
Abstract
Dengue outbreak is one of the concerning issues in Bangladesh due to the annual outbreak with the alarming number of death and infection. However, there is no effective antiviral drug available to treat dengue-infected patients. This study evaluated and screened antiviral drug candidates against dengue virus serotype 3 (DENV-3) through viroinformatics-based analyses. Since 2017, DENV-3 has been the predominant serotype in Bangladesh. We selected 3 non-structural proteins of DENV-3, named NS3, NS4A, and NS5, as antiviral targets. Protein modeling and validation were performed with VERIFY-3D, Ramachandran plotting, MolProbity, and PROCHECK. We found 4 drug-like compounds from DRUGBANK that can interact with these non-structural proteins of DENV-3. Then, the ADMET profile of these compounds was determined by admetSAR2, and molecular docking was performed with AutoDock, SWISSDOCK, PatchDock, and FireDock. Furthermore, they were subjected to molecular dynamics (MD) simulation study using the DESMOND module of MAESTRO academic version 2021-4 (force field OPLS_2005) to determine their solution’s stability in a predefined body environment. Two drug-like compounds named Guanosine-5’-Triphosphate (DB04137) and S-adenosyl-
Introduction
Dengue virus (DENV), the causative agent of illnesses like dengue fever (DF), belongs to the family Flaviviridae, where the mosquito Aedes aegypti is the primary vector. 1 DENV is divided into 4 serotypes (DENV1-4) based on amino acid-level heterogeneity, similar to serology and epidemiology. 2 The geographical location of Bangladesh in the tropical and sub-tropical regions is a suitable habitat for the survival of the dengue vector and its high transmission. In 2000, the first dengue case emerged as an epidemic in Bangladesh. 3 With DENV-3 predominance until 2002, 4 serotypes have already been detected. DENV-3 and DENV-4 were not identified in Bangladesh after that. 2 However, in 2017, DENV-3 reemerged, and in September 2019, approximately 101 354 cases and at least 179 deaths were reported, according to the Directorate General of Health Services (DGHS). DENV is almost 11 kb in length as an enveloped, positive-sense, single-stranded RNA virus. They are encoded by a single open reading frame from this genome, a 370 kDa polyprotein cleaved into 3 structural proteins (capsid, membrane, and envelope protein) and 7 non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). Structural proteins play a role in virus entry, secretion, assembly, and attachment, whereas NS proteins are engaged in enzymatic activities to favor viral replication. 4
Specific vaccinations are available for Japanese encephalitis and yellow fever viruses, which belong to the same family as the DENV (Flaviviridae). 5 Alternatively, to combat the high disease burden of DENV infection, no approved, safe, low-cost, and lifelong antiviral treatment or effective vaccine is available. 6 Inhibitors targeting host cell factors and inhibitors targeting viral components are the two approaches to designing antiviral drugs. 7 Despite providing a broad spectrum of activity, the host targeting approach has difficulties due to the higher possibility of side effects and a lack of an in-vitro model. 8 Because of minimal toxicity and side effects, one promising approach is targeting viral components. However, rapid mutations in viral components like structural protein pose a narrow spectrum of activity and show a high risk for resistance development. 7 As a conserved region, non-structural proteins can be focused on designing antiviral drugs regardless of mutational variants. 9 Out of non-structural proteins, NS3, NS4A, and NS5 are considered amenable to antiviral inhibition because of their all-enzymatic activities essential for polyprotein processing and genome replication. 6 NS3 is the second largest flaviviral protein, approximately 69 kDa, indispensable for viral replication. Being a conserved protein among the DENV, NS3 has 77% amino acid similarity in all 4 serotypes. 10 It has a C-terminal RNA helicase and an N-terminal protease domain. The C-terminal helicase collaborates with NS5 and other NS proteins to facilitate RNA synthesis and genome replication. Prior to the start of RNA synthesis, the NS3 helicase activity is crucial for the fusing of secondary structures at the untranslated regions. Prior to capping the positive-strand RNA, it is also responsible for unwinding dsRNA intermediate products produced during viral RNA synthesis. N terminal domain is responsible for breaking down the viral polyprotein precursor into individual protein. 11
The highly hydrophobic protein NS4A has a molecular weight of roughly 16 kDa and is crucial for viral replication. It’s oligomerization that provides structural stability. NS4A and NS4B, 2 integral membrane proteins, are encoded by the dengue NS4 region. These 2 proteins are connected by a 23-residue region known as the “2 K fragment,” which is cleaved by NS3 to produce the N and C terminals of NS4A. The N terminal of NS4A is found in the cytoplasm, while the C terminal is located in the ER lumen. 10 DENV replication requires NS4A protein to alter the host membrane to facilitate the replication complex’s establishment.
With 82% amino acid sequence resemblance among the 4 serotypes, NS5 is considered the most conserved protein. 12 It comprises enzyme activity necessary for viral replication and has a molecular weight of roughly 900 kDa. Its N-terminal domain encodes 2 N7 and 2-O-methyltransferase (MTase) activities in RNA cap formation. For the production of viral RNA, the C-terminus is crucial because it contains an RNA-dependent RNA polymerase. 13
Therefore, a detailed understanding of these proteins and the prevention of their role would offer helpful insight to design inhibitors that can be exploited for anti-dengue drug development.
In the case of the DENV-3 and the other 3 serotypes of dengue, repurposing FDA-approved drugs through in vitro and in silico approaches is quite familiar.14,15 To be specific, cell line-based plaque reduction assay is the gold standard for in vitro analyses, and molecular docking targeting any specific protein followed by molecular dynamics (MD) study is preliminarily accepted to identify and assess the efficacy of any drug-like compounds. 8 Some repurposing drugs are quinine, N-acetylcysteine, etc. 16 Pharmacophore-based virtual screening of antiviral compounds from different databases followed by molecular docking and dynamics simulation is also an effective way to preliminarily identify antiviral drugs against the other 3 types of DENVs. 17 In this case, the method targets any specific conserved and essential protein of DENVs. The common drug-like compounds identified through these methods are usnic acid and sulfated derivatives such as curdlan sulfate. Overall, all compounds still need to be proven effective against DENV infection. Therefore, we have targeted unique and highly conserved non-structural proteins to identify the most effective drug-like compounds through in silico approach from the DrugBank database.
As the dominant and circulating serotype, DENV-3 caused the highest case burden in Bangladesh. The main objective was to screen drugs through in silico analyses targeting non-structural proteins (experimental) against DENV-3. DENV-3 encoded specified proteins were identified using bioinformatics tools, and their sequences were retrieved for mutational analyses. Furthermore, we used different modeling methods to construct targeted proteins that best suit structures, such as NS3, NS4A, and NS5. Thus, our study was to conduct screening approaches of drugs from openly accessible drug databases that may inhibit DENV-3 infection and can be considered for further in vitro analysis.
