
Abstract
Targeted delivery of therapeutic anticancer chimeric molecules enhances the efficacy of drug by improving cellular uptake and circulation time. Engineering the molecules to facilitate the specific interaction between chimeric protein and its receptor is critical to elucidate biological mechanism as well as accuracy in modeling of complexes. A theoretically designed novel protein-protein interfaces can serve as a bottom-up method for comprehensive understanding of interacting protein residues. This study was aimed for in silico analyses of a chimeric fusion protein against breast cancer. The amino acid sequences of the interleukin 24 (IL-24) and LK-6 peptide were used to design the chimeric fusion protein via a rigid linker. The secondary and tertiary structures along with physicochemical properties by ProtParam and solubility were predicted using online software. The validation and quality of the fusion protein was confirmed by Rampage and ERRAT2. The newly designed fusion construct has a total length of 179 amino acids. The top-ranked structure from alpha fold2 showed 18.1 KD molecular weight by ProtParam, quality factor of 94.152 by ERRAT, and a valid structure by a Ramachandran plot with 88.5% residues in the favored region. Finally, the docking and simulation studies were performed using HADDOCK and Desmond module of Schrodinger. The quality, validity, interaction analysis, and stability of the fusion protein depict a functional molecule. The fusion gene IL24-LK6 after cloning and expression in a suitable prokaryotic cell might be a useful candidate for developing a novel anticancer therapy.
Introduction
The term cancer describes more than 100 malignancies which affect different tissues and cells characterized by uncontrolled proliferating abnormal cells. 1 According to the World Health Organization, the mortality rate will increase up to 80% within a decade. 2 Among different types of cancers, breast cancer is the most common cause of deaths after lung cancer. The underlying mechanism of heterogeneous breast cancer development, both histopathologically and genetically, is still uncertain. 3 The monotherapeutic treatment selection regarding cancer includes surgery, hormonal therapy, radiotherapy, and chemotherapy. Despite the success of the applied monotherapies, the chemotherapeutic drugs for cancer treatment have serious drawbacks of nonspecific toxicity. These monotherapies’ success is largely limited due to its adverse side effects and remarkable increase in resistance. 4 The conventional chemotherapeutic drugs against cancer have some limitations such as damaging the healthy growing cells and development of resistance against the drugs. 5 Some of the drugs cause secondary tumors in the body, although they do not affect the slow growing and resting cells. 6 Cytokines have potential antitumor activities and play important roles in the regulation of the immune system. Among these cytokines, the most potent interleukins (ILs) such as IL-2, IL-15, and IL-24 have been evaluated extensively for their promising anticancer drug potential.7,8 But the use of these cytokines to boost the immune activity against the cancer cell had limited success due to the requirement of high-dose administration. The vast and existing knowledge of structural and functional characteristics of protein can be exploited to design a novel peptide. An optimistic approach in cancer treatment is targeted therapy. In anticancer targeted therapy, the drugs with reduced side effects selectively target the cancer cells, without being affected by chemoresistance mechanisms 9 which lead to enhanced drug efficiency. 10 The peptide alone or in fusion with other proteins could be a choice of drug to target tumor cells. 11 The advantages of using peptide over small molecules are lower toxicity profiles and high specificity to their targets. 12 Using a chimeric protein, ie, killer peptide along with the tumor-targeting peptide, is a good approach in targeted therapy against cancerous cells.13,14 Fusion of tumor-targeting peptide and IL to make a recombinant protein may have a potential to treat the human cancer more effectively due to their synergistic effects. The benefits to use peptide therapeutics as a mean of anticancer agent include the ease of peptide modification, rapid peptide synthesis, and 10- to 20-fold high therapeutic potential compared with the individual monomeric protein when joined through recombinant DNA technology. An existing and vast knowledge base of structure and function of protein can be exploited to design a novel peptide.15,16
Interleukin 24, a member of IL-10 family, plays an important role in autoimmune diseases, inflammation, and action against infections. 17 It induces apoptotic signal when interacts specifically with its heterodimer receptors (IL-20R1/IL-20R2 and IL-22R1/IL-20R2) on MCF-7 cell and subsequently activates signal tranducer and activator of transcription (STAT), Janus kinase/signal transducer pathways.1,18,19
LK-6 is a lysine/leucine-rich cationic antimicrobial penetrating peptide known for its anticancer activity preferentially against human MCF-7 breast cancer cells. After interaction with MCF-7 cell membrane phosphatidylserine, the LK-6 peptide is internalized via clathrin-independent macropinocytosis without disrupting cell surface and induces a dramatic nuclear damage which leads to MCF-7 cell death. A recombinant protein having tumor-targeting peptide and a cytokine could have a potential to treat human cancer more effectively due to bifunctional domains of the peptide without harming normal cells. 20
Therefore, we aimed to produce the fusion protein IL24-LK6 to benefit from both selective delivery and selective activity on cancer cells to offer a novel targeted anticancer treatment. A new drug candidate is first evaluated by in silico methods for structural and functional characteristics. Further to understanding the macromolecular structure to function relationships, molecular dynamics simulation is performed. Through MD simulation, intriguing aspects such as molecular interactions, physical processes, protein minimum geometries, drug binding free energies, and precise movements of molecules or atoms over a set length of time can be examined. In addition, this method has been able to evaluate the positioning, wrapping, distance, interactions between residues, and potential involvement of hazardous and targeted moieties.21-23 So the main objective of this study was to construct a theoretical fusion peptide comprising cytokines IL-24 and a cancer-specific cell-penetrating anticancer peptides LK-6 by silico methods against the IL22RA-IL20RB receptor and evaluate its anticancer therapeutic potential.
Materials and Methods
Construction of IL24-LK6 fusion protein
FASTA format amino acid sequence of Homo sapiens IL-24 was retrieved from Universal Protein Knowledgebase (UniProtKB, http://www.uniprot.org/: Accession # Q13007) database. The amino acid sequence of LK-6 peptide was reported previously by Wang. 20 The 3-dimensional (3D) crystalline structure of soluble IL-24 heterodimer receptors (IL-22RA and IL-20RB, PDB ID: 6DF3) in PDB file format was obtained from protein data bank (RCSB PDB: 6DF3) (Table 1). For chimeric molecule designing, we excluded signal peptide sequence (1-56 amino acids) from N terminal of IL-24 and used 52 to 206 amino acids. LK-6, a peptide of 13-amino acid length, was linked to IL-24 via a previously reported rigid linker (PAPAPAPAPAP). 18
Primary sequence of molecules.

