Numerous servers—such as PSLpred, SOSUIGramN, Gneg-PLoc, DeepTMHMM 2.0, and PSORTb—have made predictions on where in the cell the HP will be found. Different cellular locations are linked to various biological processes, 101 therefore knowing where an HP is found inside the cell might provide light on its potential role. This knowledge might be useful in creating a medication that inhibits the functioning of the targeted protein. 101 As a result, the authors hypothesized the HP is a cytoplasmic protein with comparable functions to other cytoplasmic proteins (Supplementary Table 2).
Molecular docking analysis
The MD study showed that the HP and ligands had several intermolecular interactions (SAM and SAH). Docking scores of −7.4 and 7.5 (kcal/mol) for the HP indicate that SAM and SAH, 2 ligands, have a strong affinity for the HP in site-specific docking (AutoDock Vina) (Table 6 and Figure 7A and B). With a docking score of −7.7 (kcal/mol), both SAM and SAH showed strong attraction for HP in blind docking (Table 6 and Figure 7C and D). Site-specific docking, however, reveals that the HP-SAM and HP-SAH-docked complexes include 16 and 19 interacting amino acid residues of the HP, respectively (Table 6 and Figure 8A and B). The HP-SAM- and HP-SAH-docked complexes have 6 conventional hydrogen bonds. The HP-SAM had 8 van der Waals and 2 carbon-hydrogen bonds, whereas the HP-SAH-docked complexes had 9 van der Waals and 3 carbon-hydrogen bonds. Hydrogen bonds are a vital aspect in determining the specificity of ligand binding. In addition, blind docking showed that the HP has 18 interacting amino acid residues within the HP-SAM complex and 19 interacting amino acid residues within the HP-SAH complex (Table 6 and Figure 8C and D). There are