This study reveals that S-adenosyl-
Our study focused on the preliminary identification of an antiviral that may overcome the current challenges of anti-DENV drug development and further evaluate the antiviral effects of existing drugs in preclinical and clinical stages.
Materials and Methods
Identification of the sole proteins involved in the life cycle of dengue virus serotype-3
We identified the targeted non-structural proteins (non-structural protein targets) (NS3, NS4A, and NS5) through literature analysis providing importance on the activity of proteins and their involvement in the life cycle of DENV-3 to be an antiviral drug target. 4 ,21-23 These 3 non-structural proteins were used to explore antiviral drugs as these non-structural proteins hold conserved regions and play an indispensable role at the replication stage of the life cycle of DENV-3.
Mutation analyses among sole proteins of different circulatory DENV-3 strains of different time periods in Bangladesh
In different periods, amino acid sequences of identified 3 proteins (NS3, NS4A, and NS5) of 11 DENV-3 strains in Bangladesh were retrieved from NCBI to analyze the mutation in these proteins of 11 DENV-3 strains. Then, we used the multiple Sequence Alignment in conjunction with BioEdit software by using strains with the bootstrap value of 1000 of the ClustalW method and analyzed mutational change. 24 The protein structures were superimposed to determine whether there is any effect on the structure and the binding of drug-like compounds with this protein by observing whether the mutations are in the binding site. Homology modeling of NS3, NS4A, and NS5 proteins of DENV was performed using the SWISS-MODEL database considering 5yw1.1A, 1qsd.1.A, and 5jjs.1.A as templates for NS3, NS4A, and NS5 to determine the 3-dimensional (3D) structure with sequence identity around 80.10%, 15.38%, and 96.74%, respectively. The modeled proteins were stored in Protein Data Bank (.pdb) format and taken to PyMOL 3D viewer for further analysis. 25 PyMOL was used for 3D structural visualization. In PyMOL, specific modeled proteins were superimposed individually with the proteins of BBH51325.1 strain to determine structural change where mutations occurred, and root-mean-square deviation (RMSD) values were noted. The superimposition was conducted by the “super” command, allowing a residue-based pairwise alignment and a structural superposition in PyMOL. Then, the refinement cycle was set to zero to obtain the RMSD value of all atoms.
Protein modeling and validation
The modeling of 3 identified proteins (NS3, NS4A, and NS5) was performed based on 3 different methodologies (homology modeling, ab initio, and threading) with Robetta, 26 HHpred, 27 and SWISS-MODEL 28 server. In the case of modeling with Robetta, threading (RoseTTAFold) and ab initio methods were used for each protein. On the contrary, best-suited template models (Table 3) were used in the case of protein modeling with the HHpred and SWISS-MODEL databases. The amino acid sequences of 3 non-structural proteins (NS3, NS4A, and NS5) were retrieved from NCBI (NCBI ID BBH51325.1). We selected the sequence of the recently submitted predominant circulatory DENV-3 (NCBI ID BBH51325.1) strain sequence, from where we retrieved the amino acid sequence of non-structural proteins before modeling. Three models of each protein were generated to select the best model based on bioinformatic tools. The protein’s 3D structure quality was assessed by PROCHECK 29 and validated with SAVES-Verify 3D, 30 and MolProbity scores. 31 Ramachandran Plot was derived from PROCHECK for each of the 3 proteins to evaluate the stereochemistry of the protein structures. 32 The torsion angles (psi ψ) of the main chain of each protein were determined to assess the stereochemistry of the proteins in the Ramachandran plot.
Moreover, the best model for each protein was visualized and selected for further analyses, and the 3D image was generated with the UCSF Chimera visualization tool. 33 Eventually, the physicochemical properties of these proteins were determined by performing ExPASy: ProtParam tools. 34 Finally, the overall charge of proteins was determined by Atomic Charge Calculator. 35
Retrieval of drug-like compounds from DRUGBANK
Drug-like compounds reacting with these 3 non-structural proteins were retrieved from the DRUGBANK database (https://go.drugbank.com). 36 In the case of identifying the drug-like compounds, the amino acid sequences of proteins were used as target sequences with an expectation value (E) of 0.00001, and −3 was set as a penalty score for each mismatch. In addition, the 3D structure of drug-like compounds (experimental or approved) that can interact and interfere with non-structural proteins were retrieved. From the DRUGBANK database, 4 drug-like compounds interacting with 3 non-structural proteins were retrieved in 3D-SDF format. After retrieving, the data format of these compounds was converted to PDB format using Open Babel GUI. 37 These compounds were subjected to dock with specified proteins to determine the binding efficacy with those proteins. Moreover, admetSAR 38 was deployed to delineate the ADMET profile of these compounds.
Molecular docking and statistical analysis
Molecular docking followed by MD simulation techniques were deployed to determine the binding efficiency of drug-like compounds with specified proteins. 39 Before the molecular docking study, the energies of 3 non-structural proteins were minimized with the YASARA energy minimization server. 40 Avogadro 41 and Open Babel 42 tools were used to optimize the geometry of drug-like compounds after minimizing energy to minimize the atomic clashes. The AutoDock Vina tool 43 was used under the common platform of PyRx 44 to determine the binding energy of inhibitor drug-like compounds. Protein dehydration, protonation, and a 1-grid point spacing grid box have all been set. The protein and ligand sequences were then formatted using Open Babel. 42 Afterward, the molecular docking experiments were conducted with an exhaustiveness value of 8. Binding energy (KJ/mole) was released based on protein-ligand interaction. We also performed the molecular docking experiment of drug-like compounds with specified proteins using the PatchDock server. 45 The top 10 docked solutions of protein-ligand derived from PatchDock were sent to the FireDock server 46 to refine the solutions. We also used the SWISSDOCK server 47 to correlate the molecular docking experiments. In this circumstance, the binding energies derived from SWISSDOCK and AutoDock were expressed in the same unit (KJ/mole). Therefore, Pearson’s product-moment correlation test 48 was conducted, and the overall binding energy concordance curve was also generated with the help of R Programming at RStudio. 49 In addition, the paired t test was performed between these 2 energy sets to check the extent of identity that the data sets retain; at the same time, the overall concordance between these 2 energy sets was also observed. Finally, we used PyMOL 3D viewer, Chimera, and BIOVIA Discovery Studio Visualizer software to speculate protein-ligand 3D and 2D interactions. Once molecular docking analyses have been performed, the interacting amino acid residues in the protein binding pocket with the specified types of interactions were also revealed by BIOVIA Discovery Studio Visualizer software. 50 The X, Y, and Z coordinates of the binding amino acids of each targeted protein were observed by PyMOL. Finally, the type and quantity of charges of proteins were also determined by Atomic Charge Calculator II. 51
Molecular dynamics simulation study of protein-ligand complexes
After all the above evaluations, the best response provided protein-drug-like compound solutions subjected to a MD simulation study to determine the stability of the complexes in the body environment. The stability of the docked complexes is evaluated by performing the MD simulation 52 to understand how they would behave in a predefined environment through atomic movements.53,54 The protein complexes were taken to simulate 100 ns MD simulation using the DESMOND module of MAESTRO software academic version 2021-4 (Schrödinger Release 2021-4: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2021. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2021). 55 The protein-ligand complexes are minimized before building the system. The System Builder panel was used to prepare an orthorhombic box for simulation with a 10 Å distance from the protein with an explicit Single Point Charge (SPC) water model where the force field was OPLS_2005. The system was neutralized by adding 0.15 M NaCl to maintain Na and Cl counter ions, and the pH was maintained at 7.0 ± 2.0. Molecular dynamics of 100 ns were considered in the MD simulation where constant temperature, constant pressure (NPT) settings were used. The physiological condition of the simulation was 310 K and 1.013 bar, which mimics the physiological state of the human cell.56,57 The energies and structures were recorded every 100 ps and saved in the trajectory, where 1000 frames were generated throughout the simulation. The Simulation Interaction Diagram module then examined the trajectories to investigate the root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), 3D structure, and protein-ligand contact. These were used to evaluate the stability of the ligand-protein complexes based on the simulated trajectories. The average distance caused by the displacement of selected atoms for a specific time frame relative to a reference time frame was measured using root mean square deviation (RMSD) in MD simulation. RMSD values for specific protein structures like C, backbone, side chain, and heavy atoms were computed first. The RMSD of the protein fit ligand calculated from all time frames at the reference time (in our example, 100 ns) was then calculated.