Secondary structure of chimeric protein
For secondary structure prediction of chimeric protein, GOR IV (http://gor.bb.iastate.edu/) online server was used and further functional characteristics such as secondary structure, coiled-coil domain, regions lacking normal structure, solvent-accessible surface area, low-complexity sections, and location of disulfide bridges were assessed. 24
Homology modeling to predict 3D structure and validation
By using the primary sequence of protein, 3D structure was generated by 3 different software programs, including AlphaFold 2 (https://colab.research.google.com), 25 I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/),26-28 and trRosetta (https://yanglab.nankai.edu.cn›trRosetta).29,30 A template (PDB ID: 6DF3) was used by these software programs to build the model. Next, the quality and validity of newly constructed models were evaluated on different programs such as Verify3D (http://services.mbi.ucla.edu/Verify_3D) 31 which determines amino acid sequence compatibility to 3D atomic model, ERRAT2 (https://saves.mbi.ucla.edu/results?job=1033540&p=errat) which checks overall quality factor (OQF), and Rampage server (http://mordred.bioc.cam.ac.ukrapper/rampage.php) 32 which can generate the Ramachandran (RC) plot. Further, the galaxy refine server (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) 33 was employed before docking studies to refine and minimize the energy of modeled proteins to improve the structural geometry and to diminish significant steric clashes produced during the model building.
Physiochemical parameters of IL24-LK6 chimeric protein
The physical and chemical characteristics of chimeric protein were assessed theoretically by ProtParam (https://web.expasy.org/protparam/). This software calculated the isoelectric point (pI), molecular weight, amino acid composition (%), extinction coefficient, aliphatic index, and grand average of hydropathicity (GRAVY) using bioinformatics algorithms. 34 The solubility of chimeric protein was predicted on protein-sol online server (https://prosa.services.came.sbg.ac.at/prosa.php).
Toxicity, antigenicity, and allergenicity prediction
To predict the toxicity, antigenic, and allergenic nature of chimeric protein, the FASTA format amino acid sequence was submitted to online servers of ToxinPred, VaxiJen, and AlgPred, respectively. ToxinPred (https://webs.iiitd.edu.in/raghava/toxinpred/index.html) predicted the regions of toxic amino acid residues in the submitted sequence. AlgPred (https://webs.iiitd.edu.in/raghava/algpred2/batch.html) is based on the similarity index of any protein region with known epitope. VaxiJen server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) calculated antigenicity depending on the physical and chemical properties of the submitted protein sequence. We used a threshold of 0.5 to differentiate between the antigenic and nonantigenic proteins.35,36
Molecular docking and interactions
Because IL-24 generates apoptotic signal when binds to its receptor therefore to observe the interaction and the binding pattern between modeled protein and its specific receptor, HADDOCK v.2.4 was engaged for docking. 37 PyMOL molecular graphic system was adopted for removal of water molecules and attached IL-24 from receptor in the PDB downloaded file format of crystalline structure (PDB ID: 6DF3) along with active site prediction of receptor protein before proceeding to docking. The protein-protein interactions were analyzed on PDBePISA and PDBsum which served to identify interacting interfaces, hydrogen bonds, Gibbs free energy (ΔGint, kcal mol−1), salt bridges, nonbonded contacts, tunnels, and pores of complex.38,39 Finally, the binding affinity was calculated from prodigy online server (https://wenmr.science.uu.nl/prodigy/). 40
Molecular dynamics simulation
The Desmond module of Schrodinger was exploited to conduct MD simulation studies. The dynamic behavior and stability of the protein-protein complexes were investigated using their docked poses. The protein-protein complex was preprocessed using Protein Preparation Wizard of Maestro, which included complex optimization and minimization. All the systems were prepared using the System Builder tool. Solvation of the complexes was performed with the simple point-charge (SPC) water model with orthorhombic box, along with a 10-Å distance from the edge of the box, and the system was neutralized with Na+/Cl− ions. To mimic physiological conditions, 0.15-M sodium chloride (NaCl) was added. The potential energy of the protein complex was minimized by employing the constant- temperaure, constant-pressure (NPT) ensemble. The molecular dynamics simulations were performed at 300-K temperature and 1-atm pressure for 100-ns and NPT production ran under the OPLS4 force field. The models were relaxed before the simulation. The short-range electrostatic interactions were calculated using the particle mesh Ewald method. 41 The cutoff radius in Coulomb interactions was 9.0 Å. The water molecules were explicitly described using the SPC model. 42 The Martyna-Tuckerman-Klein chain coupling scheme with a coupling constant of 2.0 ps was used for the pressure control and the Nosé-Hoover chain coupling scheme 43 for the temperature control. The trajectories were saved for examination after every 100 ps, and the simulation’s stability was verified by comparing the root mean square deviation (RMSD) of the protein complex over time. The projected changes in their conformation from the initial structure over the entire simulation period were expressed as RMSD and root mean square fluctuation (RMSF) for MD simulations. A detailed description of the methodology can also be found elsewhere.44-46
Results and Discussion
Engineering of IL24-LK6 fusion protein
The sequence of IL-24 and LK-6 was retrieved from the National Center for Biotechnology Information and fused via a rigid linker to construct a single peptide of 179 amino acids (Figure 1). The presence of rigid linker ensures the separation of both functional peptides, provides stability, and prevents the formation of disulphide bond. The specific restriction sites were added to both the ends, N and C terminals, of the fusion construct to be cloned into a suitable vector for in vitro expression studies.

Schematic representation of IL24-LK6 fusion protein. IL indicates interleukin.
