
Abstract
Introduction:
Epidermal growth factor receptor (EGFR) is a transmembrane protein that belongs to the ErbB/HER-family of tyrosine kinase receptors. Somatic mutations and overexpression of EGFR have been reported to play a vital role in cancer cell development and progression, including cell proliferation, differentiation, angiogenesis, apoptosis, and metastatic spread. Hence, EGFR is an important therapeutic target for the treatment of various types of epithelial cancers. Somatic mutations have led to resistance to clinically approved synthetic EGFR inhibitors. Furthermore, synthetic EGFR inhibitors have been associated with several side effects. Thus, there is a need to develop novel EGFR inhibitors with an acceptable biosafety profile and high efficacy.
Methods:
Herein, we employed structural bioinformatics and theoretical chemistry techniques via molecular docking, molecular mechanics generalized Born surface area (MM-GBSA) calculation, density functional theory analysis (DFT), and pharmacokinetic study to identify novel EGFR inhibitors.
Results:
The stringent molecular docking and MM-GBSA calculations identified MET 793, LYS 745, PHE 723, ASP 855, ARG 411, and THR 854 as principal amino acid residues for EGFR-ligands interactions. Furthermore, Colocasia affinis Schott compounds exhibited higher binding energy and more stable interactions than the reference compound (gefitinib). DFT analysis also ascertains better bioactivity and chemical reactivity of C. affinis Schott with favorable intramolecular charge transfer between electron-donor and electron acceptor groups. The pharmacokinetic profile of C. affinis Schott bioactive compounds satisfies Lipinski’s rule of five assessment.
Conclusion:
Collectively, C. affinis Schott compounds demonstrated higher inhibitory potentials against EGFR and better pharmacological properties when compared with gefitinib. C. affinis Schott compounds are therefore suggested as promising therapeutic EGFR inhibitors for cancer treatment.
Introduction
The epidermal growth factor receptor (EGFR) is a transmembrane protein that belongs to the ErbB/HER-family of receptor tyrosine kinases and exerts an essential physiological role in the epithelial cells.1,2 EGFR is structurally characterized by the presence of an extracellular ligand-binding domain, structural motifs including glycosylation and immunoglobin-like sites, an EGF-like domain composed of tyrosine rich C region, and the tyrosine kinase domain. Ligand binding triggers structural conformational changes in homo- and hetero-dimerization associated with EGFR and other activated HER family analogs. Therefore, EGFR function and catalytic activities are activated by this multi-step interaction. 3 Following EGFR dimerization, various residues of the intrinsic EGFR kinase domain are autophosphorylated, leading to the activation of downstream signaling cascades such as RAS/MAPK, PI(3)K/Akt, PLCc/PKC, and Jak/iSTAT pathways 4 (Figure 1). Several investigators have reported the deregulation and overexpression of EGFR in different epithelial tumors. This is also consistent with the previous hypothesis that deregulated EGFR expression have been associated with clinical manifestations of various human cancer cells, including prostate, breast, lung, head and neck squamous cell carcinoma (HNSCC), colon, and pancreatic cancer.5-7
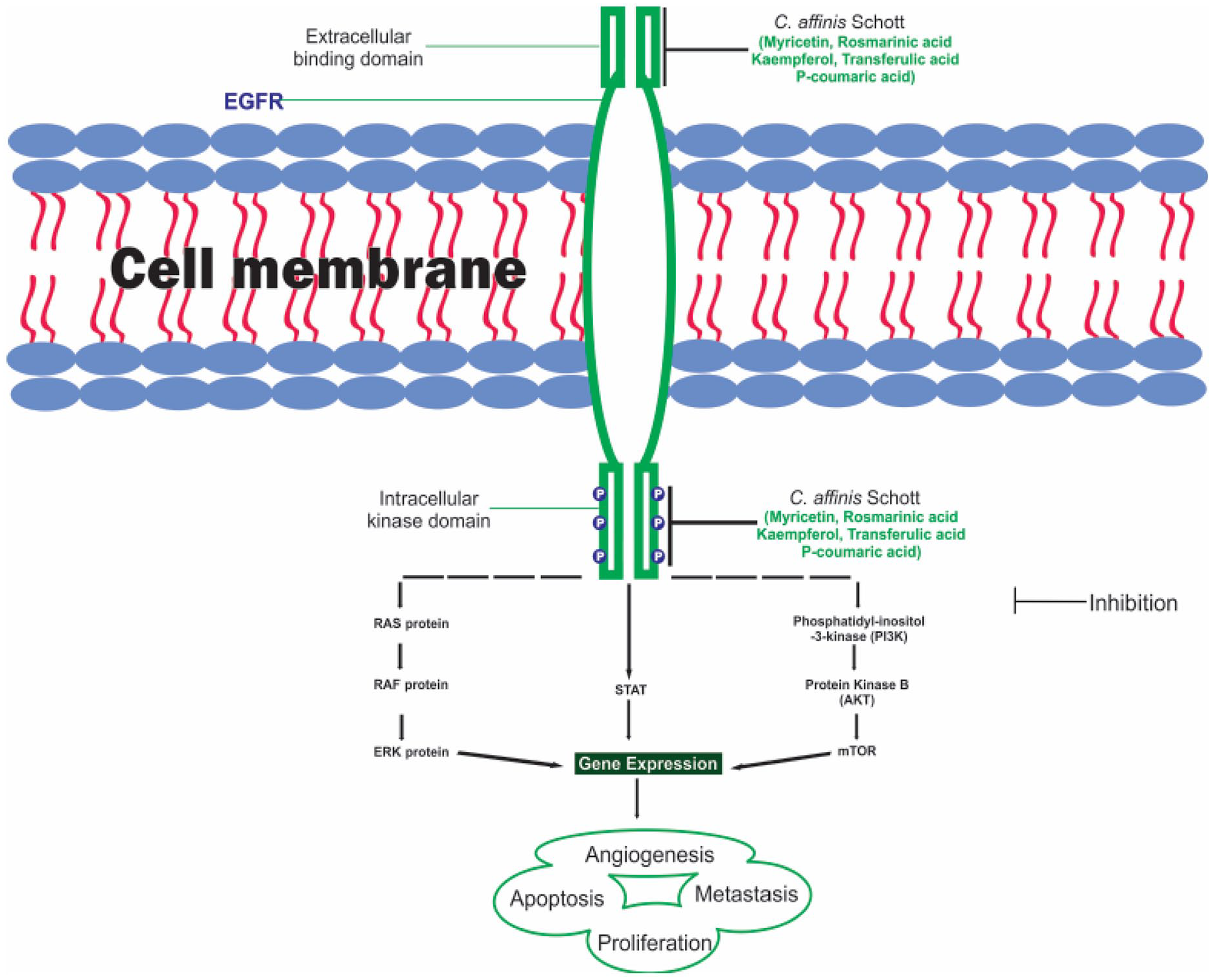
EGFR signaling pathway.