a total of 6 conventional hydrogen bonds in the docked complexes of HP-SAM and HP-SAH. The HP-SAM contained 11 van der Waals and 1 carbon-hydrogen bonds, whereas HP-SAH docked complexes had 8 van der Waals and 11 carbon-hydrogen bonds. Notably, the amino acid residues LYS-21, PHE-22, VAL-24, GLN-195, and ASP-196 are all documented in both site-specific and blind docking to interact with the SAM and SAH. Docking scores of −7.1 (kcal/mol) and −7.3 (kcal/mol) were obtained from the SeamDock server for the HP-SAM and HP-SAH complexes, respectively, validating the predictions of AutoDock Vina (Table 6 and Supplementary Figure 4). Results from the functional domain and MSA analyses suggested that the HP may act as a variant of SAM-dependent MTase; this hypothesis was confirmed by the following docking study. Therefore, we decided to conduct our molecular dynamic simulation research on docked complexes generated using site-specific docking (AutoDock Vina).
The MD analysis of the HP with the SAM and SAH.
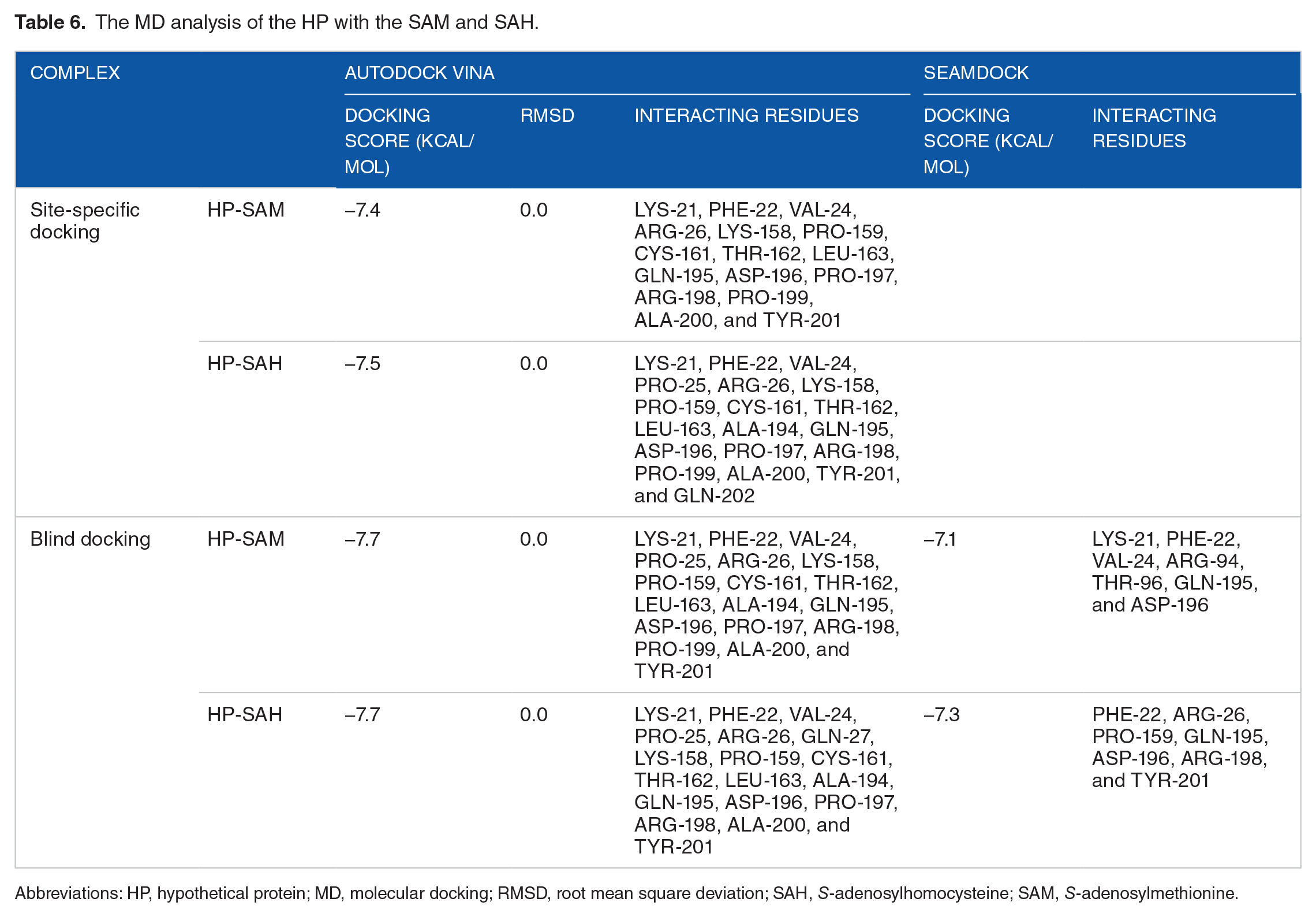
Abbreviations: HP, hypothetical protein; MD, molecular docking; RMSD, root mean square deviation; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine.

The molecular docking analysis of the HP with the SAM and SAH. The figure depicted both the site-specific (A and B) and blind docking (C and D) studies, where the ribbon indicates the HP and the sticks indicate the ligand (green color).

The interacting amino acid residues of the HP-ligand complexes, including HP-SAM (A and C) (site-specific and blind) and HP-SAH (B and D) (site-specific and blind) complexes predicted by AutoDock Vina software. The yellow color sticks depicted the ligands, whereas the disk represents the interacting amino acids.
Molecular dynamic simulation
The stability and performance of the docked protein complexes have been assessed using an MDS to investigate the atomic dynamic movements inside the complexes. Using a time-dependent MDS at 50 ns with the Gromacs forcefield on the Webgrow server, we have assessed the anticipated stability and flexibility of docked complexes such as HP-SAM and HP-SAH generated by AutoDock Vina. The RMSD and RMSF plots have been used to evaluate the complexes’ residual fluctuations and changes. To assess the equilibrium and stability of the HP-SAM and HP-SAH complexes, we calculated their average potential. It has been calculated that the average potential energy of the HP-SAM is −25 4405 kJ/mol, whereas that of the HP-SAH is −25 4919 kJ/mol (Supplementary Figure 5). The root mean square error, Rg, SASA, kinetic energy, enthalpy, volume, and density were all reported throughout the simulation. Changes in protein structure may be evaluated using RMSD by looking at how far C atoms deviate from the average orientation (Figure 9A to D). The average RMSF of all residues has also been counted to assess the local structural flexibility of the HP-SAM and HP-SAH (Figure 9E and F). Because of the correlation between a protein’s Rg and its SASA, the Rg of the HP-SAM and HP-SAH has been determined in the context of structural compactness evaluation (Figures 9C and D and 10A and B). The SASA of the HP-SAM is lower than that of the HP-SAH up to 50 ns. Structural stability has also been predicted for the HP-SAM and HP-SAH, based on their average intramolecular hydrogen bonds (Figure 10E and F). For energy, it is estimated that the HP-SAM has an average kinetic energy of 52 481.3 kJ/mol and an average enthalpy of −60 9650 kJ/mol. The average kinetic energy and enthalpy of the HP-SAH, however, are much higher than those of the original SAH, coming in at 52 575.6and −610 548 kJ/mol, respectively. The subsequent analysis of the docked complexes, HP-SAM and HP-SAH, revealed their stability and flexibility through the parameters such as RMSD, RMSF, Rg, SASA, and hydrogen bond analysis. The graphical depicts of all these parameters conveyed that the docking complexes are well stable and flexible, which imparts the HP to be a probable SAM-dependent MTase as well.

Molecular dynamic (MD) study of the HP-ligand complexes. The RMSD and RMSF of the HP-SAM (A, B, and C) and HP-SAH (D, E, and F) complexes were depicted as 50 ns run and up to 246 amino acid residues, respectively.

The Rg, SASA, and hydrogen bond analysis of the HP-SAM (A, C, and E) and HP-SAH (B, D, and E) docked complexes by MD simulation.
Conclusions
It has been established that the HP of PM strain HN06 is a valuable and stable protein, and one of the protein’s functional domains is tRNA (adenine(37)-N6)-methyltransferase TrmO. Surprisingly, the HP is an essential component in preventing the spread of pasteurellosis as it is a modified form of SAM-dependent MTase, namely, Class VIII SAM-dependent MTase. The biocomputational examination, in particular by MD and simulation studies, established the HP to be a Class VIII SAM-dependent MTase. It is possible to draw the conclusion that HP has great potential to progress research on Pasteurella infection, for example, by creating medications to treat this particular illness. We advise additional research into the protein’s comprehensive characterization, in vitro and in vivo assessment to assess its potential as a new Pasteurella infection research tool.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322231184024 – Supplemental material for In Silico Functional Characterization of a Hypothetical Protein From Pasteurella Multocida Reveals a Novel S-Adenosylmethionine-Dependent Methyltransferase Activity
Supplemental material, sj-docx-1-bbi-10.1177_11779322231184024 for In Silico Functional Characterization of a Hypothetical Protein From Pasteurella Multocida Reveals a Novel S-Adenosylmethionine-Dependent Methyltransferase Activity by Md. Habib Ullah Masum, Sultana Rajia, Uditi Paul Bristi, Mir Salma Akter, Mohammad Ruhul Amin, Tushar Ahmed Shishir, Jannatul Ferdous, Firoz Ahmed, Md. Mizanur Rahaman and Otun Saha in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
The authors acknowledge the Department of Microbiology, Noakhali Science and Technology University and Research Cell, NSTU for providing the research facilities and fundings respectively.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MHUM, OS, SR, and UPB carried out the studies (data collection, curation, molecular, and data analysis) and participated in drafting the manuscript. MHUM, MSA, MRA, TAS, JF, FA, and MMR critically reviewed and drafted the manuscript. MHUM and OS visualized figures, interpreted data and results, and critically reviewed and edited the manuscript. OS developed the hypothesis, supervised the whole work, and helped to prepare and critically revise the manuscript. All authors read and approved the final manuscript
Data Availability
No data were used to support this study.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.