Secondary structure analysis
The secondary structure was predicted by GOR IV, that defines the alpha helix and beta sheets of protein, and it is the intermediate structure between primary and tertiary structure. Our modeled protein contains 54.75% (98 residues) alpha helix, 9.5% (17 residues) beta sheets, and 35.75% (64 residues) random coils (Figure 2). The higher percentage of alpha helix imparts structural stability to the molecule. The predicted structure of the rigid linker is a coil that will surely separate IL-24 and LK-6 molecules apart ensuring their functional capabilities.
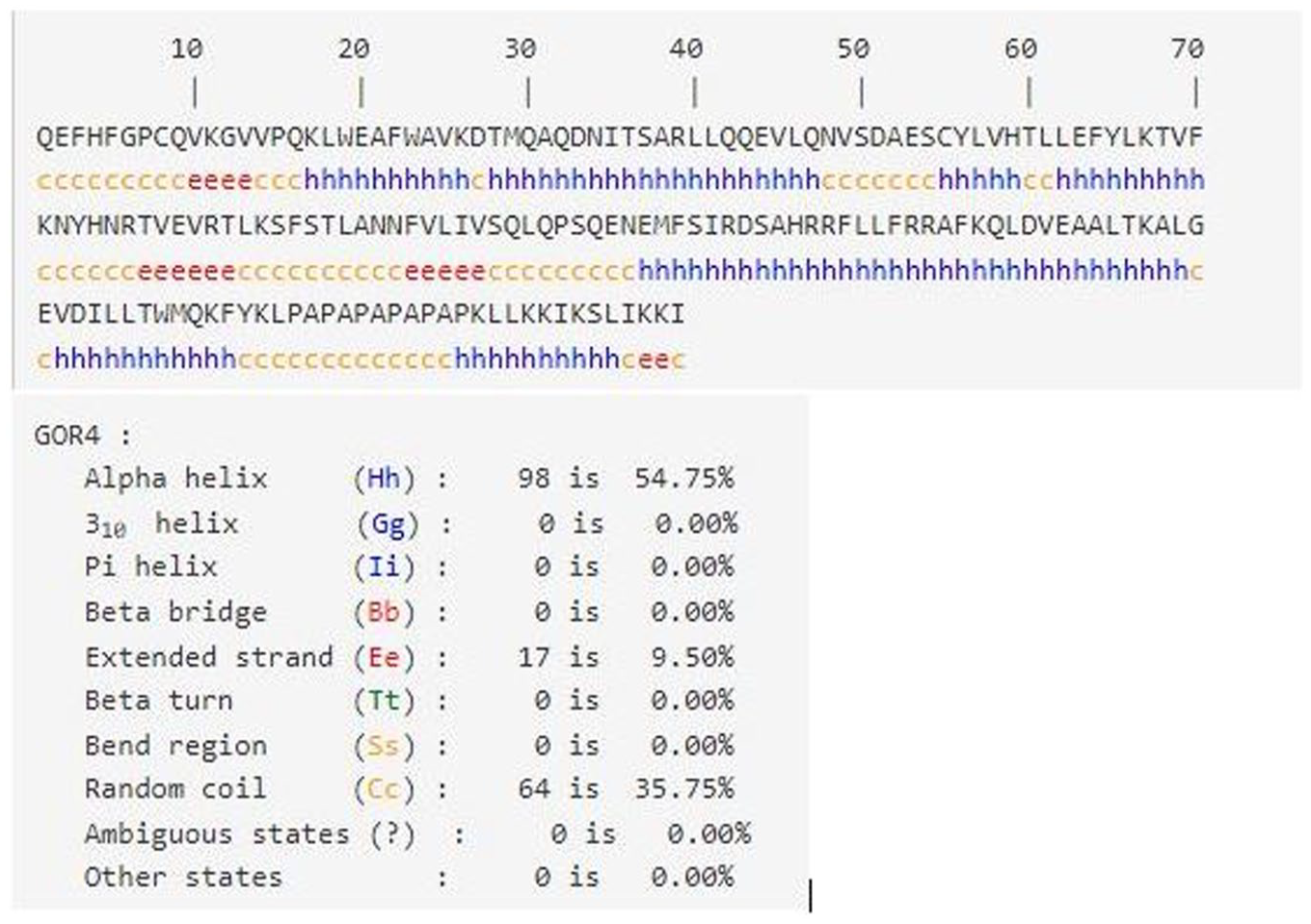
Schematic illustration of primary and secondary structure prediction by GOR IV.
Tertiary structure evaluation
The tertiary structure of the chimeric protein was prepared by operating homology modeling. FASTA sequence was submitted to 3 different 3D protein builder online servers AlphaFold 2 colab, I-TASSER, and trRosetta to get the most accurate and stable configuration of fusion construct. AlphaFold 2 predicts the 3D protein structure based solely on its primary amino acid sequence with high accuracy of side chains. The output file of AlphaFold 2 contains 5 different models ranked on the basis of local model quality of the protein structure. The top AlphaFold 2 model from the output file was selected for comparison with other models. I-TASSER produced 5 top models based on the C-score with a higher model value signifying more confidence. 47 So the top I-TASSER model with the highest C-score of −0.39, TM-score of 0.66, and RMSD of 5.9 was selected for comparison with other models. Like I-TASSER, trRosetta also constructed 5 top models based on highest TM-scores with restrains from both homologous templates and deep learning. A model with the highest TM-score of 0.745, Z-score of 37.8, and E-score of 1.2E-4 was selected for comparison with other models. After comparison of all top models obtained from 3 different servers, such as I-TASSER, AlphaFold 2 colab, and trRosetta, the best 3D model was presented by AlphaFold 2 so that the structure was selected and proceeded for further analysis (Figure 3).