Broad spectrum homology analysis with Homo sapiens
NCBI pBLAST tool 58 was deployed to determine whether other pathogenic viruses contain proteins similar to these non-structural proteins. Furthermore, the presence of ortholog proteins in Homo sapiens was also determined with 0.0001 as the threshold (E) value.
Results
Crucial role of sole non-structural proteins in viral multiplication
Three non-structural proteins play an indispensable role in the life cycle of the DENV-3 virus. These 3 proteins are NS3,59,60 NS4A, 61 and NS5. 62 NS3 protein performs multiple essential functions, particularly the helicase and protease activity required for genome replication;21,59 NS4A maintains the integrity of the membrane and forms a replication complex component. 63 In contrast, NS5 protein conducts the methyltransferase and RNA-dependent RNA polymerase activity (RdRp). 12 These determine the suitability of the 3 non-structural proteins for the drug target. Moreover, NS5 retains one of the most conserved regions in all virus proteins. 12
Mutation analysis among sole proteins
We observed several mutations in sole proteins after aligning sequences of DENV-3 proteins from the 2002-2019 timeline circulating in Bangladesh. In NS3 protein (Figure 1A), we found amino acid mutation Tyrosine to histidine at 40 number position, Valine to Isoleucine at 106 number position, Lysine to Arginine at 235 number position, Alanine to Threonine at 290 number position, Aspartic acid to Glutamic acid at 304 & 330 number positions, Alanine to Valine at 336 number position, Valine to Alanine at 432 number position, and Methionine to Threonine at 456 number position in 2002-2006 timelines serotypes compared with BBH51325.1 of 2019 as the reference sequence. In NS4A (Figure 1B), we found mutations such as Valine to Alanine at 68 number position, Lysine to Arginine at 76 number position, Valine to Isoleucine at 78 & 89 number position, Alanine to Valine at 90 number position, and Isoleucine to Valine at 100 number position in 2002-2006 timelines sequence. Whereas in NS5 (Figure 1C), we found mutations such as Arginine to Lysine at 32, Threonine to Methionine at 97, Lysine to Arginine at 115, Alanine to Threonine at 155, Histidine to Tyrosine at 254, Isoleucine to Valine at 370, Threonine to Alanine at 382, Isoleucine to Threonine at 395, Lysine to Glutamic acid at 401, Leucine to Phenylalanine at 430, Alanine to Serine at 514, and Valine to Alanine at 536 number position in 2002, 2006 variants of DENV-3.

Mutation analysis in the amino acid level of 3 non-structural proteins in dengue virus serotype-3 (DENV-3): amino acid mutation determination of (A) NS3, (B) NS4A, and (C) NS5 proteins over the time in Bangladesh.
These proteins’ 3D structures were constructed by SWISS homology modeling. After superimposing modeled proteins in PyMOL, the deviation of the target protein structure from the reference structure is expressed by the RMSD value. The lower RMSD means higher structural similarity, and the higher RMSD means higher structural difference. RMSD values of all proteins show <1 Å, which depicts minor or negligible structural changes (Table S1).
Protein modeling
All 3 non-structural proteins (NS3, NS4A, and NS5) were modeled with ROBETTA, HHpred, and SWISS-MODEL database. In the case of SWISS and HHpred modeling, templates with the best suites are used to build the models. For modeling with SWISS-MODEL, 5YW1.1.A (identity 80.1 %), 6HUM.1.A (identity 9.09 %), and 4HHJ.1.A (identity 96.69 %) were used as a template to model NS3, NS4A, and NS5 protein, respectively (Table 1). Likewise, 2WV9_A (probability 100%), 3HR7_B (probability 91.6%), and 5JJS_A (probability 100%) were used as templates in HHpred to model NS3, NS4A, and NS5 proteins, respectively.
Protein modeling templates used for homology modeling of non-structural proteins.

For protein modeling with ROBETTA, we performed threading (RoseTTAFold) and the ab initio method to build models. Therefore, no template was required to build these models. Above all, we got 12 protein models, of which 4 models for each protein (Table 2). The 3D structures of these proteins were visualized, and PNG files (publication grade) with high resolution were derived by the UCSF Chimera visualization tool.
Overall scores of protein models by PROCHECK, MolProbity, and VERIFY-3D.

Validation of protein models
All 12 protein models were subjected to validation with different validation software tools and databases to determine the best suite model for each protein. We deployed the PROCHECK and SAVES-VERIFY 3D, Ramachandran plotting, and Z score determination from MolProbity. These parameters are used to determine the structural integrity of protein models. The Z score of each protein model was determined to evaluate the structure’s normality compared with high-resolution structures. 64 A Z score between −3 to +3 denotes a good-quality structure.
Moreover, Z score around zero determines the best standard structure of the protein. All of the above scores of the 12 protein structures were incorporated in Table 1. According to the result, we have selected protein models for NS3 and NS5, which were modeled with the RoseTTAFold method with the aid of ROBETTA. In addition, the ab initio modeling method leading protein model was selected for NS4A protein. These 3 models’ 3D structures were visualized and delineated in Figure 2. The overall validation scores of selected proteins are visualized in Figures 3 and 4.

Crystal 3D structure visualized by chimera and structural stability determination by ProSA: crystal structure of (A) NS3, (C) NS4A, and (E) NS5 protein; structural stability assessment of (B) NS3, (D) NS4A, and (F) NS5 protein of DENV-3.
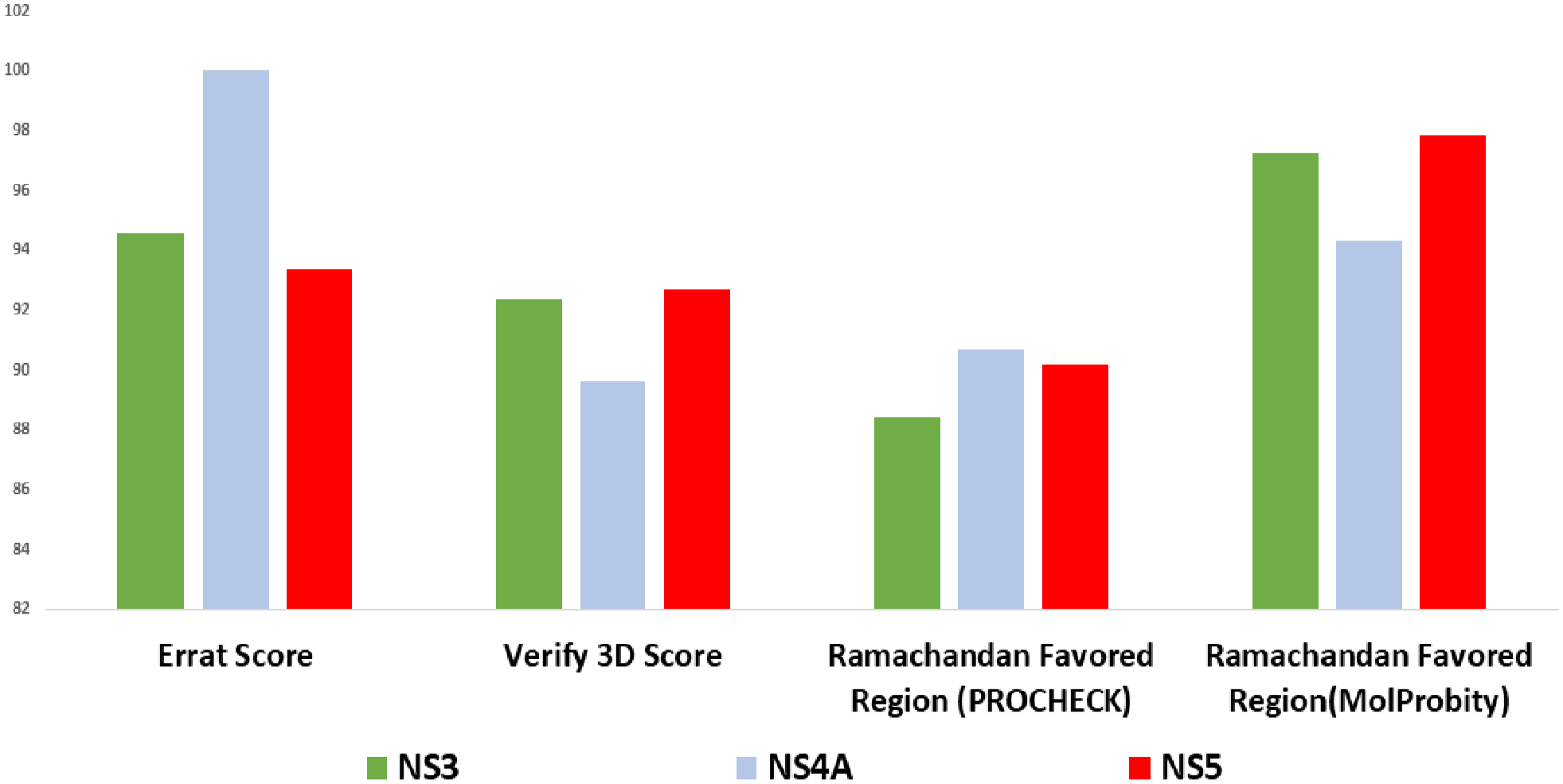
Overall 3-dimensional quality assessment profile of 3 selected NS3, NS4A, and NS5.
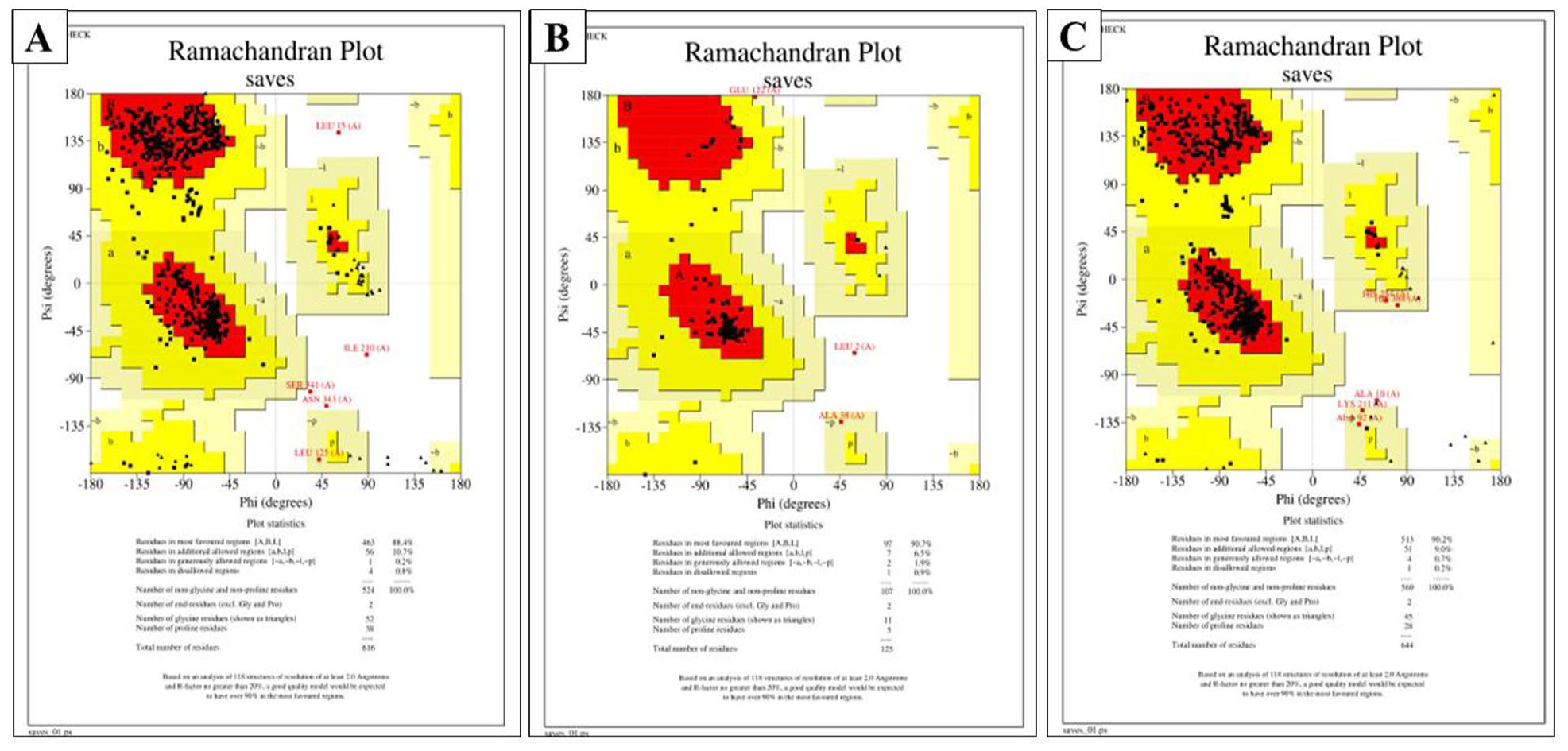
Structural integrity of amino acid residues determination in proteins by Ramachandran plot with PROCHECK: (A) NS3 protein showed 88.4% amino acid residues in the favored region, (B) NS4A have 90.7%, and (C) NS5 protein have 90.2% amino acids in the favored region.
Physicochemical properties of proteins
Among the 3 non-structural proteins, NS4A contains a lesser number of amino acids (125) compared with NS3 (616 amino acid residues) and NS5 (644 amino acid residues). Isoelectric points (pI) of NS3, NS4A, and NS5 proteins are 8.68, 5.92, and 7.96, respectively. All 3 proteins retain zero physiological charges. Moreover, the cellular location of NS3 and NS5 is cytoplasmic, and NS4A is in the plasma membrane of the host (Table 3).
Physicochemical properties of 3 non-structural proteins of DENV-3.

Abbreviation: DENV-3, dengue virus serotype-3; pI, isoelectric point.
Four drug-like compounds to inhibit the non-structural proteins
We have selected 4 drug-like compounds, which will react and theoretically inhibit the function of 3 non-structural proteins through a target sequence-based search (in silico) at the DRUGBANK database. These 4 compounds are Alpha-
Physiochemical properties of drug-like compounds.

Molecular docking analyses
Four different drug-like compounds were subjected to dock against 3 specified non-structural proteins. Energy minimized protein was retrieved as a .sce file from YASARA and converted to .pdb format. The energies of drug-like compounds were minimized, and the geometry was optimized by Avogadro and Open Babel software. At first, we performed docking with AutoDock Vina, then SWISSDOCK, and PatchDock, followed by refinement in FireDock. SWISSDOCK and AutoDock Vina provided binding energy of drug-like compounds with proteins in the KJ/mole unit.
Moreover, FireDock provided global energy for each complex, and the docking scores were observed from PatchDock. All of the scores and energy were delineated in Table 5. In light of the docking scores and binding energies, it is evident that 2 drug-like compounds named Guanosine-5’-Triphosphate (GTP) and S-adenosyl-
Molecular docking scores and binding affinity determination.

Abbreviation: NCBI, National Center for Biotechnology Information; ACE, Atomic Contact Energy.
Guanosine-5’-Triphosphate’s global binding energies for NS3, NS4A, and NS5 are −31.11 KJ/mole, −31.94 KJ/mole, and −23.59 KJ/mole, respectively. Likewise, the PatchDock scores are 5188, 4352, and 5468, respectively (Table 5). Moreover, the estimated binding energies (∆G) derived from docking with AutoDock Vina of Guanosine-5’-Triphosphate with NS3, NS4A, and NS5 are –36.81 KJ/mole, −35.06 KJ/mole, and −28.03 KJ/mole. Likewise, the estimated binding energies (∆G) derived from docking with SWISSDOCK are –44.55 KJ/mole, −36.23 KJ/mole, and −40.96 KJ/mole (Table 5). Therefore, the binding energy (∆G) above −33.47 KJ/mole is considered an efficient binding. 65 Here, the negative sign indicates that the binding energies are exothermic.
The interacting amino acid residues of NS3 with Guanosine-5’-Triphosphate are ASP (288), ASP (407), GLU (510), LEU (441), ARG (385,597), SER (600), PRO (361), LEU (427), and LYS (428). The types of bonds are pi-Alkyl, hydrogen, and attractive charge bonds (Table 6). Moreover, the interacting amino acid residues of NS4A with Guanosine-5’-Triphosphate are LYS (123), GLU (120), ARG (125), GLU (122), GLN (124), HIS (30), and GLY (35) (Figure 5).
Interacting amino acids in protein and their types of interactions with drug-like compounds.


Interaction (3D and 2D) of guanosine-5’-triphosphate with 3 non-structural proteins of DENV-3: the 3D binding of guanosine-5’-triphosphate with (A) NS3, (C) NS4A, and (E) NS5 protein; 2D interaction with bonding types of guanosine-5’-triphosphate with (B) NS3, (D) NS4A, and (F) NS5 protein.
The types of bonds are Pi-anions and hydrogen bonds (Table 6). The interacting amino acid residues of NS5 with Guanosine-5’-Triphosphate are ASP (415), GLU (209), ARG (222), CYS (460), SER (461,547), ARG (488,480), TYR (357), and THR (545). The types of bonds are hydrogen and attractive charge bond (salt bridges or ionic interaction).
In the case of S-adenosyl-
X, Y, Z coordination of binding site amino acids in protein with specified drug-like compounds.


Interaction (3D and 2D) of S-adenosyl-
The X, Y, and Z coordination of those mentioned above interacting amino acids are incorporated in Table 7.
Statistical analyses revealed the correlation of binding energies
The t test was conducted between 2 data sets of binding energy derived by docking with AutoDock and SWISSDOCK. The null hypothesis is “both data sets have an identical central tendency.” After performing the paired t test with R programming, the null hypothesis was rejected with a P value of .0004695 at a 95% confidence interval. These 2 data sets were also subjected to conduct Pearson’s product-moment correlation test. The correlation value was .805528 with a P value of .001564 at a 95% confidence interval. The correlation curve was generated with RStudio (Figure 7).
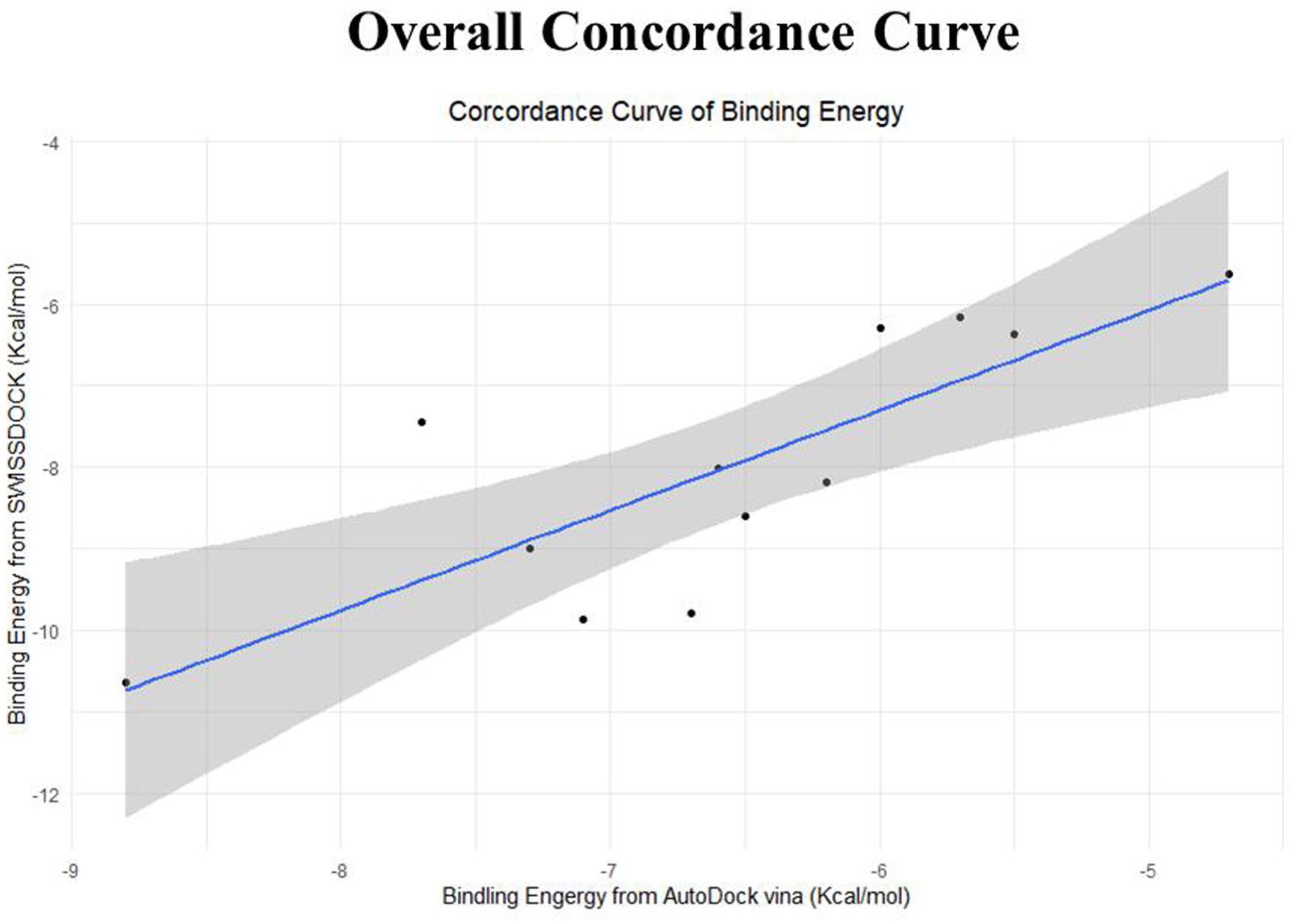
The overall concordance analysis of docking energies that are derived from SWISSDOCK and AutoDock Vina: the curve of the standard error determines the good correlation (.8) of docking energy sets derived from SWISSDOCK and AutoDock.
No ortholog was found in Homo sapiens
Broad-spectrum pBLAST was used to determine whether the human being contains an ortholog of these 3 non-structural proteins of DENV-3. However, Homo sapiens do not have any homologous proteins. Finally, this finding facilitates the acceptability of these drug-like compounds as lead molecules in drug discovery research.
Molecular dynamics simulation
Analysis of our 6 protein–ligand docking complexes found alpha Carbon (Cα) atoms of the NS5 protein with GTP and AHC showed fluctuations less than 3.5Å. The NS4A protein also showed RMSD near 4Å for both drugs. The NS3 protein exhibited a maximum fluctuation of more than 9Å observed in the NS3-GTP complex during the 100 nanoseconds (ns) simulation run (Figure 8B). The NS3-AHC complex has shown fluctuations near 7Å at 60 ns and 95 ns. From the data, we can assume that NS5 protein were found to be equilibrated after 50 ns and NS4A proteins after 75 ns, and both exhibited RMSD values near the acceptable limit except for NS3 proteins, which can be considered unstable. In the case of the protein-ligand complex, analysis of RMSD from the data obtained from protein fit ligands showed the complex of the NS5 protein with AHC was less than 3 Å and for complex with GTP was more than 5Å during the 100 ns simulation interval (Figure 8E). However, it was mentionable that the complex of NS3 with GTP showed RMSD near 5Å until 70 ns, and then it jumped to more than 7Å, and the complex with AHC showed RMSD near 7Å and also more than 8Å at some points (Figure 8A). The complex of NS4A with GTP has shown an RMSD value near 5Å and came to nearly 4Å after 90 ns of simulation, and the complex of NS4A with AHC showed an RMSD of more than 12Å at 67 ns and was more than 7Å most of the simulation runtime, but there was a major change at 50 to 65 ns where the RMSD was near 3Å (Figure 8C).

Molecular dynamics (MD) simulation study of proteins and docked protein-ligand complexes: the root alpha carbon (Cα) mean square deviation (RMSD) value delineation of (A) NS3 protein, (C) NS4A, and (E) NS5 protein in docked complexes with guanosine-5’-triphosphate (GTP) and S-adenosyl-
The RMSF is effective for determining and evaluating local conformational changes in protein chains and ligands. The variations produced by residue index C were used to compute the local structural fluctuations of the NS3, NS4A, and NS5 proteins associated with GTP and AHC chemicals. Surprisingly, NS5 protein residues exhibit low RMSF values, except for the N- and C-terminals, which have significant RMSF values (Figure 9C), which is standard in the case of both terminals. For the NS3 protein, the RMSF value was more than 5Å at 4 residues, which denote high fluctuations (Figure 9A). In the case of NS4A protein, the RMSF value mainly was low, but at residue 75, it was more than 5.5Å (Figure 9B). NS4A with GTP complex has shown lower RMSF than the NS4A with AHC complex.

RMSF value non-structural proteins in docked complexes at MD simulation: RMSF value of (A) NS3, (B) NS4A, and (C) NS5 proteins throughout the 100 ns simulation run at complexes with Guanosine-5’-Triphosphate (GTP) as well as S-adenosyl-
Initially, we considered 4 drug-like compounds to be potential antiviral compounds against DENV-3. However, performing all of the bioinformatic analyses, we have narrowed it down to 2 drug-like compounds for further research based on their binding efficiency and stability with non-structural proteins of DENV-3. We evaluated and found that S-adenosyl-
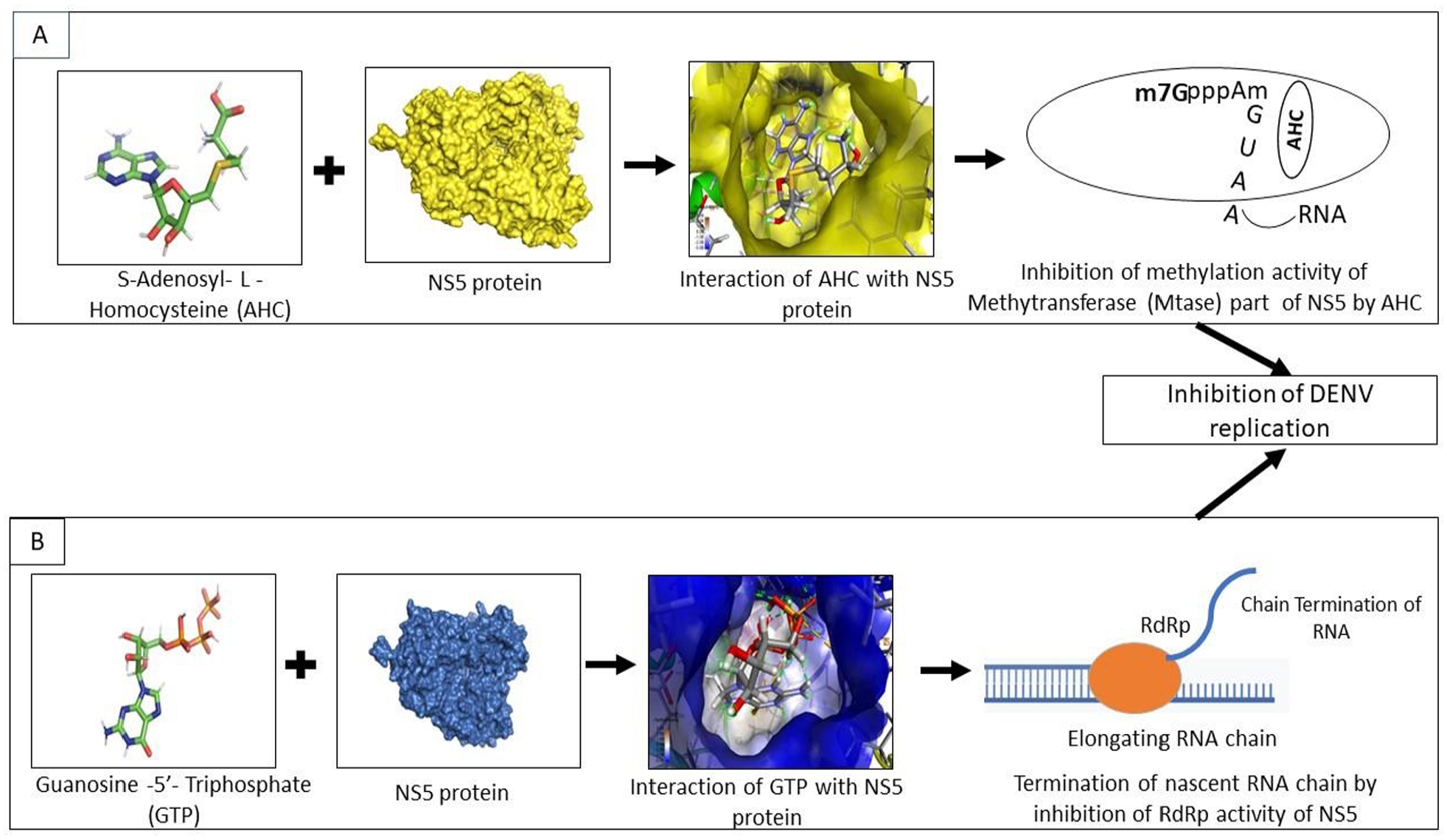
Illustration of the possible sequential inhibition of DENV-3 replication by S-adenosyl-
Discussion
No recognized vaccine or drug is available to treat DENV-infected patients. Bangladesh is one of the worst-affected endemic countries in the world. DENV-3 is the most prevalent serotype in Bangladesh. 66 In addition, the reemergence of DENV-3 caused the worst form of dengue outbreak from 2017 to 2019. 2 Therefore, we aim to detect antiviral drug-like compounds that can inhibit the multiplication of DENV-3 by interacting with non-structural proteins as they uphold structural conserveness regardless of mutational genotypes.67,68 We targeted 3 non-structural proteins named NS3, NS4A, and NS5, which serve an indispensable role in the replication of DENV-3. Mainly, NS3 is involved in helicase and protease activity, NS4A is involved in maintaining virus particles’ integrity by conducting oligomer formation, and NS5 plays methyltransferase activity along with RNA-dependent-RNA polymerase activity.
We retrieved sequences of targeted non-structural proteins of DENV-3 from different timelines in Bangladesh to observe mutations. We performed Multiple Sequence Alignment (MSA), which suggests 2.58% amino acid mutation in NS3 protein, 5.3% in NS4A, and 2.22% in NS5 protein among those variants of DENV-3 (Figure 1). Moreover, we haven’t found any mutation in the binding sites of drugs. The impact of mutations was determined using SWISS-MODEL homology modeling and superimposed individually with the reference structures in PyMOL, which performs a residue-based pairwise alignment. The RMSD value of NS3, NS4A, and NS5 protein is 0.686 Å, 0.126 Å, and 0.956Å, respectively (Table S1). These nominal values indicate that mutations do not cause any conformational changes, particularly in the binding sites of drugs. Therefore, antiviral drug targeting non-structural protein’s may provide long-term protection against DENV-3 regardless of regional variants. ROBETTA database, HHpred, and SWISS-MODEL were used to speculate the best models. HHpred and SWISS-MODEL used the homology modeling method, and ROBETTA deployed RoseTTAFold and the ab initio strategy to model the proteins. Both homology and threading (RoseTTAFold and ab initio) methods were considered for the protein modeling. We found 12 protein models (4 models for each protein). The protein model quality was assessed through PROCHECK (Ramachandran Plotting), SAVES-VERIFY 3D, ERRAT score, and MolProbity (z score). We have selected the best model for each protein based on these scores. We selected 3 models based on the overall evaluation performance. These 3 models are NS3 (RoseTTAFold), NS4A (ab initio), and NS5 (RoseTTAFold). For the NS3 protein model (RoseTTAFold), the Z score was 0.9 (Table 2), which indicates good native quality as a Z score between −2 to +2 expresses a good native structure of the protein. 64 ERRAT score determines the overall protein’s good structure quality if the score resides greater than 50. 69 Here, the ERRAT score for NS3, NS4A, and NS5 proteins were 94.56, 100, and 93.35, respectively. The VERIFY-3D score reveals the protein’s structural integrity, and scores above 80% represent the excellent structural integrity of the protein models. 69 We found that the VERIFY-3D scores of these 3 proteins are 92.37% (NS3), 89.6% (NS4A), and 92.7% (NS5). According to PROCHECK, the amino acid residues under the Ramachandran Favored region were 88.4%, 90.7%, and 90.2 for NS3, NS4A, and NS5, respectively (Figures 2 and 4). Likewise, Ramachandran plotting by MolProbity reveals the percent of amino acid residues in the favored region are 97.23%, 94.3%, and 97.82% for NS3, NS4A, and NS5, respectively. All of the scores determine the regularity of the protein structures. 70 The physicochemical properties of proteins were obtained with the ProtParam tool of ExPASy (Table 3). The instability index values are 30.79, 33.55, and 35.47 for NS3, NS4A, and NS5, respectively. The instability index value below 40 states the stability of proteins. 71 Therefore, we can assume the strength of our protein structures. In addition, the protein’s GRAVY (grand average of hydropathy) score determines the protein’s hydrophobicity or hydrophilicity. A GRAVY score below zero indicates hydrophobic and globular, but a score above zero determines hydrophilic. For example, GRAVY scores of NS3, NS4A, and NS5 are –0.532, 0.574, and –0.658. It makes evident that the NS3 and NS5 are hydrophobic and globular, but NS4A is hydrophilic. Furthermore, the theoretical pI of NS3, NS4A, and NS5 are 8.68, 5.92, and 7.96. It means NS4A is acidic, and NS3 and NS5 are alkaline. Moreover, the physiological charges of all 3 proteins are zero. Interestingly, NS3 and NS5 protein’s cellular location is cytoplasmic, and NS4A is in the plasma membrane. Above all, these physiological properties lead these 3 proteins to form hydrogen bonds promptly, resulting in an ideal target for antiviral drug-like compounds.
According to the DRUGBANK database, 4 drug-like compounds can play an inhibitory role against NS3, NS4A, and NS5 of DENV-3. Such as Alpha-
Molecular docking was performed to analyze drug-like compounds’ binding efficiency and screen out the best drug-like compound among the suggested 4. The energy was minimized to optimize the geometry of proteins and drug-like compounds. These energy-minimized compounds were considered for molecular docking analyses to get more authentic results.
72
Four different docking tools/servers were used to conduct the molecular docking analyses and validate the outcomes. Based on the scores and binding energies, we have sorted 2 compounds as promising lead compounds to proceed with further analyses (Table 5). These 2 compounds are Guanosie-5’-Triphosphate (GTP) and S-adenosyl-
Moreover, in the case of bond formation, S-adenosyl-
In addition, an MD simulation study determined the stability of interaction. These results support the effective binding ability of S-adenosyl-
An MD simulation was conducted to determine the stability of the complex between protein and drug-like compounds in the human body environment. Based on the RMSD result, it can be determined whether the simulation has equilibrated. Fluctuations between 0 and 3 Å within a reference protein structure are perfectly acceptable, where a much larger value indicates a significant conformational change of the protein and the system is not stable. 77 Cα atoms of the NS5 protein with GTP and AHC showed fluctuations very close to the acceptable limit. Both NS3 complexes are not found to be equilibrated during the 100 nanoseconds (ns) simulation time, whereas both complexes of NS4A are assumed to be equilibrated after 75 ns. The RMSD of the protein-ligand complex of NS5 with AHC was within the acceptable limit (< 3Å), which indicates it was very stable during the simulation. But the complex of NS5 with GTP cannot be considered as durable as the RMSD was beyond the acceptable limit (>5Å). All the NS3 and NS4A, including the complex of NS5 with GTP, are unstable, whereas the complex of NS5 with AHC was significantly stable and equilibrated during the 100 ns simulation. In the case of the RMSF result, both complexes of NS5 were found to be having lower fluctuation than the other complexes of NS3 and NS4A proteins.
Upon binding of S-adenosyl-
Finally, the MD study revealed that the binding of NS5 with S-adenosyl-
Conclusion
There is no specific antiviral drug available to treat the infection of dengue. Therefore, our investigation explored that S-adenosyl-
Supplemental Material
sj-docx-1-bbi-10.1177_11779322231158249 – Supplemental material for S-Adenosyl-l-Homocysteine Exhibits Potential Antiviral Activity Against Dengue Virus Serotype-3 (DENV-3) in Bangladesh: A Viroinformatics-Based Approach
Supplemental material, sj-docx-1-bbi-10.1177_11779322231158249 for S-Adenosyl-l-Homocysteine Exhibits Potential Antiviral Activity Against Dengue Virus Serotype-3 (DENV-3) in Bangladesh: A Viroinformatics-Based Approach by Dipok Kumer Shill, Shafina Jahan, Mohammad Mamun Alam, Md Belayet Hasan Limon, Muntasir Alam, Mohammed Ziaur Rahman and Mustafizur Rahman in Bioinformatics and Biology Insights
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research study was funded by core donors who provide unrestricted support to icddr,b for its operations and research. Current donors providing unrestricted support include the Governments of Bangladesh, Canada, Sweden, and the United Kingdom. We gratefully acknowledge our core donors’ support and commitment to icddr,b’s research efforts.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Mohammad Mamun Alam generated the idea, supervised all analyses, and monitored manuscript writing. Dipok Kumer Shill and Shafina Jahan performed the proteins modeling, data analysis, molecular docking, and all of the simulations studies and prepared the manuscript. Md Belayet Hasan Limon conducted molecular dynamics experiments. Muntasir Alam reviewed the manuscript. Mohammed Ziaur Rahman and Mustafizur Rahman guided the study analysis and secured funding.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.