Since the discovery of EGFR for its versatile role in tumorigenesis during the last 4 decades, EGFR has received intense consideration as a therapeutic target for cancer treatment. One effective way to block EGFR signaling activity is by inhibition with small molecules that bind competitively with the ATP binding site of the EGFR tyrosine kinase. 8 Erlotinib, gefitinib, osimertinib, and lapatinib are small molecules EGFR inhibitors and FDA-approved drugs for cancer treatment. EGFR inhibitors function either through a reversible or non-reversible mechanism. The reversible inhibitors bind competitively to the ATP binding site while non-reversible inhibitor displayed inhibitory potential against allosteric sites or Cys 797 residues. 9 Gefitinib binds reversibly to the binding site of EGFR thereby competing for the ATP binding pocket. 10 Another approach in cancer therapy uses monoclonal antibodies to inhibit natural ligand binding at the extracellular binding domain of EGFR.9,10 Cetuximab is a monoclonal antibody that hampers receptor dimerization and activation, ultimately downregulating the downstream effectors. 11 Cetuximab can either be used as chemotherapy, monotherapy, and/or radiotherapy and has proven effective in HNSCC and advanced metastatic colorectal cancer. 12
Previous studies have reported the resistance of cancer cells to synthetic drugs. Thus, multidrug combination therapy would be a practical treatment approach in comparison to single drugs. However, the major setback in multidrug therapy is excessive cytotoxicity. 13 In addition, several different structural scaffolds that bind to the hydrophobic region of EGFR could help prevent cancer treatment. 14 Somatic mutations have led to resistance to gefitinib and other synthetic EGFR inhibitors. Thus, there is a need to develop novel EGFR inhibitors with an acceptable biosafety profile and high efficacy. The acquired resistance and associated side effects of synthetic drugs prompted the search for natural products with pharmacological potential for cancer therapy. 15 Colocasia affinis Schott is a perennial plant that belongs to the Araceae family. It is commonly known as the Dwarf elephant’s ear, and it is found abundantly in the metropolis of Asian countries and other parts of the world. 16 Mondal et al 17 reported the antioxidant, anti-inflammatory, antidiarrheal, and antimicrobial activity of the methanolic extract of C. affinis Schott. Phytochemical screening of C. affinis Schott revealed various classes of natural chemical groups such as phenols, terpenoids, saponins, and flavonoids. 18 To the best of our knowledge, there has been no attempt to evaluate the anticancer effect of C. affinis Schott. Hence, this is the first computational study to identify hit compounds from C. affinis Schott for cancer treatment.
Here, we investigated the therapeutic potential of bioactive compounds from C. affinis Schott in the treatment and management of cancer. Structural bioinformatics and advanced theoretical chemistry techniques were utilized through molecular docking, Prime MM-GBSA (Molecular Mechanics-Generalized Born Surface Area), density functional theory analysis, and pharmacokinetic study. Based on the results, C. affinis Schott bind firmly and effectively inhibited EGFR. Strong, stable interaction, and coordination of C. affinis Schott with the amino acid residues at the binding site of EGFR are the probable mechanism of its inhibition. Overall, we identify new promising small molecules that may serve as EGFR inhibitors.