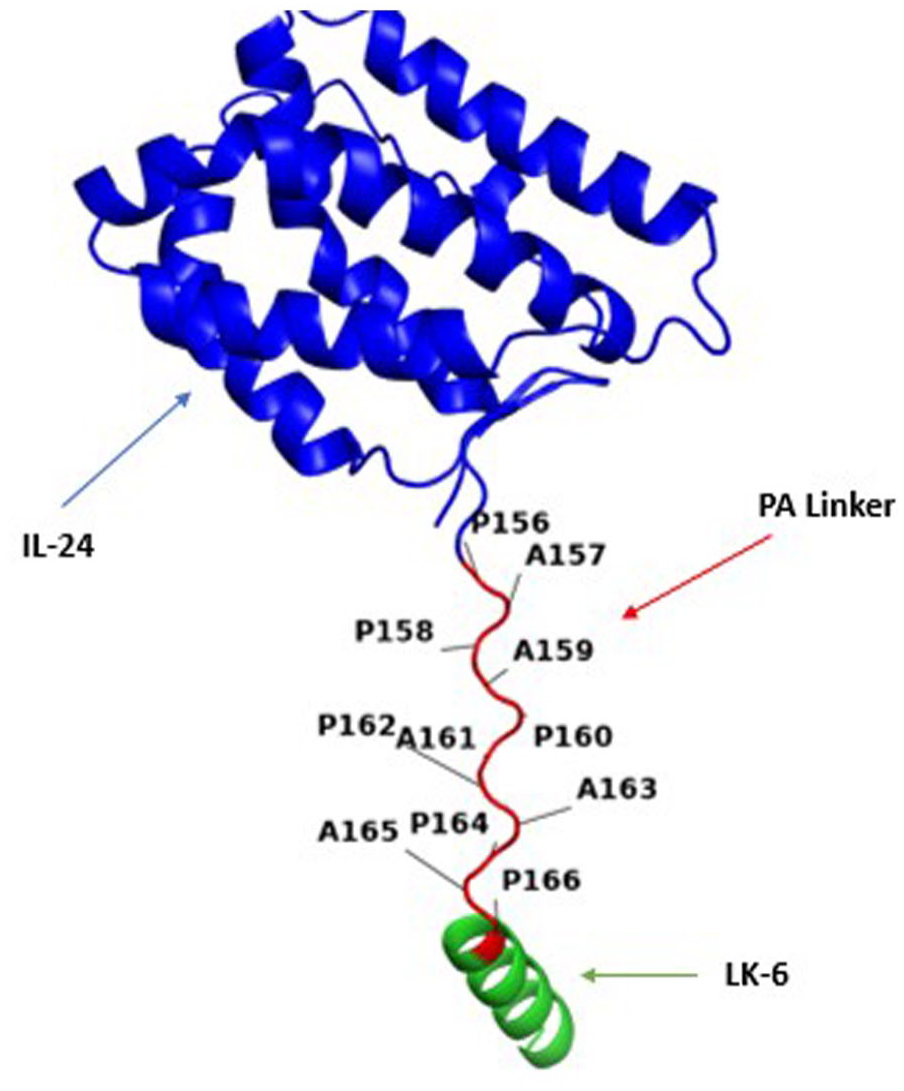
The ribbon structure visualization of IL24-LK6 fusion protein on PyMOL illustrates the 2 protein domains are completely separated by a rigid linker with no interaction which ensures the integrity and functional capability of both segments. IL indicates interleukin.
Reliable 3D structure selection, quality check, and validation
The Ramachandran plot provides a distribution of phi and psi angles of each amino acid residue with a higher percentage of amino acid in the preferred region and in the additionally allowed region suggests the overall structural reliability, accuracy, and validation. 48 A reliable hit from 3 predicted models was selected based on the quality score and stereo chemical validation (Table 2).
Quality comparison of homology modeling on different software programs.

Abbreviation: RC, Ramachandran plot values in the favored region.
All values are expressed in %.
The selected modeled fusion construct showed 88.5% residues in the allowed region, 10.3% in the additionally allowed region, and 1.2% in the generously allowed region of the RC plot. The only amino acid glycine is known by its symmetrical alpha carbon property and sometimes giving unusual behavior in the RC plot (Figure 4).

Ramachandran plot of IL24-LK6 fusion protein shows the presence of 88.5% amino acid residues in the favorable region (red), 10.3% in the allowed region (yellow), 1.2% in the generously allowed region (light brown), and no amino acid in the disallowed region (white). IL indicates interleukin.
Furthermore, Verify 3D program (https://saves.mbi.ucla.edu/results/job=1;032202&p=verify) was used to measure the compatibility of modeled protein with the primary sequence using a 3D profile. 41 Residues having averaged 3D to 1D score ⩾0.2 with the overall result “Pass” are considered for further studies (Figure 5).

Verify 3D graphical representation.
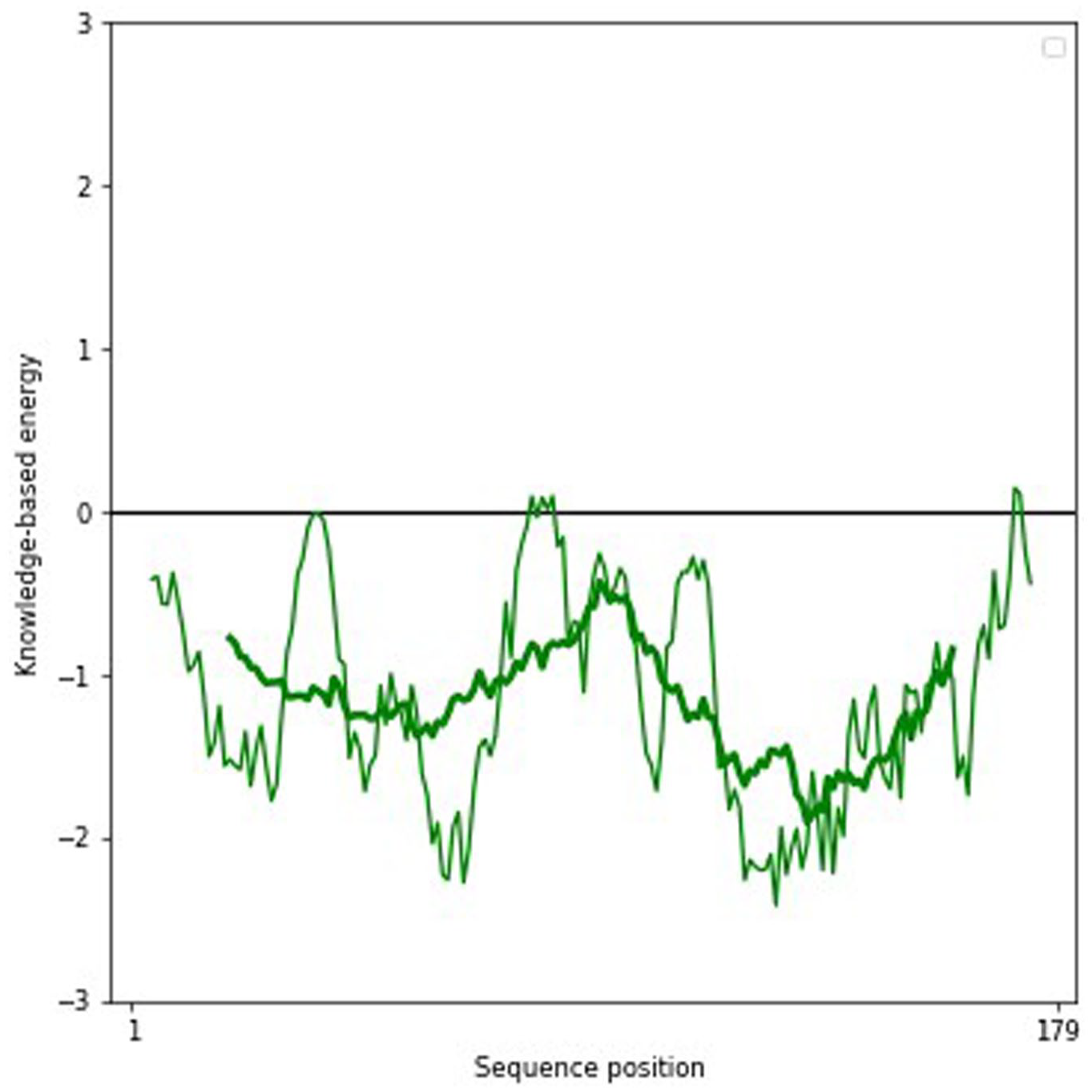
Finally, ERRAT2 program (website) predicts the OQF which expresses the percentage of protein for which the calculated error value falls below the 95% rejection limit, with equal or more than 95% indicating good high-resolution structure (Figure 6). According to the results, the most precise 3D structure was modeled by AlphaFold 2 with ERRAT OQF of 98.63, Verify 3D score of 95.48%, and 95.2% residues in the favorable region of the RC plot (Figure 3). The selected model’s energy plot was generated by proSA-web online server to indicate the erroneous residues of protein. The positive residue energy represents invalid or wrong amino acid position (Figure 7). The AlphaFold 2 fusion construct was further proceeded to assess solubility and docking studies.
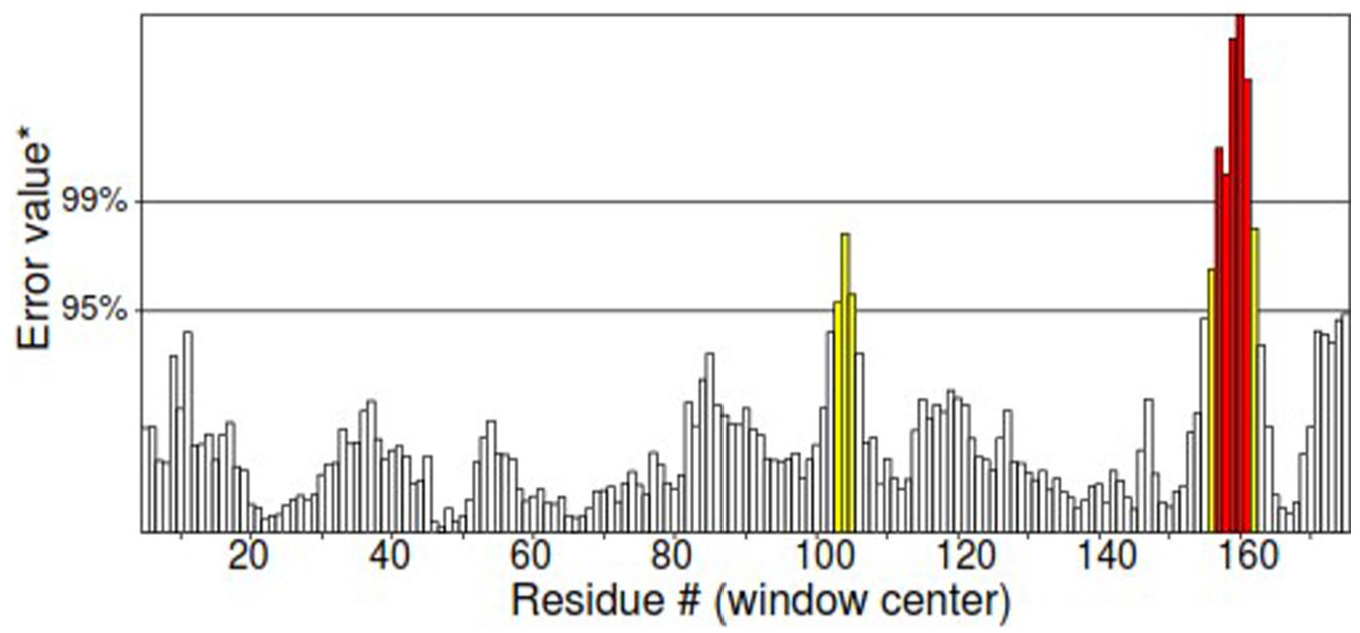
Graphical representation of quality error prediction by ERRAT2 program for each amino acid in IL24-LK6 fusion protein. IL indicates interleukin.
Graphical representation of local quality estimate of each amino acid in fusion protein.
Predicted physicochemical parameters of fusion protein
ProtParam online server computed the physicochemical parameters shown in Table 3. The presence of many basic amino acids (Arg + Lys = 24) as compared with acidic (Asp + Glu = 16) imparts alkaline nature to the chimeric molecule with a theoretical pI of 9.57. The extinction coefficient of the modeled protein is 22 585 M−1 cm−1, an important factor in protein-protein interaction. The in vitro estimated half-life in a mammalian cell is 0.8 hour and the instability index is 46.71.