Methodology
Protein preparation
The 3-dimensional crystal structure of EGFR was retrieved from the protein data bank with PDB ID: 5D41. The protein co-crystallized with a native ligand was prepared using the protein preparation module of Schrödinger Maestro 11.5. The target protein (EGFR) was refined by assigning bond orders, adding hydrogen atoms. Furthermore, prime tool was utilized for filling missing loops and side chain. EGFR was further optimized through the generation of tautomeric states at a neutralized pH, restrained minimization using the OPLS3 force field.19,20 The prepared EGFR was selected for molecular docking.
Ligand preparation
The bioactive compounds from C. affinis Schott and the reference compound were obtained from published literatures,10,16 and their 2D structures were retrieved from the NCBI PubChem database. The ligands were prepared using the LigPrep of Schrodinger suite by employing OPLS3 forcefield. Epik module was utilized to generate the compounds ionization states at a pH of 7.0 ± 2.0. 21
Receptor grid generation
Receptor grid generation defines the binding orientation and the size of the active site for protein-ligand docking. The scoring coordinates of the EGFR binding pocket was determined based on the co-crystallized ligand using the receptor grid generation module of Schrödinger Maestro 11.5. The x, y, z grid are −33.613, 27.672, and 18.59, respectively. 19
Molecular docking procedure
The prepared ligands were docked into the defined active site of EGFR via Glide-SP (standard precision) followed by XP (extra precision) to correct false-positive results. 22 The van der Waals scaling factor was set at 0.80 for the ligands atoms. The docking protocol was validated by splitting the co-crystallized ligand from the protein, prepared and re-dock into the binding site of EGFR. The calculated root means square deviation (RMSD) of 1.54 Å (normal range: 1-2 Å) confirms the reliability and reproducibility of the docking approach. 23
Binding free energy calculation/thermodynamics calculation
The molecular mechanics generalized Born surface (MM-GBSA) tool integrated with prime of the Schrödinger Maestro 11.5 was employed to calculate the binding free energy of the docked complexes. The relative free energy of the docked complexes was computed using the OPLS3 force field, VSBG solvent, and the rotamer search algorithm.24,25
The binding free energy was calculated using the equation below.
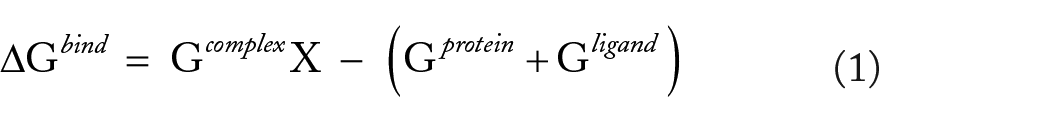
Density functional theory analysis
The theoretical methods employed to compare the chemical and biological activities of compounds have become widespread nowadays. A quantum chemical calculation via density functional theory (DFT) was used to investigate the physicochemical properties of selected bioactive compounds from C. affinis Schott and predict compounds with prominent biological activities. Firstly, the conformer distribution search was performed on each bioactive compound and the most stable conformer was selected for full.
DFT calculation with B3LYP functional method 26 and 6-31G* basis set 27 as implemented in Spartan 14 computational software on an Intel (R) computer with 2.60 GHz, 500 G hard disc, and 6.00 GB ram specifications. As a result of the calculations performed using this method, many parameters can be obtained. Several parameters obtained from the calculations are highest occupied molecular orbital energy (EHOMO), lowest unoccupied molecular orbital energy (ELUMO), energy band gaps (Eg), ionization energy (I), electron affinity (A), chemical hardness (η), chemical softness (δ), chemical potential (μ), electronegativity (χ), electronic energy, enthalpy, Gibb’s free energy, and dipole moment (D).
The energy bandgap (Eg) was calculated from the difference between ELUMO and EHOMO (2)

electron affinity (A) and ionization potential (I) are related to ELUMO and EHOMO using Koopman’s theorem, 28 as shown in equations (2) and (3), respectively.


The electronegativity (χ) and chemical hardness (η) of the compounds were calculated using Parr and Pearson. 29


Additionally, chemical softness (δ) is the inverse of chemical hardness

Evaluation of ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties
The ADMET study was carried out to evaluate the pharmacokinetic profile of the docked compounds including drug-likeness properties, Lipinski’s rule of five violations, and toxicity. The ADMET properties were analyzed using the admetSAR web server (http://lmmd.ecust.edu.cn/).30,31 This is a web-based platform where the screened compounds were assessed for their pharmacokinetics, aqueous solubility, and pharmacodynamics using various models.
Results and Discussion
Molecular docking, drug-like properties, and interaction profiling of EGFR-ligand complexes
Computational methods are often used for molecular docking to predict the ligand-receptor complex structure; this is usually achieved through sampling conformations of the ligand in the protein’s active site and a score ranking of the conformations. The computational study includes binding affinity (kcal/mol) prediction, the interaction of the ligands within the binding pocket of epidermal growth factor receptor (EGFR), MM-GBSA, and their pharmacokinetic profile, as shown in Table 1. EGFR is a receptor tyrosine kinase (RTK); the elevated gene expression of EGFR has been reported to be associated with poor prognosis in oral squamous cell carcinoma (OSCC), 32 its’ inhibition have been reported in several cases of cancer, including breast and oral cancers.
Molecular docking and MM-GBSA results of bioactive compounds from C. affinis Schott and reference compound.
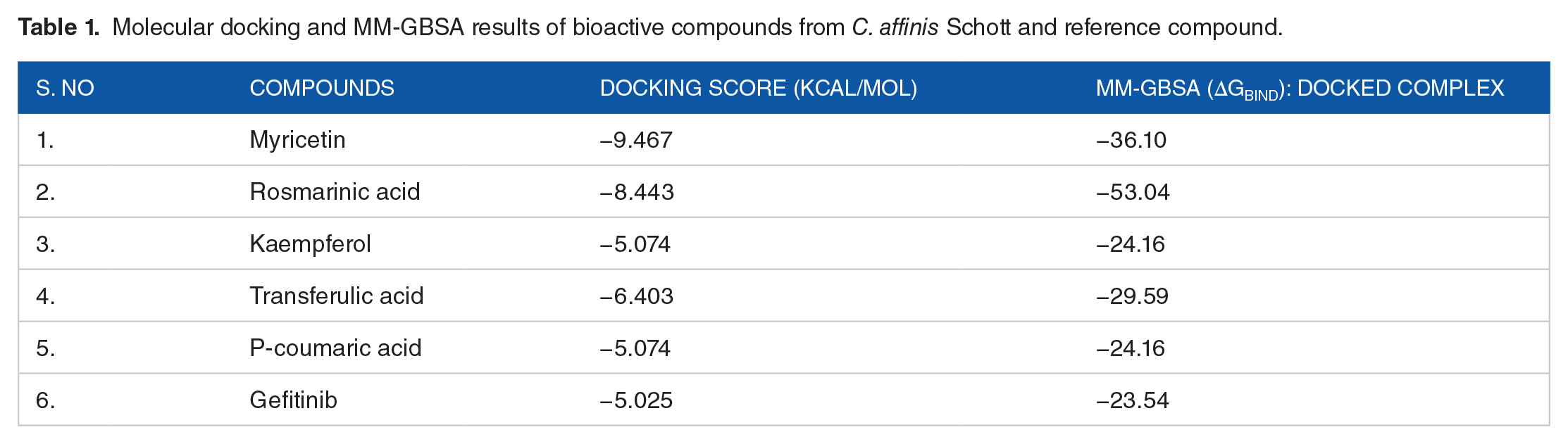
The bioactive compounds from C. affinis Schott showed a favorable binding affinity and optimally saturated the active site of EGFR, ranging from P-coumaric acid to Myricetin with the binding energy of −5.074 and −9.467 kcal/mol, respectively. A more negative binding energy corresponds to sturdier binding. Following the docking approach, the lead compounds bind firmly within the active site of EGFR while forming principal amino acid interactions with the following amino acid residues: MET 793, LYS 745, PHE 723, ASP 855, ARG 411, and THR 854 (Figure 2). 33 These amino acid residues play a fundamental role in predicting the EGFR binding site and the mechanism of catalysis. The docked compounds interacts with MET 793, LYS 745, PHE 723, ASP 855, ARG 411, and THR 854 in the EGFR binding pocket through H-bond formation with the nitrogen atom of the quinazoline ring and Van der Waals interactions. Understandably, the ligand-EGFR complexes result in inter and intra molecular interactions such as hydrogen bonding, pi-pi stacking, pi-cation, and salt bridge.

2D molecular contacts profiling of docked compounds with amino acid residues (4.00 Å) at the active site of EGFR: (a) myricetin, (b) rosmarinic acid, (c) kaempferol, (d) transferulic acid, (e) p-coumaric acid, and (f) gefitinib.
Myricetin was the best bioactive molecule of C. affinis Schott, with the highest binding energy of −9.467 kcal/mol. It interacts with the hydrophobic and polar amino acids MET 793 ARG 841 and established pi-cation interaction with ASP 855. Rosmarinic acid with the binding energy of −8.443 kcal/mol is suggested to interact primarily with the surrounding amino acids via hydrophobic, pi-pi stacking, or van der waals forces: PHE 723, LYS 745, THR 854, ASP 855, and MET 793 (Figure 3).

Interaction profile of the EGFR-ligand complexes after molecular docking studies in 3D. Interactions are shown in dotted lines: (a) myricetin-EGFR complex, (b) rosmarinic acid-EGFR complex, (c) kaempferol-EGFR complex, (d) transferulic acid-EGFR complex, (e) P-coumaric acid-EGFR complex, and (f) gefitinib-EGFR complex.
Kaempferol comprised of a ring system that is believed to occupy the EGFR binding site completely with a binding energy of −8.425 kcal/mol. Transferulic acid and P-coumaric acid exhibited binding energy of −6.403 and −5.074 kcal/mol respectively while establishing hydrogen bonding interaction with MET 793 and polar interaction with THR 854. C. affinis Schott molecules have better binding energy than Gefitinib which is a standard drug used as a positive control for the ligands. Gefitinib showed binding energy of −5.025 kcal/mol; this shows that C. affinis Schott has a high potential to bind EGFR for the treatment of several kinds of cancer.
The ∆Gbind for EGFR-hit ligand complexes were calculated using the MM-GBSA module integrated with the prime program of the Schrodinger suite. The ∆Gbind was utilized for advanced mechanics calculation of the binding energy for the screened compounds following the docking analysis. Several investigators have documented that, MM-GBSA approach is a reliable post docking method for calculating the binding position of docked complexes. 34 Based on the MM-GBSA output (Figure 4), myricetin, rosmarinic acid, kaempferol, transferulic acid, p-coumaric acid demonstrated binding energy of −36.10, −53.04, −44.67, −29.59, and −24.16 kcal/mol, respectively (Figure 4). Gefitinib had the lowest binding affinity of −23.54 kcal/mol. Hence, the ∆Gbind results further ascertain the C. affinis Schott compounds’ better binding energy compared to the positive control ligand (gefitinib).

Graphical representation of the molecular docking score and Prime/MM-GBSA binding energy (∆Gbind) of C. affinis Schott compounds and the reference ligand. The left frame (blue) signifies the docking score, while the right frame (red) shows the MM-GBSA binding energy.
Density functional theory analysis
Thermochemical analysis
Thermodynamic properties are crucial parameters in determining the spontaneity of a chemical reaction and the chemical stability of a reaction. Gibbs free energy is an essential thermodynamic quantity used to describe the ligand-receptor interaction. It represents the probability of bio molecular events occurring. A positive value of free energy indicates that binding will not occur without adding the required external energy while negative free energy shows that binding will occur spontaneously. 35 The extent of interaction of the ligand with the receptor is determined by the magnitude of the negative free energy. Also, enthalpy is a measure of the total energy of a thermodynamic system. The binding enthalpy reflects the energy change of the system when the ligand binds to the receptor. Table 2 shows Gibb’s free energy calculated for the studied compounds. All the compounds have negative free energy indicating that binding with the target receptor can occur without supplying any external energy. Rosmarinic acid and Myricetin show the highest free energy (−1297.057 and −1179.140 Hartree), which indicate that Rosmarinic acid will interact more than other studied compounds. The dipole moment provides information about the polarity of a compound and the distribution of electrons in the compound. 36 It enhances binding affinity, non-bonded interactions, and hydrogen bond formation with the receptor protein. As shown in Table 2, Rosmarinic acid shows the highest dipole moment (4.11 debye).
Molecular weight, electronic energy, enthalpy, Gibb’s free energy, polar surface area (PSA), and polarizability values obtained via DFT at the B3LYP/6-31G* level.

Frontier molecular orbital (FMOs)
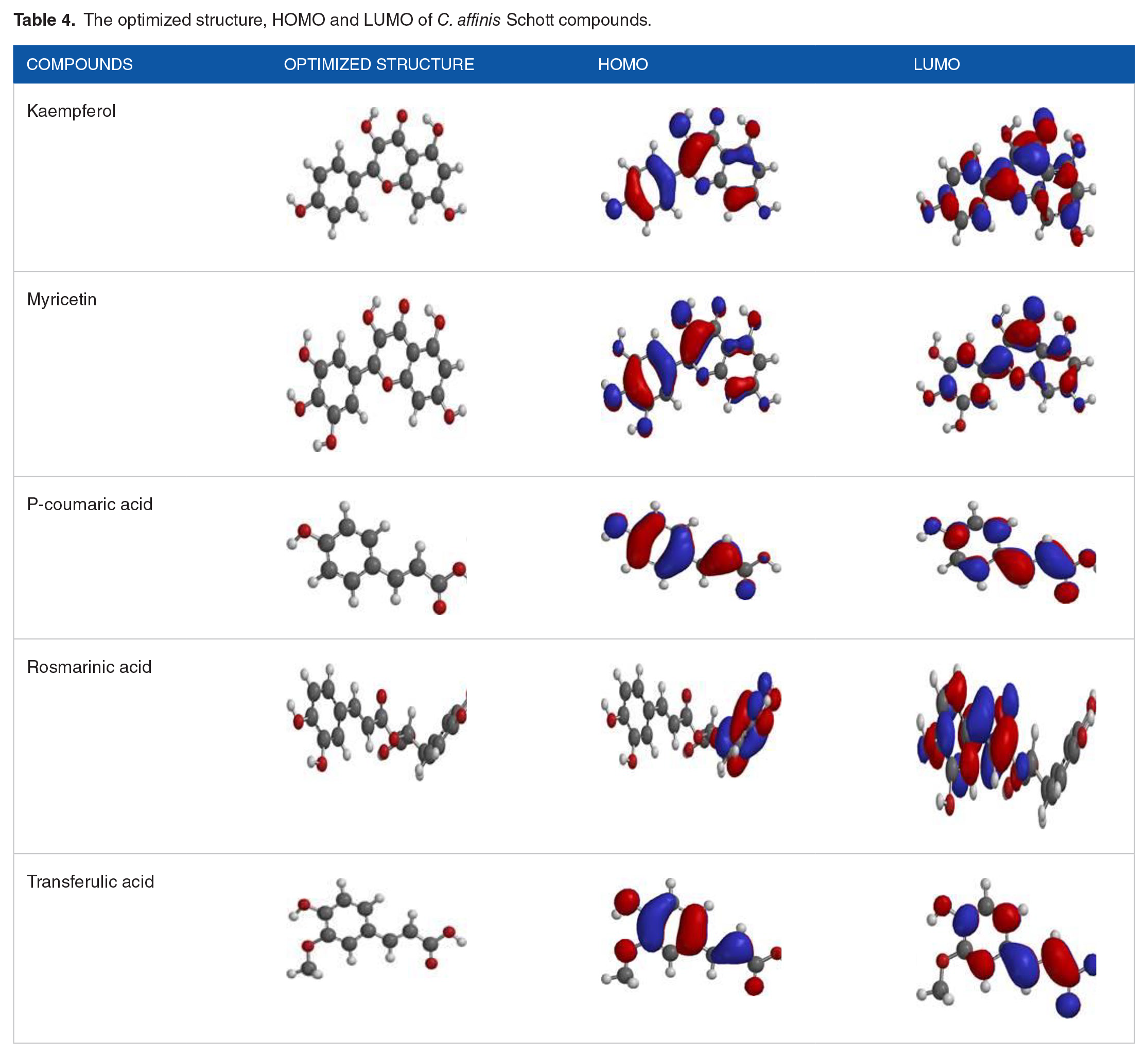
The FMOs, HOMO, and LUMO, are the most crucial orbitals in the molecule. They play an essential role in the optical, electric properties, 37 UV-Vis spectral, and quantum chemistry. The FMOs describe how the molecule interacts with other molecules and provides information about the transfer of electron in a molecule and the chemical reactivity and stability of a molecule. The HOMO energy describes the electron-donating ability; higher values of EHOMO indicate a better tendency of the molecule to donate electron. 38 The ELUMO determines the power of a molecule to accept an electron, lower value of ELUMO of a molecule increases the probability of accepting electrons. Therefore, higher values of EHOMO and lower values of ELUMO are responsible for the low stability and high reactivity of a molecule. From Table 3, the EHOMO values for the studied compounds increase in order; Coumaric acid < Transferulic acid < Rosmarinic acid < Kaempferol < Myricetin. Myricetin (−5.45 eV) shows the highest value of EHOMO, which indicates a better tendency to donate an electron to the target receptor than other compounds. Also, the ELUMO values calculated are shown in Table 3. Myricetin displays the lowest value of ELUMO, indicating the ability to accept electron than other studied compounds. Furthermore, both the EHOMO and ELUMO are entirely spread over the molecular structure as seen in Table 4, significant overlapping of HOMO-LUMO is expected, leading to strong charge transfer behavior. The band gap energy between the EHOMO and ELUMO is vital in predicting the chemical reactivity of a molecule. The values of band gap energy reflect the chemical reactivity and stability of a molecule. The larger the band gap energy, the harder and more stable and less reactive the molecule. A decrease in the energy band gap signifies high reactivity and low stability. The values of the energy band gap are in order; Myricetin < Kaempferol < Rosmarinic acid < Transferulic acid < Coumaric acid. Myricetin shows the lowest band gap among the isolated compounds, indicating it is more reactive toward the target receptor than other compounds.
Chemical parameters obtained via DFT at the B3LYP/6-31G* level.

Abbreviations: (Eg), energy band gaps; (EHOMO), highest occupied molecular orbital energy; (ELUMO), lowest unoccupied molecular orbital energy; (I), ionization energy; (δ), chemical softness; A, electron affinity; η, chemical hardness; μ, chemical potential; χ, electronegativity.
The optimized structure, HOMO and LUMO of C. affinis Schott compounds.
Global reactivity descriptors
Global reactivity descriptors (GRD) were calculated to acquire a deep understanding of the chemical stability and the reactivity of the bioactive compounds toward the target receptor. The GRD calculated are ionization energy, electron affinity, chemical hardness, chemical softness, chemical potential, and electronegativity. Ionization energy (I) describes the chemical reactivity and stability of a molecule. It is defined as the energy needed to remove an electron from a molecule. High ionization energy means high stability and chemical inertness, while small ionization energy indicates high reactivity and low chemical inertness. 39 Myricetin from Table 3 has the lowest ionization energy (5.45 eV), marking the best reactive compound toward the target receptor (EGFR). Electron affinity (A) is the energy liberated when an electron is added to a neutral molecule. A molecule with high electron affinity is prone to accept electron easily than one with low electron affinity. 40 Kaempferol and Myricetin show the highest electron affinity indicating the most reactive compound.
Chemical hardness and softness are essential in understanding the reactivity of the chemical system. Chemical hardness expresses the resistance toward the electron cloud deformation of a molecule. 41 A hard molecule has large band gap energy, while a soft molecule has small band gap energy. The soft molecule will be more and easily polarizable than the hard molecule. 42 As shown in Table 3, coumaric acid has the highest hardness value (2.18 eV), indicating the hardest molecule. Myricetin has the lowest softness value (0.54 eV), indicating the softest molecule. Electronegativity (χ) represents the ability of the molecule to attract electron electrons toward itself. 40 From Table 1, coumaric acid has the highest electronegativity (3.81 eV) compare to all other compounds.
Evaluation of ADMET properties
The ADME properties are used to predict the pharmacokinetics potentials of the bioactive molecules. 43 The ADME properties tested are revealed as models in Table 5. This model is suitable for testing compound suitability for oral dosing, assuming intestinal permeability and determining drug efflux. The pharmacokinetic study of the bioactive compounds from Colocasia affinis Schott shown an acceptable biosafety profile and can be suitable for oral prescription. The bioactive molecules have lower toxicity effects than Gefitinib (standard drug). Gefitinib shows blood brain barrier permeation, hepatotoxicity, and also serve as inhibitors of CYP1A2, CYP2C19, CYP3A4. Furthermore, gefitinib is a substrate of CYP2D6, CYP3A4CYP, and p-glycoprotein.
Pharmacokinetics predictions for myricetin, rosmarinic acid, kaempferol, transferulic acid, p-coumaric acid, and gefitinib.
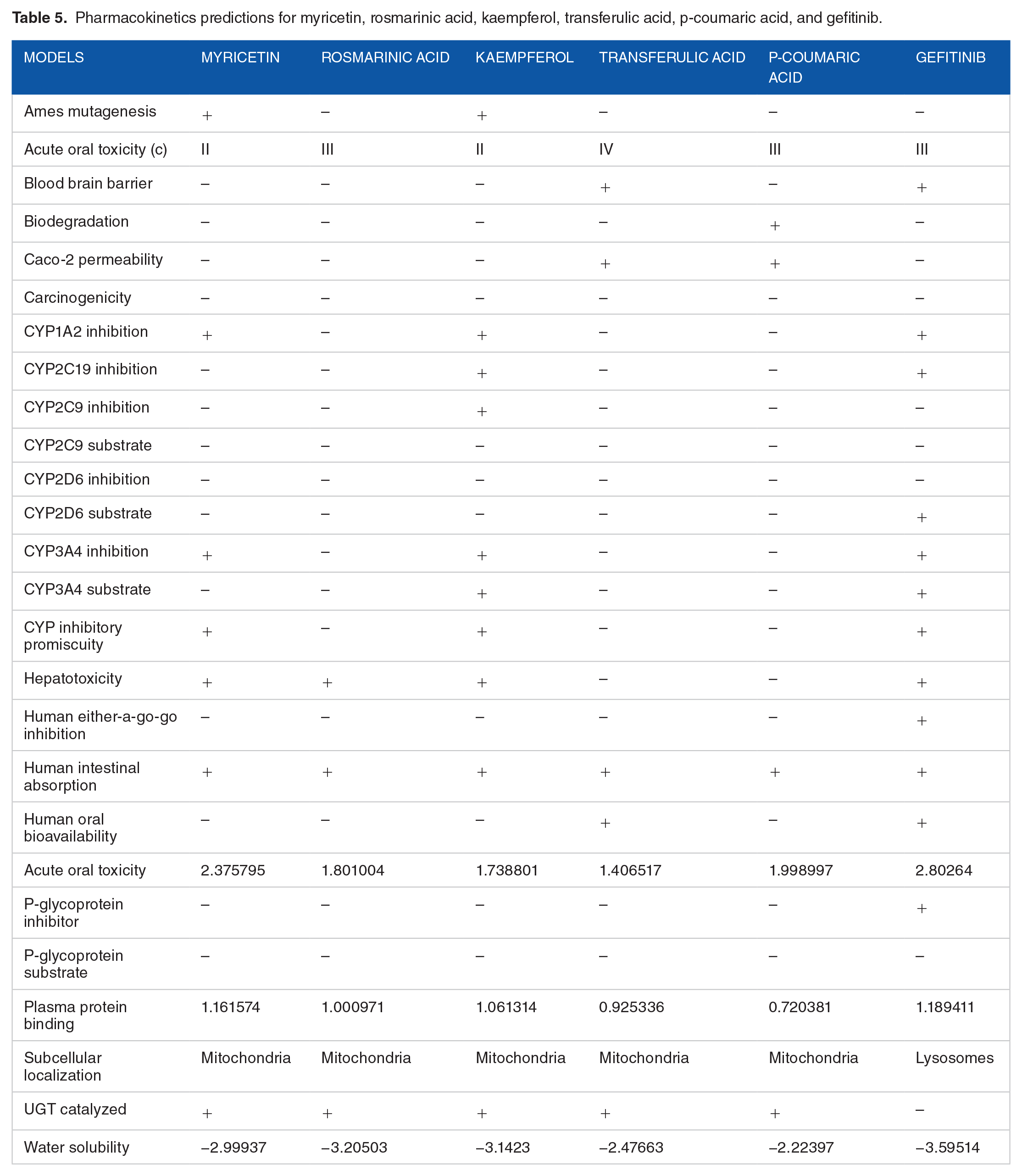
The pharmacokinetic analysis of Myricetin showed that it has very low toxicity; it has a negative blood-brain barrier, cannot be easily biodegraded, it is less carcinogenetic and does not inhibit CYP2C19, CYP2C9, CYP2D6, Human either-a-go-go, P-glycoprotein, and can easily be absorbed by the human intestinal tract with low acute oral toxicity value. Myricetin was proved to have psychological properties as a suitable candidate to treat type-II diabetes mellitus through insulin production after the administration of glucose; the insulinotropic characterization of Myricetin performed on isolated islets and in Wistar rats revealed its’ glucoregulatory activity. 44 Li et al 45 have proved Myricetin as an active anticancer compound with pharmacological effects on tamoxifen and its metabolites.
Rosmarinic acid pharmacokinetic properties are a good recommendation for oral prescription as it has a low acute toxicity value. It does not cause Ames mutation, and it has a negative blood-brain barrier, it is not carcinogenic, it does not inhibit CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4, CYP, Human either-a-go-go, P-glycoprotein; it has moderate water solubility value and can easily be absorbed by the human gastrointestinal tract. Rosmarinic acid was revealed to destroy cancer cells by inhibiting the Aurora kinases, which plays an essential role in cell cycle regulations. 46 Rosmarinic acid has anti-inflammatory properties, and they have analgesic, antipyretic, and platelet-inhibitory actions. They inhibit cyclooxygenase, which blocks the synthesis of prostaglandins; this accounts for their pharmacological properties.47,48
Kaempferol also has some novel ADME properties, which makes it an active compound of Colocasia affinis; it cannot be easily biodegraded, it has low acute oral toxicity value with a negative blood-brain barrier; it is not carcinogenic; it does not inhibit CYP2D6, Human either-a-go-go, P-glycoprotein, and a moderate water solubility value. Devi et al 49 reviewed the anti-inflammatory effects of Kaempferol through its chemical composition, toxicity, and bioavailability. The inspected properties show it promising outcomes in treating inflammations. 49 Furthermore, Calderón-Montaño et al 50 reviewed Kaempferol to be antioxidant, anti-inflammatory, antimicrobial, anticancer, cardioprotective, neuroprotective, antidiabetic, anti-osteoporotic, estrogenic/antiestrogenic, anxiolytic, analgesic, and antiallergic activities.
Transferulic acid also has ADME properties that make it a promising component for an oral prescription; it does not cause Ames mutation and have no blood-brain barrier; it cannot be biodegraded; it has negative carcinogenicity and does not inhibits CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4, and Human either-a-go-go; it is not toxic to the liver and can easily be absorbed by the intestinal tract; it also has positive human bioavailability. Transferulic acid was proved to inhibit EGFR, which aids cell proliferation and DNA synthesis; the molecular docking result showed that trans ferulic acid forms hydrogen bond interaction with Lys 745 and Met 793 and exhibits stronger hydrophobic interactions with multiple amino acid residues at the EGFR kinase domain; this reflects it ADME properties as it has been proved to be used as an agent of formulating cancer drugs. 51
P-coumaric acid has advantageous pharmacological properties: it does not act as Ames mutagen, it has a negative blood-brain barrier; it is not carcinogenic and does not inhibits CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4, P-glycoprotein, and Human either-a-go-go; it has negative hepatotoxicity with moderate water solubility value. Tyrosinase (TYR) catalyzes rate-limiting steps of melanogenesis, and therefore its inhibitors can be used as hypopigmented agents. P-coumaric acid (p-CA) has been proved to interfere with the pro-melanogenic actions of tyrosine due to its structural similarity. Potent antimelanogenic effects of p-CA were observed in human epidermal melanocytes that were exposed to UVB radiation. 52
The bioactive compounds of Colocasia affinis conform to the Lipinski’s rule of five violations, which makes it a promising drug, especially for cancer treatment, as the docking results revealed that they have a high affinity for EGFR, thereby rendering the cancer cells inactive and leading to apoptosis. According to Christopher Lipinski’s rule of five, the molecules of Colocasia affinis Schott can be proposed to be an active oral drug; the rule is based on the determination of pharmacokinetic properties of the biomolecules including their absorption, distribution, metabolism, and excretion (ADME). Lipinski’s rule states that orally bioactive drug should not violate more than one of the following criteria: not more than 5 hydrogen-bond donors (HBD ⩽ 5), not more than 10 hydrogen-bond acceptors (HBA ⩽ 10), the molecular mass of not more than 500 Da (MW ⩽ 500 Da) and octanol-water partition coefficient not exceeding 5 (log P ⩽ 5). 53 From Table 6, all the compounds including the standard drug do not violate the Lipinski’s rule of five except myricetin with 1 valuation due to 6 hydrogen bond donors.
Drug-likeness predictions of compounds.

Abbreviations: HB, hydrogen bond; iLog P, implicit log P; Mol. Wt., molecular weight; ROF, rule of five.
Conclusion
This study was carried out to identify selective inhibitors of EGFR protein which can disrupt EGFR catalytic activity of EGFR by docking in the bioactive compounds from Colocasia affinis Schott at the interface of the EGFR kinase domain. After stringent molecular docking, quantum chemicals (MM-GBSA) calculations and density functional theory analysis, C. affinis Schott compounds were identified with stable interaction, higher binding energy, and better chemical reactivity than the reference compound (gefitinib). Pharmacokinetic models predicted C. affinis Schott as a novel therapeutic candidate. Overall, C. affinis Schott is an excellent therapeutic intervention in cancer treatment. However, in vitro and/or in vivo investigations are required to validate C. affinis Schott compounds in EGFR-targeted drug development.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors Contributions
TAB and NI conceptualized the study and wrote the original draft of the article. OTA, SIM, OAS, and WFA assisted with data collection and editing. ACJ and EAO also assisted with the editorial works. DAO supervised the project. All authors read and approved the final manuscript.