Physical and chemical properties of fusion construct.

Abbreviations: GRAVY, grand average of hydropathicity; IL, interleukin; pI, isoelectric point.
A high aliphatic index of 96.98 indicates a wide temperature range for protein stability. The reactivity of chimeric protein in water is estimated by a low GRAVY index of −0.135. Moreover, ProtPram calculated the highest percentage of Leu (12.8%) in the chimeric construct. For solubility prediction, protein-sol software was employed which compares the solubility of the query FASTA sequence of amino acid to its database and any value greater than 0.45 is predicted as soluble. 49 Our selected model has the scaled solubility value of 0.562 which can be predicted as soluble (Figure 8).
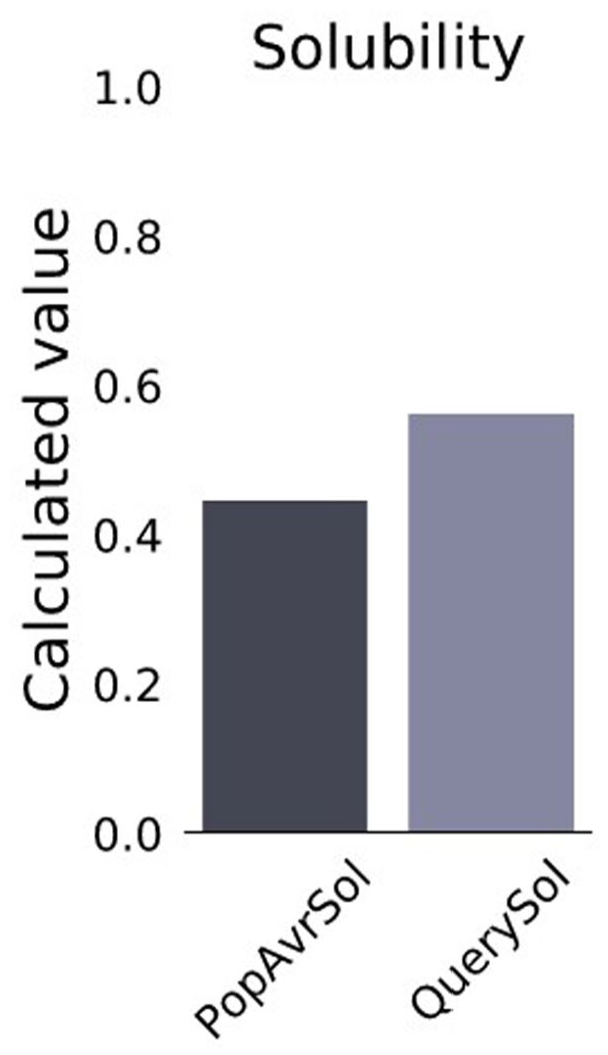
The population average for the experimental dataset (PopAvrSol) on protein-sol.
Toxicity, allergenicity, and antigenicity evaluation
ToxinPred, AlgPred, and VaxiJen designated the IL24-LK6 fusion protein as nontoxic, nonallergen, and nonantigenic, respectively. None of the peptide residue exhibits toxicity (data not shown) with an overall score of 0.4478 for protective antigen prediction. The results suggest a nonimmunogenic nature of chimeric protein and propose its use as a good anticancer drug candidate.
Docking analysis
To elucidate the mechanism of interaction between fusion protein and its receptor, HADDOCK protein-protein docking was performed. For interaction studies, the top structure, based on score, electrostatic energies, and van der Waals attraction, has been selected. Among protein-protein docking, the major interactions are electrostatic and van der Waals which mutually find accurate results along with promising levels as scoring function. 50 HADDOCK server clustered a total 159 structures in 13 clusters representing 79% of water-refined models generated by HADDOCK. Table 4 shows the statistical parameters of top 7 clusters. The Z-score signifies the number of standard deviations from the mean of the cluster location of score (negative value indicates better results). The more negative value of electrostatic and van der Waals interactions depicts more stable docked complex. Moreover, the binding strength of the complex depends on 2 factors, the total number of interacting amino acid residues and area of corresponding interface. Larger interface area depicts larger binding energy (van der Waals + electrostatic + H-bond) and more stable structure. 51
Protein-protein docking at HADDOCK server.

Abbreviation: RMSD, root mean square deviation.
Residue interaction analysis
Computational estimation of binding energy in protein-protein interface is a challenging problem, although dozens of tools are available based on analytical or empirical approaches; however, the performance of each tool varies according to the particular protein and we cannot pinpoint a single module which provides a reliable quantification. 18 The interaction studies of docked complex analysis were resolved by PDBePISA and PDBsum servers (Table 5). In addition, salt bridges, mapping of interacting hydrogen bonds, and ΔGint were also evaluated. ΔGint value expresses free energy of solvation attained during formation of assembly (total energy of solvation in assembled structure − isolated structure solvation energies) (Table 6). 52 The docked complex revealed a total of 7 interactions mediated by 2 salt bridges and 5 hydrogen bonds with IL-22 alpha 1 subunit of IL-24 heterodimer receptor. According to PDBePISA, the formation of complex is primarily contributed by the interaction between fusion protein and IL-22R1 subunit (Figure 9(B)) which is in agreement with previous studies. 37 The residues at interface between these chains extend over 1181.5 Å with stabilization energy of −8.6 kcal mol−1 involving Ser105, Met109, Glu56, Ser105, Arg112 and Ser110, Asn106, Asp51, Ser110, and Lys154 in hydrogen bonding contributed by fusion construct and IL-22 alpha 1 subunit, respectively. Surprisingly, no interaction was observed between fusion construct and IL-20 receptor beta subunit. Figure 9(A) and (C) illustrates the binding residues and interacting surfaces. The potential salt bridges were accomplished between Glu 2 , Lys154 and Arg112, and Asp111 from fusion construct and IL-22 alpha 1 subunit, respectively. The scores for binding affinity were predicted as a temperature function. The significant increase of Kd value from 25°C to 40°C indicates a decreased binding affinity for receptor protein having a similarity with the previous study (Table 6). 53
Interface statistics (chains A, B, and C have been labeled in Figure 9).
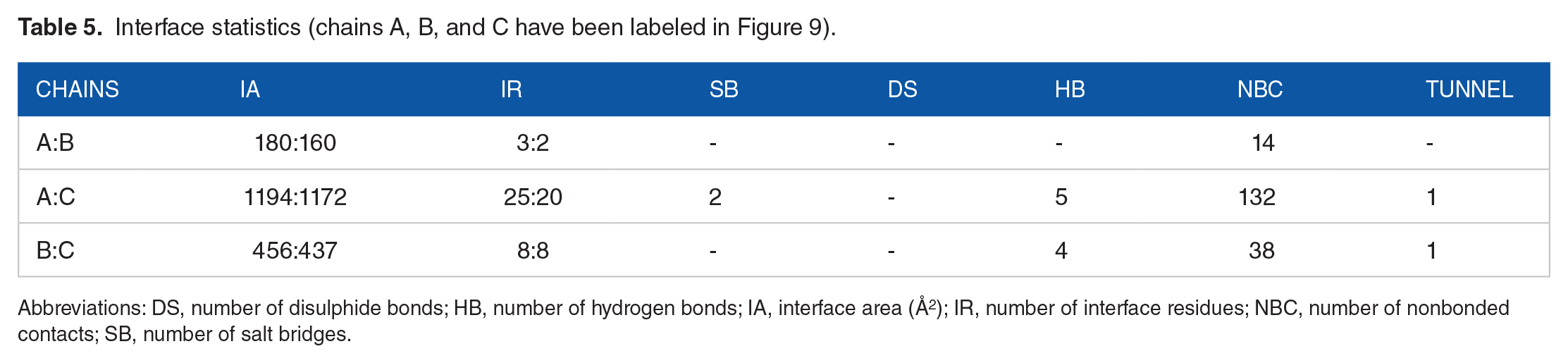
Abbreviations: DS, number of disulphide bonds; HB, number of hydrogen bonds; IA, interface area (Å 2 ); IR, number of interface residues; NBC, number of nonbonded contacts; SB, number of salt bridges.
Binding affinities of fusion construct.

Abbreviation: IL, interleukin.
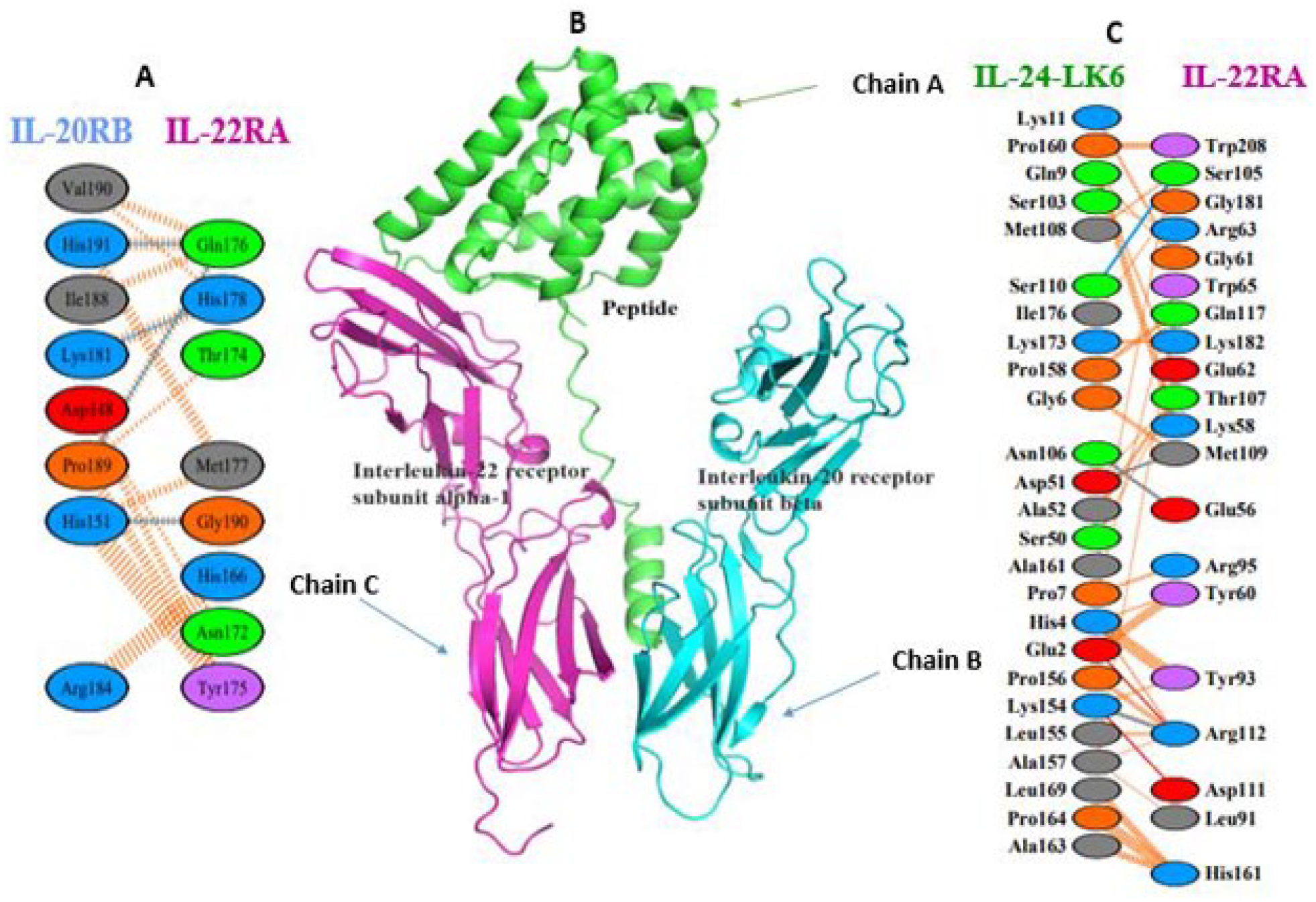
(A, C) PDBsum’s interaction plot of chimeric molecule docked with receptor protein. The key indicates hydrogen bond (blue line), salt bridges (red line), disulphide bonds (yellow line), and nonbonded contact (orange tick-mark) for amino acid residues at protein-protein interface. (B) Docked peptide with IL-22 alpha-1 receptor and IL-20 beta receptor. IL indicates interleukin.
Molecular dynamics simulation
Molecular dynamics simulations were performed on the top hits containing high binding energies. Over the simulation period, the projected conformational changes from the initial structure were presented in RMSD. Moreover, structural stability, atomic mobility, and residue flexibility at times of interaction of protein hit were expressed with RMSF values. The peaks of RMSF graph represent the fluctuation portion of the protein through the simulation. The N- and C-terminals show more changes than any other portion of the fusion protein. Alpha helices and beta strands show less fluctuation, as they are stiffer than the unstructured part of protein, than loop portion. The RMSD of the complex showed small deviation until almost 10 ns and then there was deviation of 1 Å system throughout the simulation. It indicates the stability of the IL24-LK6 fusion protein/receptor complex and whether the simulation has equilibrated (Figure 10). Similarly, the RMSF is useful for characterizing local changes along the protein chain.

Root mean square deviation plot of IL24-LK6 fusion protein with receptor complex. IL indicates interleukin.
Peaks reflect sections of the proteins that fluctuate the most during the simulation on the RMSF. IL24-LK6 fusion protein tails (both N- and C-terminals) are more likely to change than other regions of the protein. Alpha helices and beta strands, for example, are usually stiffer and less fluctuating than the unstructured component of the protein. The residues with higher peaks, according to MD trajectories, belong to loop areas or the N- and C-terminal zones. For RMSF, there was fluctuation of almost 4.17 Å of ILE 35, 3.44 Å of GLU 79, 4.14 Å of GLU 104, 3.98 Å of GLY 61, and 3.13 Å of SER 119; the remaining structure was made stable comparatively (Figure 11). The radius of gyration is the distribution of atoms in a protein around its axis (Rg). Rg denotes the distance between the rotating point and the location where the energy transfer has the greatest impact. This concept can also be used to identify different polymer types, such as proteins. The calculation of Rg and distance calculations are the 2 most essential markers for predicting a macromolecule’s structural activity. The rate at which a protein fold is proportional to its compactness can be measured using a sophisticated computer method for calculating the gyration radius. The value of Rg increases from 31.5 to 32.6 until almost 5 ns; after that, there was almost equilibrium until 100 ns (Figure 12). The analyses of energy parameters for complex indicate that the total energy of the system was decreased and hence the complex gets stable (Figure 13).

Root mean square fluctuation plot of IL24-LK6 fusion protein with receptor complex. IL indicates interleukin.
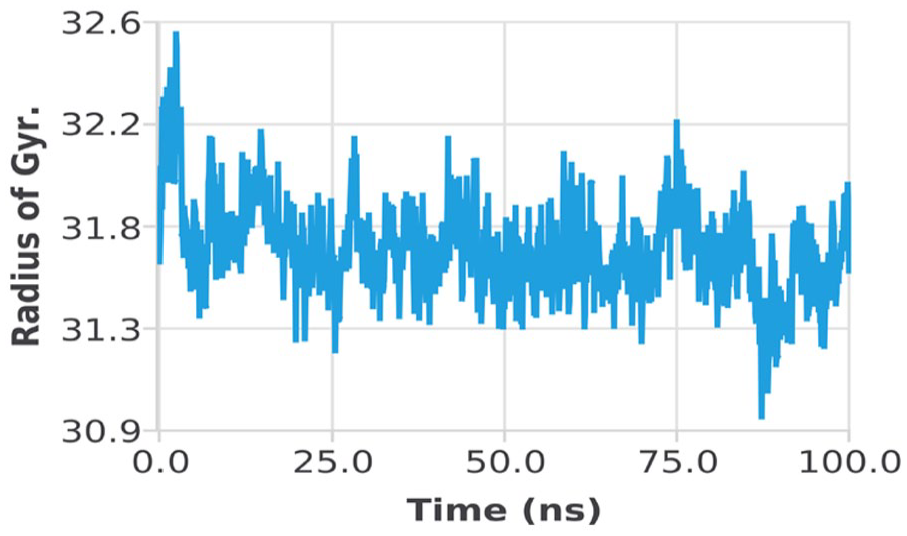
Radius of gyration of IL24-LK6 fusion protein with receptor complex. IL indicates interleukin.

The total energy of docked complex.
Conclusion
In silico approach is deemed as an amenable alternate method for designing antitumor fusion protein. The use of computational methods can fairly decrease the time and cost of peptide designing and it also avoids the ethical aspects of in vivo experiments. Herein, by in silico approach, we have designed IL24-LK6 fusion protein exhibiting the potential of enhanced activity against breast cancer. The predicted model was assessed for quality, validation, physicochemical properties, solubility, docking, interaction studies, and molecular dynamics simulation. The findings suggest that our chimeric protein could be a stable antitumor candidate. The concept is to fuse a targeting peptide with a penetrating peptide via a rigid linker to keep both moieties intact, which could enhance the antitumor activity by dual action. This theoretically presented model would lay the groundwork for our in vitro studies in future to reveal the better insights of its anticancer activity.
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by grants from the Higher Education Commission (HEC) Pakistan under NRPU 2021 (Project Number 16935) and the University of the Punjab, Lahore, Pakistan. Furthermore, the authors are grateful to HADDOCK 2.4 online server team for providing such a great platform to perform state of the art protein-protein docking analysis.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
HMR, HB, and HMR helped in methodology, formal analysis, and validation; MN and MTK contributed to investigation resources; SA helped in data curation; and HMR, HB, and HMR prepared the original draft. All authors have read and agreed the published version of the article.
Data Availability
Data could be provided from Dr Hamid Bashir, email: