
Abstract
Despite the rapid developments and advancements to improve treatments, Breast cancer remains one of the deadliest health challenges and the most frequently diagnosed tumor. One of the major problems with treatment is the unique difference that each cancerous cell exhibits. As a result, treatment of breast cancer has now become more personalized based on the specific features of the tumor such as overexpression of growth factor receptors (Epidermal growth factor receptor (EGFR), Human Epidermal Growth Factor Receptor 2 (HER2)), hormone receptors (Human Estrogen receptor alpha (ER)) and kinases involved in pivotal signaling associated with growth (Phosphatidylinositol 3-kinase (PI3K)). Several chemotherapeutic agents have been developed to curb the menace, but the associated adverse drug effects cannot be overlooked. To this end, this study employed a molecular modeling approach to identify novel compounds of natural origin that can potentially antagonize the receptors (mentioned above) associated with the pathophysiology of breast cancer and at the same time pose very little or no side effects. The results of the molecular model of biological interactions between a library of 118 anthocyanins and the binding pockets of the protein targets identified 5 compounds (Pelargonin, Delphinidin 3-O-rutinoside, Malvin, Cyanidin-3-(6-acetylglucoside), and Peonidin 3-O-rutinoside) with good binding affinities to the protein targets. Further MM-GBSA calculations returned high binding energies. The specific molecular interactions between the compounds and the targets were analyzed and reported herein. Also, all the compounds exhibited good pharmacokinetic profiles and are therefore recommended for further analyses as they could be explored as new treatment options for a broad range and personalized breast cancer treatments.
Background
The burden of breast cancer has continued to rise globally, becoming the most frequent form of tumor diagnosed 1 and is the foremost origin of cancer-related death among women. Reports has shown that out of 9 women, at least 1 will have the disease in her lifetime. 2 Despite advancements in several clinical trials to improve treatment, the mortality rate continues to increase. Breast tumor is usually linked with dysregulation of estrogen and progesterone hormone receptors.3,4 Additionally, overexpression of the Epidermal growth factor receptor (EGFR1), Human epidermal growth factor receptor 2 (HER2), dysregulation of the Estrogen receptor-positive (ER+), and Phosphatidylinositol-3-kinase a (PI3Ka) signaling pathways have been extensively studied in breast cancer.4,5 Hence, it is imperative to keep unraveling new methods and molecules targeting these proteins.
Estrogen receptor-positive (ER+) breast tumor is responsible for roughly 75% of all breast tumor incidences in women. 6 The ER-α signaling pathway is the primary driving mechanism in the pathophysiology of many breast cancer cases. The dysregulation of protein co-regulators involved in the ER-α signaling pathway performs a critical function in breast carcinogenesis, promoting the expression of oncogenic proteins such as cyclin D1 and c-Myc.7,8 Estrogens are the primary sex hormones regulating the menstrual cycle and the development of female sexual organs. 8 Estrogen functions by binding to estrogen receptors (ER), the major subtypes being ER-α and β.6,8,9 ER-α is majorly present in the ovary, uterus, bone, mammary gland, and male reproductive organs but plays vital roles in the uterus and mammary gland. Synthesized estradiol (E2) from aromatase enzyme binds to the estrogen receptor and undergoes receptor dimerization to form an estradiol-ER complex. The complex formed is moved to the nucleus, which regulates the transcription of certain target genes in the cell nucleus by binding to DNA, specifically via interaction with estrogen response elements (ERE), which are regulatory elements. 6 ER-alpha has 3 distinct domains; AF2 domain (Activator Function-2 domain), Activator Function-1 domain, and DBD domain (DNA binding domain). The formed multiprotein complex activate transcription via their domains.6,10 Thus, the ER-α receptor is the fundamental target for treating ER-positive breast cancers via endocrine therapy to block ER transcription.6,9
Human epidermal growth factor receptor 2 (HER2) is a transmembrane protein receptor encoded by the HER2 gene positioned on the chromosome 17 long arm, and accounted for roughly 20% to 25% of all breast cancers.11,12 HER2 is a part of the EGFR family comprising 4 HER receptors: HER4, HER3, HER2, and HER1.11,13 Upon HER2 receptor activation, specific tyrosine kinase residues are phosphorylated and signaling proteins are activated which results in the initiation of downstream signaling mechanisms.14,15 The mitogen-activated protein kinase (MAPK) and phosphatidylinositol triphosphate kinase (PI3K) signaling mechanisms are the essential pathways triggered by the HER2 receptor, controlling cell cycle progression, angiogenesis, apoptosis, cell growth, and survival.11,15 HER2 receptor overexpression is recognized as a HER2 activation mechanism in HER2+ breast malignancies. 11 Tyrosine kinase activity in normal cells is a strictly regulated process. However, in tumor cells, overexpression of HER2 results in prolonged tyrosine phosphorylation of the kinase domain. Due to this, there is a sustained signaling pathways activation, which causes uncontrolled proliferation of cells.11,15 Thus, the HER2 receptor is the primary target for treating HER2-positive breast cancers, blocking the activation of downstream signaling pathways.11,14
The epidermal growth factor (EGFR) is a transmembrane protein receptor housed by the EGFR gene. The EGFR is the first of the EGFR family, which consists of 4 transmembrane tyrosine kinases: Her4/ErbB4, Her3/ErbB3, Her2/ErbB2, EGFR (ErbB1, HER1). 16 Although these members are on separate chromosomes, they all have a similar framework.16,17 The EGFR family plays essential functions in cell cycle regulation, survival, differentiation, and cell proliferation.16,18 The activation of EGFR by binding its specific ligands results in receptor dimerization, which is essential for the phosphorylation of tyrosine kinases. Subsequently, these phosphorylated tyrosine kinases activate different downstream signaling mechanisms.18,19 EGFR is a commonly mutated oncogene in solid cancers and is overexpressed in approximately 14% of all breast tumors. In addition, half of all triple-negative breast cancer (TNBC) (ie, cancer cells without ER, PR and HER2) overexpress EGFR.5,20 Hence, targeting the EGFR via EGFR inhibitors is necessary to treat breast cancer.
Phosphatidylinositol-3-kinase a (PI3Ka) is a class of enzymes (lipid kinases) that participates in cellular activities regulation, such as differentiation, survival, angiogenesis, apoptosis, DNA repair, motility, and cell proliferation. 21 These enzymes function as signaling substances and are well known in the PI3K/AKT/mTOR signaling pathway.21,22 Although PI3K activity is tightly controlled in normal cells by internal signal, research shows that dysregulation of the PI3K signaling pathway is linked to one-third of human tumor development. 21 Furthermore, it has been observed that in breast tumor patients, the phosphoinositide 3-kinase (PI3K) signaling pathway is commonly upregulated and is associated with breast tumorigenesis and progression.23,24 The class IA PI3Ks (PIK3CA) are the most repeatedly modified class in breast tumors, with approximately 40% of HR-positive breast cancers and 9% of triple-negative breast cancer (TNBC) having PIK3CA mutations.25,26 Thus, it is essential to investigate potential PI3K inhibitors in breast cancer treatment.
Anthocyanins (ACNs) are natural compounds and the most abundant flavonoids in flowers, fruits, vegetables, and cereals.27,28 Anthocyanins are bioactive compounds with numerous therapeutic activities, including inflammatory, anti-cancer, and antioxidant.28,29 Anthocyanins exert antioxidant effects by scavenging free radicals. They have also been reported to regulate the expression of the inflammatory factors via inhibition of Nuclear factor kappa B (NF-kB).29,30 Studies have reported anthocyanins’ inhibitory and anti-metastatic potential on breast cancer.31,32 Thus, anthocyanins have piqued the interest of researchers, especially in oncotherapy. Herein, an integrated molecular modeling approach including molecular docking, molecular mechanics, generalized Born surface area, absorption, distribution, metabolism, excretion studies, and pharmacophore modeling, was employed to estimate the inhibitory activities of Anthocyanins against the targets and identify novel compounds that can be employed in treating ER+, HER2+, and EGFR+ breast cancer.
Methods
Protein targets and ligands
Anthocyanins were mined from Phytohub and PubChem in 2D SDF format (Figure 1) to identify potential inhibitors of Human Estrogen receptor alpha, Human Epidermal growth factor receptor 2, Epidermal growth factor receptor, and Phosphatidylinositol-3-kinase. 33 Following that, the crystal structures of the Human Estrogen receptor alpha (PDB ID: 3ERT), Human Epidermal growth factor receptor 2 (PDB ID: 3PP0), Epidermal growth factor receptor (PDB ID: 3POZ), and Phosphatidylinositol-3-kinase a (PDB ID: 4JPS) were procured from the Protein Data Bank (http://www.rscb.org/).

The chemical structures of the top-scoring compounds: (A) Pelargonin, (B) Delphinidin 3-O-rutinoside, (C) Malvin, (D) Cyanidin-3-(6-acetylglucoside), and (E) Peonidin 3-O-rutinoside.
Preparation of ligands
The LigPrep tool was used to prepare the bioactive Anthocyanin compounds used in molecular docking. At pH = 7.2 ± 0.2, their ionization states and tautomers were generated, followed by optimization using the OPLS 2005 force field (Schrodinger release 2017).
Preparation of protein and receptor grid generation
The protein structures were imported into Maestro, then subsequently prepared using the protein preparation wizard. Missing side chains were incorporated using prime, Waters, and other bound moieties (non-standard ligands) were removed, hydrogen positions were fine-tuned, and the proteins were subjected to restrained energy minimization. 34 Grid boxes were also created with respect to the position of the co-crystallized ligand of the proteins to guide the automated docking procedure.
Molecular docking
The Glide script was used on maestro 11.1 to perform the molecular docking procedure. 35 To detect significant inhibitory interactions with the proteins, the compounds were docked into a prepared grid of protein targets. 36 The most stringent screening (XP) results were downloaded for further evaluation.
ADMET/Tox screening
The lead compounds’ pharmacokinetics, drug-likeness, and toxicity were ascertained using the swissADME (http://www.swissadme.ch) and Pro-Tox II online servers (https://tox-new.charite.de/protoxII).
Pharmacophore modeling
The Schrodinger suite’s PHASE graphical user interface was used to gather information on the Molecular orientation of vital functional groups primarily involved in the top-scoring ligands’ characteristic binding to the protein target. 37
MM/GBSA
The Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) continuum solvent model evaluated the docked protein-ligand complex binding free energy.
Prime rotamer search techniques were merged with the OPLS3 force field and the VSGB solvent model to finish this study.
Binding energy calculations
The one-average molecular mechanics generalized Born surface area (MM/GBSA)38,39 methods developed in the MOLAICAL code 40 were employed to calculate the relative binding energy which the ligand (L) combines with the protein receptor (R) to produce the complex (RL).
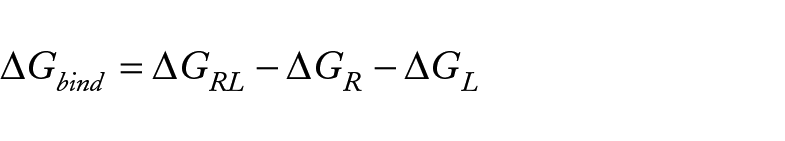
This can be denoted by the contribution of various interactions,
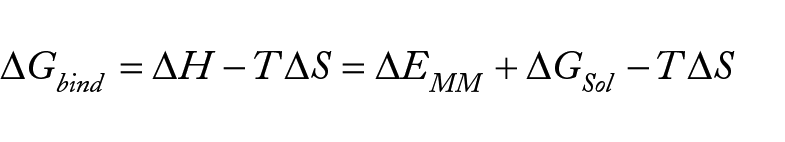
Where the changes in the gas phase molecular mechanics
Results
Molecular docking result
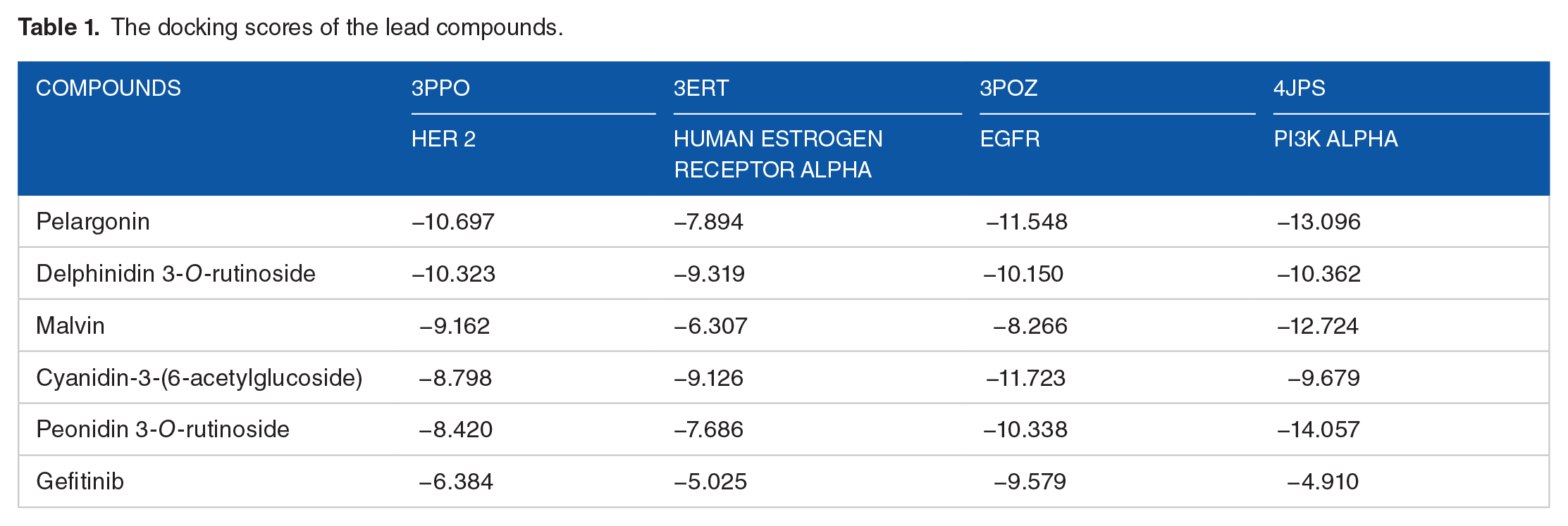
See Table 1
The docking scores of the lead compounds.
Molecular interactions
Molecular interactions between the lead compounds and the ligand binding sites of the protein targets are shown in Figures 2 to 5.
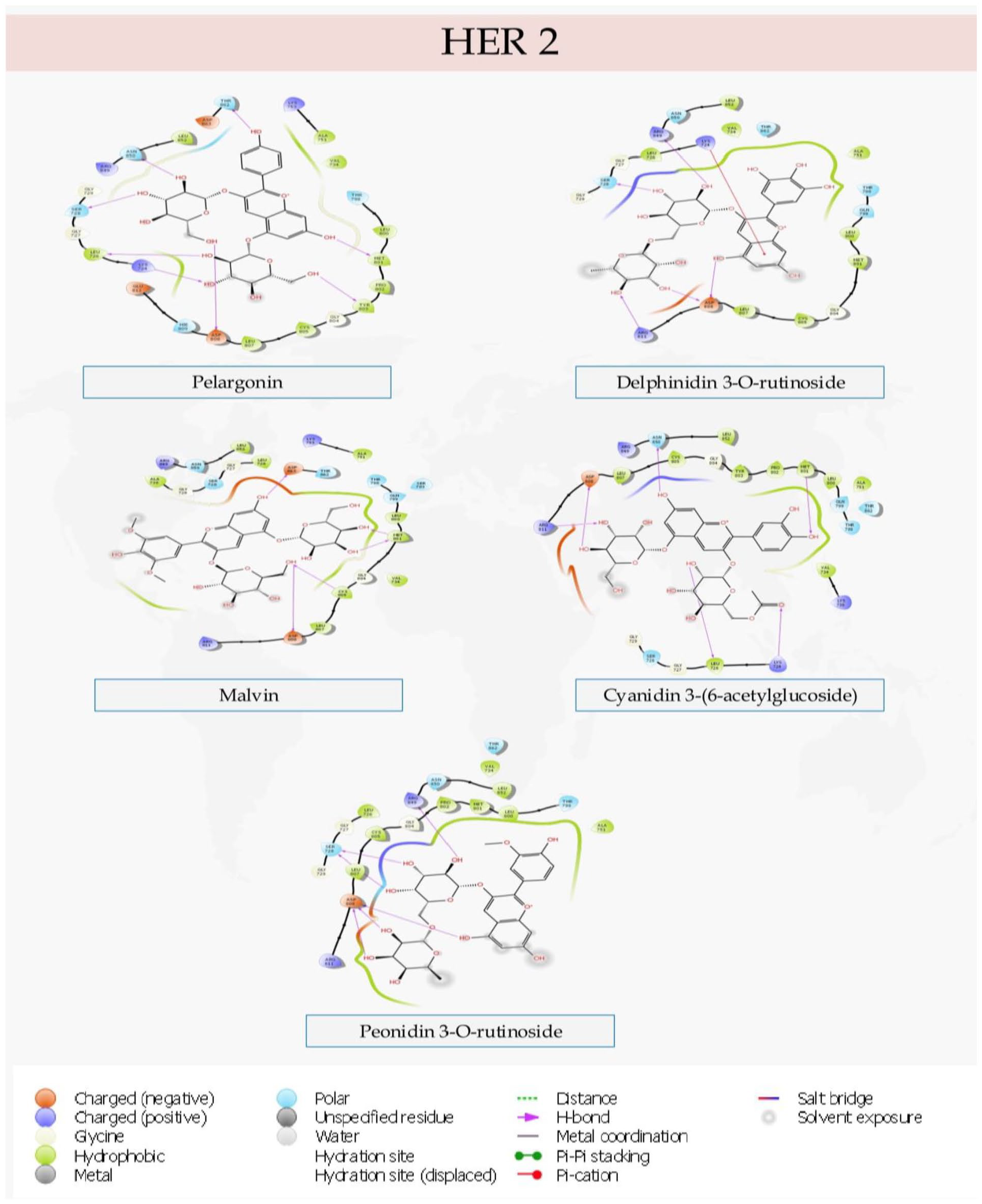
Specific interactions of the lead compounds with the binding pocket of Human Epidermal Growth Factor Receptor 2 (HER2).
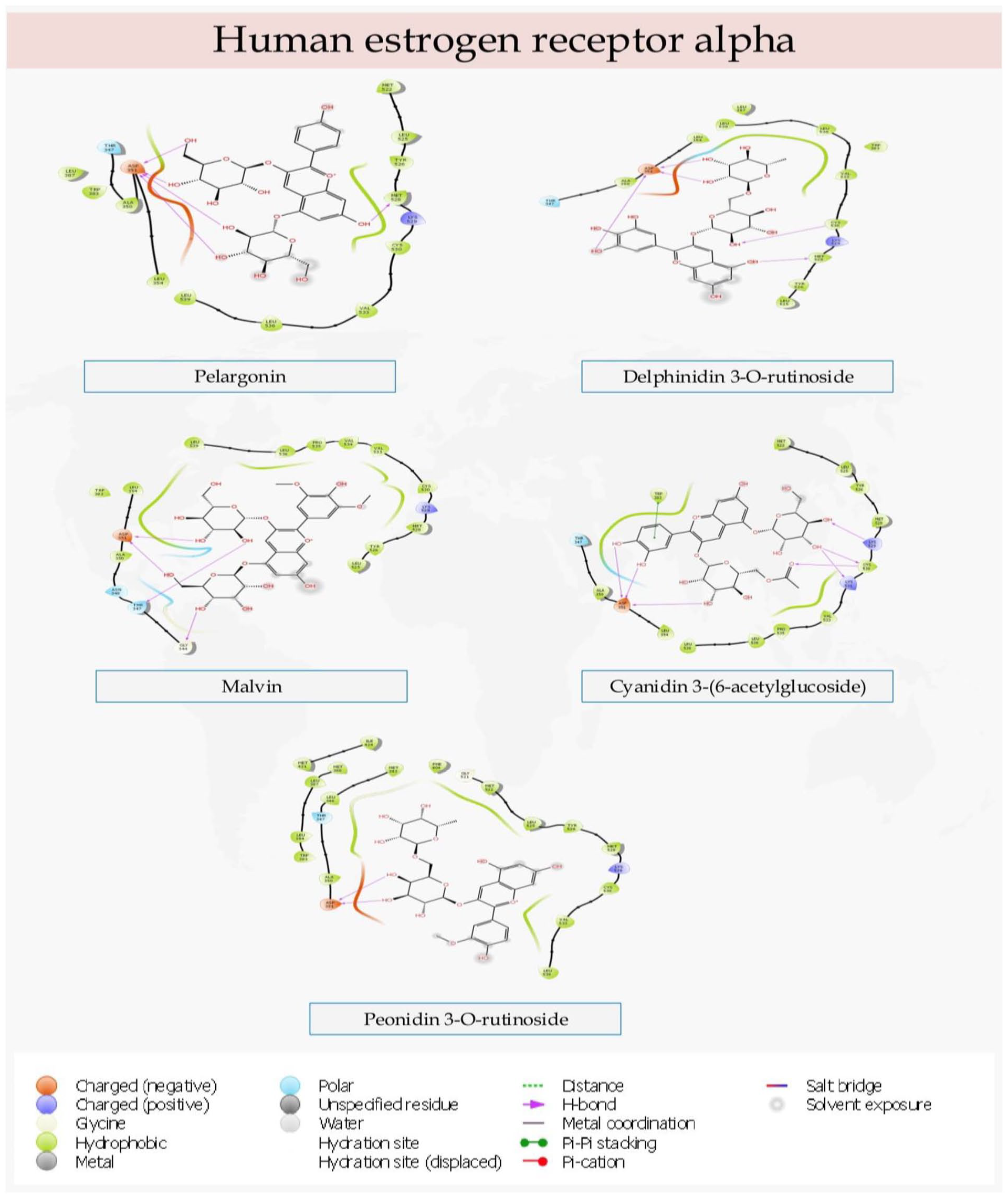
The interactions of the lead compounds with the amino acid residues of the binding pocket of Human estrogen receptor alpha.

2D diagram of the specific interactions of the top-scoring compounds with the ligand-binding domain of Epidermal Growth Factor Receptor.

The molecular interactions of the lead compounds with the kinase domain of PI3K alpha.
Free binding energy calculation
The MMGBSA-based free binding energies are shown in Figure 6.

The binding energies of the top-scoring compounds with the target proteins.
Lead compound-receptor based pharmacophore model
The developed pharmacophore hypothesis based on the complexes between our hit compound and the binding sites of the protein target is shown in Figure 7

Pharmacophore models of Cyanidin 3-(6-acetylglucoside) and the protein targets.
ADMETox screening
The predicted absorption, distribution, metabolism, excretion and toxicity profiles (ADMET) of the lead compounds are presented in Tables 2 to 4.
The physicochemical properties of the test compounds.

C1, Pelargonin; C2, Delphinidin 3-O-rutinoside; C3, Malvin; C4, Cyanidin-3-(6-acetylglucoside); C5, Peonidin 3-O-rutinoside.
Distribution, metabolism, and bioavailability score of test compounds.

C1, Pelargonin; C2, Delphinidin 3-O-rutinoside; C3, Malvin; C4, Cyanidin-3-(6-acetylglucoside); C5, Peonidin 3-O-rutinoside.
The toxicity profiles of the test compounds.
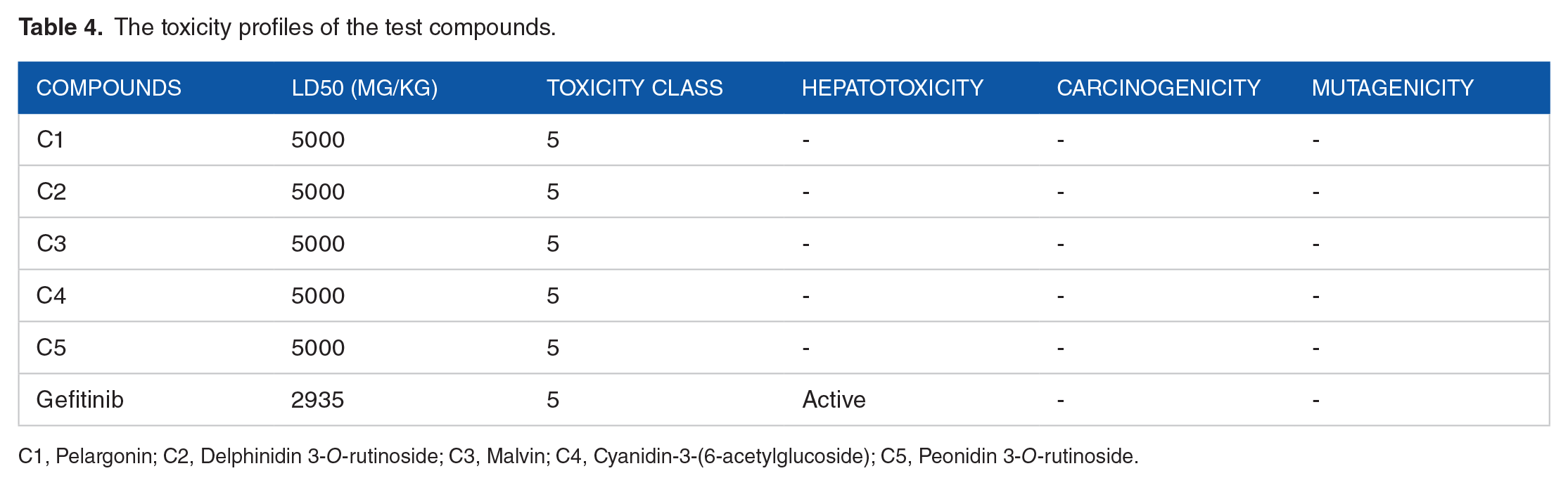
C1, Pelargonin; C2, Delphinidin 3-O-rutinoside; C3, Malvin; C4, Cyanidin-3-(6-acetylglucoside); C5, Peonidin 3-O-rutinoside.
Discussion
Due to the unique differences portrayed by cancerous cells, treatment of breast cancer has become more personalized. Treatment is given based on the specific features of the tumor such as overexpression of growth factor receptors (Epidermal growth factor receptor, Human Epidermal Growth Factor Receptor 2, HER2), hormone receptors (Human Estrogen receptor alpha), and kinases involved in pivotal signaling associated with growth (Phosphatidylinositol 3-kinase). In this study, novel compounds from plant origin that are capable of antagonizing the receptors associated with different types of breast cancer were identified using an integrated molecular modeling approach.
Molecular Model of Biological Interactions
The results of the molecular model of the essential biological interactions between the test compounds and the proteins showed that the reported compounds exhibited impressive binding data to all protein targets under study. A library of several anthocyanins (118) was engaged in the study since anthocyanins have been continuously associated with huge health benefits. The molecular docking results showed that a reasonable number of the compounds showed very good binding affinity to at least 1 of the protein targets (Table 1). Five lead compounds with good binding affinities to all 4 targets were identified and reported herein.
Pelargonin had the highest binding affinity (−10.697) to the ligand-binding site of Human epidermal growth factor receptor 2 (HER 2). Delphinidin 3-O-rutinoside also showed high ranking (−10.323) with respect to the binding affinity it exhibits to the protein target, HER 2. Malvin, Cyanidin-3-(6-acetylglucoside), and Peonidin 3-O-rutinoside had docking scores of −9.162, −8.798, and −8.420 respectively. The obtained docking scores are adjudged to be excellent based on the fact that they are a lot better than the score obtained from the binding of the standard drug Gefitinib to the protein target. The development of therapies targeted at HER2 has undoubtedly improved the life expectancies of patients with HER2-positive early-stage breast cancer cases. 41 Also, about 25% of all breast cancer cases have been found to overexpress HER2, and overexpression of this protein has been linked to more deadly pathophysiology. 42 HER2-positive breast cancer cases belong to a type of breast cancer that is known to be malignant, likely to grow more rapidly, and tend to spread quickly or extensively. 43 The impressive binding affinities exhibited by the lead compounds show that they are well suited to antagonize HER2.
Delphinidin 3-O-rutinoside exhibited the highest binding potential to Human estrogen receptor alpha with a docking score of −9.319. Cyanidin-3-(6-acetylglucoside) ranked lower (−9.126) but very close to the top-scoring compound. Pelargonin, Peonidin 3-O-rutinoside, and Malvin (−7.894, −7.686, and −6.307 respectively) also showed good binding prospects to the protein target. Interestingly, all the reported anthocyanins showed better binding data to the binding pocket of the protein than the standard drug Gefitinib (−5.025) used in the treatment of breast cancer. These findings suggest that these compounds have the potential to serve as antagonists of Human estrogen receptor alpha. Hormone-dependent estrogen receptor-positive (ER+) breast cancer has been the subject of extensive research geared toward developing novel effective therapeutic compounds for the treatment of hormone-dependent breast cancer. This is because ER+ breast cancer constitutes about 75% of all breast cancer cases. The female sex hormone estrogen plays a fundamental role in the pathogenesis and pathophysiology of breast cancer, this makes estrogen receptor a cardinal target for the treatment of breast cancer. 6
Cyanidin-3-(6-acetylglucoside) (−11.723) and Pelargonin (−11.548) exhibited the most robust binding to the ligand-binding pocket of Epidermal Growth Factor Receptor (EGFR). EFGR is one of the foremost established targets of new anti-cancer molecules. 20 Delphinidin 3-O-rutinoside and Peonidin 3-O-rutinoside had docking scores of −10.150 and −10.338 respectively. However, the standard, Gefitinib (−9.579) had a higher docking score than Malvin (−8.266). It is known that about half of all triple-negative breast cancer (TNBC) cases and inflammatory breast cancer (IBC) cases are characterized by an overexpression of Epidermal growth factor receptor and small molecule inhibitors of EGFR have been continuously evaluated for the treatment of breast cancer. 20
The simulation of the biological interactions between the test compounds and the kinase domain of phosphatidylinositol 3-kinase alpha showed positive results for all the test compounds. The phosphatidylinositol-3-kinase pathway is the most frequently occurring signal Cascades abnormally activated in breast cancer progression, this makes it a pivotal therapeutic target. 44 Peonidin 3-O-rutinoside showed the highest binding affinity to PI3K-α with a docking score of −14.057. Pelargonin had a close binding result by returning a docking score of −13.096. Malvin (−12.724) also showed impressive binding data alongside Delphinidin 3-O-rutinoside (−10.362) and Cyanidin-3-(6-acetylglucoside) (−9.679). Notably, all the reported compounds ranked higher than Gefitinib (−4.910).
In drug design and medicinal Chemistry, it is quite beneficial to analyze the interactions exhibited by test compounds with the ligand-binding sites of protein targets. This is done to identify specific interactions that are optimizable. The identified interactions can then be optimized to strengthen the binding interactions between the ligands and the protein targets and ultimately increase the inhibitory potentials of the interactions. To this end, the specific molecular interactions of the test compounds with the binding pockets of the protein targets were analyzed and presented in Figures 2 to 5.
Pelargonin showed hydrogen bond interactions with Leu726, Lys724, Ser728, Met801, Tyr802, Asn850, Thr852, and Asp808 in the binding pocket of Human Epidermal growth factor receptor 2 (HER2). Also, Delphinidin 3-O-rutinoside had a pi cation interaction with Lys724 and hydrogen bond interactions with Ser728, Arg849, Asp808, and Arg811. Malvin had hydrogen bond contacts with Met801, Cys805, Asp808, and Asp863. Furthermore, Cyanidin-3-(6-acetylglucoside) exhibited hydrogen bond contacts with Leu726, Lys724, Asp808, Arg811, Met801, and Asn850. Peonidin 3-O-rutinoside however showed interactions with Arg849, Ser728, and Asp808. All the compounds exhibited interactions with Asp808. Additionally, Pelargonin, Delphinidin 3-O-rutinoside, and Cyanidin-3-(6-acetylglucoside) each had at least one interaction with Lys274 and Leu276. Interestingly, Pelargonin and Cyanidin-3-(6-acetylglucoside) were found to exhibit similar binding pose and interactions with the binding site of the protein. Both compounds had contacts with Leu726, Lys724, Asp808, Met801, and Asn850. In the same vein, Delphinidin 3-O-rutinoside and Peonidin 3-O-rutinoside exhibited similar binding in form of interactions with Arg489, Ser728, and Asp808 with Delphinidin 3-O-rutinoside having additional interactions with Arg811 and Lys724.
In the molecular engagements with Human estrogen receptor alpha, Pelargonin interacted with Asp351 and Met528. Delphinidin 3-O-rutinoside similarly had interactions with Asp351, Met528, and Cys530. Malvin had hydrogen bond contacts with Gly344, Thr347, and ASP351. Also, Cyanidin-3-(6-acetylglucoside) had hydrogen bond interactions with Lys529, Cys530, Lys531, and ASP351, as well as a π stacking with the aromatic amino acid residue Trp383. Whereas, Peonidin 3-O-rutinoside had a singular interaction with Asp351. Notably, all the compounds made hydrogen bond contacts with Asp351. Pelargonin and Delphinidin 3-O-rutinoside had relatively s similar binding character. Both compounds, in addition to Asp351, also had interactions with Met528. However, Delphinidin 3-O-rutinoside had an additional hydrogen bond contact with Cys530.
Furthermore, Pelargonin made hydrogen bond interactions with Cys797, Asp800, Ala722, Gly724, Asn842, and Lys745 in the binding pocket of Epidermal growth factor receptor. Also, Delphinidin 3-O-rutinoside exhibited interactions with Asp800, Cys797, Asp837, Arg841, and Ser720. Malvin showed hydrogen bond interactions with Ser720, Thr854, Asp855, Lys745, Asn842, and Asp800, as well as a π-π stacking interaction with Phe997. Cyanidin-3-(6-acetylglucoside) also had interactions with Lys745, Asn842, Asp855, Met793, Cys797, Asp800, and Leu718. While Peonidin 3-O-rutinoside exhibited interactions with Asp800, Met793, Asp855, Asn842, and Ala722. All the compounds had hydrogen bond interaction with amino acid residue Asp800. Pelargonin, Delphinidin 3-O-rutinoside, Malvin, and Cyanidin-3-(6-acetylglucoside) each had at least 1 contact with Cys797 and Lys745.
Finally, in binding to the ligand-binding site of PI3K alpha, Pelargonin exhibited hydrogen bond interactions with Asp810, Asp933, Val851, Ser774, Gln859, Ser854, and Glu849, as well as a π-π interaction withTyr836, an aromatic amino acid residue. Also, Delphinidin 3-O-rutinoside had hydrogen bond contacts with Asp810, Asp933, Tyr836, Ser919, Ser774, and Val851. Malvin showed interactions with Ser774, Asp805, Asp810, Asp933, and Val851. Cyanidin-3-(6-acetylglucoside) also had interactions with Asp810, Tyr836, Asp933, Ser774, Gln859, Ser854, and Val851. Meanwhile, Peonidin 3-O-rutinoside exhibited specific hydrogen bond interactions with Ser919, Ser774, Thr856, Asp933, Lys802, Tyr836, Asp810, and Val851. It is noteworthy that all the lead compounds exhibited interactions with Asp810, Asp933, Val851, and Ser774. Also, all the compounds but Malvin had additional contact with the aromatic amino acid residue Tyr836. Delphinidin 3-O-rutinoside, Cyanidin-3-(6-acetylglucoside), and Peonidin 3-O-rutinoside exhibited hydrogen bond interactions this residue (Tyr836) while π-π stacking was the basis of interaction with Pelargonin.
Binding Energy
The top-scoring compounds were rescored using MM-GBSA approach to estimate the binding energies of the compounds with the ligand-binding pockets of the protein targets. Most times in drug design, it is relevant to employ molecular docking in generating the molecular pose of a compound bound to the binding pocket of a protein target and subsequently engaging MM-GBSA to calculate the strength of that particular interaction. This approach is continuously gaining relevance in molecular drug design and development. The ∆G bind of the complexes complements the reliability of the docking scores.
The MM-GBSA-based binding energy calculations reveal that in binding to HER2, Cyanidin-3-(6-acetylglucoside) has the highest binding energy with a ∆G bind value of −65.506 kcal/mol. The standard drug Gefitinib had higher ∆G bind (−56.038 kcal/mol) than the other compounds. Delphinidin 3-O-rutinoside returned a value (−55.546 kcal/mol) slightly lower than the standard drug. Meanwhile, Pelargonin, Malvin, and Peonidin 3-O-rutinoside had ∆G bind values of −52.932, −54.234, and −53.762 kcal/mol respectively.
Similarly, Cyanidin-3-(6-acetylglucoside) had the highest binding energy (−63.967 kcal/mol) with Estrogen receptor. Peonidin 3-O-rutinoside and Malvin also showed impressive binding relative to the standard drug with ∆G bind values of −57.726 and −52.464 kcal/mol respectively. Gefitinib (−49.304 kcal/mol) notably had higher value than Pelargonin (−44.242 kcal/mol) and Delphinidin 3-O-rutinoside (−35.499 kcal/mol).
Delphinidin 3-O-rutinoside had the highest binding energy with EGFR (−68.276 kcal/mol). Peonidin 3-O-rutinoside comes very close with a ∆G bind value of −67.104 kcal/mol. Further, Cyanidin-3-(6-acetylglucoside) and Pelargonin similarly returned impressive values of −61.449 and −61.041 kcal/mol. However, Gefitinib (−63.659 kcal/mol) had a higher value than Malvin (−57.718 kcal/mol).
Finally, in the characteristic binding to PI3K, Cyanidin-3-(6-acetylglucoside) once again had the highest ∆G bind value (−79.406 kcal/mol). Notably, all the test compounds returned higher values than Gefitinib (−46.582 kcal/mol). Pelargonin had the closest binding energy to Cyanidin-3-(6-acetylglucoside) with a value of −60.521 kcal/mol. Whereas Delphinidin 3-O-rutinoside, Malvin, and Peonidin 3-O-rutinoside had the values of −56.754, −56.549, and −56.248 kcal/mol respectively.
Cyanidin 3-(6-acetylglucoside) is adjudged to be the most ideal candidate for a broad range cancer treatment based on the MM-GBSA ∆G bind calculations (Figure 6). The compound showed the highest binding energy of all compounds in complex with Human Epidermal growth factor receptor (−65.506 kcal/mol), Human estrogen receptor alpha (−63.967 kcal/mol), and Phosphatidylinositol-3-kinase alpha (−79.406). On a whole, all the compounds showed robust binding affinities and energies to the protein targets and could be explored in personalized breast cancer treatment.
Pharmacophore Model
A pharmacophore is a collection of structural and electronic features that is essential for the firm molecular interaction between a ligand and a protein target. In this study, complex-based pharmacophore models were developed based on the complexes formed between Cyanidin-3-(6-acetylglucoside) (adjudged to be the most ideal candidate) and the protein targets using Phase. This was done to identify the spatial and physicochemical properties essential for the firm binding of the compound to the targets. As shown in Figure 7, the results showed that a combination of hydrogen bond acceptors, hydrogen bond donors, and aromatic rings is essential for strong molecular interaction between Cyanidin 3-(6-acetylglucoside) and the binding pockets of the protein targets. Therefore, in line with the predicted hypothesis, hydrogen bond acceptors, hydrogen donors and aromatic rings are the basis with which the hit compound interacted with the ligand binding sites of all proteins. The high binding affinities exhibited by the compound suggest that the developed hypothesis may be instrumental in the identification of compounds with anti-cancer potential on a molecular modeling level. Although the accuracy of a pharmacophore model based on experimentally active compounds is deemed to be higher, yet predictive models are also significant. The structural information obtained from the pharmacophore model can be employed as a backbone in identifying a wide range of inhibitors of the protein targets under study.
Pharmacokinetic Profiles
Investigation of ADMET behaviors of compounds has become indispensable in drug design and development. Molecules under development must meet some stipulated criteria for them to be considered drug candidates. Compounds have been the subject of rejection during development due to the unfavorable ADMET profiles they exhibit. On this note, the ADMET behaviors of the test compounds were predicted to identify the major strengths and possible ADMET weaknesses that can be optimized. Also, the ADMET behaviors of the top-scoring compounds were compared with Gefitinib. Structure-based ADMET profiling was carried out on the compounds using SwissAdme and Pro-tox II web servers. The predicted physicochemical properties are presented in Table 2. The molecular weight of the reported anthocyanins ranges from 491.42 to 655.58. Gefitinib returned a molecular weight of 446.9 g/mol which is relatively similar to that of Cyanidin-3-(6-acetylglucoside) (491.42 g/mol). Of all compounds, Malvin exhibits the highest molecular weight with a value of 655.58 g/mol.
To gain insights into the absorption of the compounds in living systems, lipophilicity was calculated. This was done to give an idea of how quickly and efficiently they are absorbed in human cells. The calculated lipophilicity per SwissAdme is the partition coefficient of n-octanol to water. In this study, the value of consensus log P which is the arithmetic mean of 5 models of lipophilicity was adopted for better accuracy. Of all the test compounds, Gefitinib exhibited the highest lipophilicity with a value of 3.92. Notably, Cyanidin 3-(6-acetylglucoside) showed the highest lipophilic levels of all the lead compounds while Malvin is adjudged the least lipophilic per SwissAdme predictions. An oral drug is expected to have a good level of lipophilicity to aid its movements across membranes. According to the prediction, Gefitinib and Cyanidin 3-(6-acetylglucoside) have higher tendencies than the other compounds to cross the lumen of the intestine during absorption and also the lipophilic membrane of cells during distribution.
In addition to lipophilicity, water solubility is also one of the cardinal factors that dictate the ADMET behaviors of drug candidates. The values of the Silicos-IT model of water solubility for the test compounds showed that Pelargonin is the most soluble of all compounds tested with a value of 0.27. Interestingly, all the reported anthocyanins showed optimal solubility values and are thus classified as soluble. However, Gefitinib which had a value of −7.94 is considered poorly soluble. This may be a result of its high lipophilicity. A drug candidate is expected to have optimal levels of lipophilicity and water solubility. An administered compound must dissolve in water to aid its gastrointestinal absorption. Low levels of lipophilicity may reduce the efficiency of absorption. Also, during distribution, drugs are transported in the hydrophilic condition of system circulation. By implication, the low level of water solubility might give an edge to the reported anthocyanins which had sufficient levels of water solubility.
Furthermore, Gefitinib was predicted to have the structural orientation to permeate the blood-brain barrier. In opposition to this, all the reported anthocyanins cannot permeate the blood-brain barrier (Table 3). Blood-brain barrier permeation is a property that is mostly peculiar to drugs whose target site of action is the brain. Administration of drugs that are not targeted at the brain and can permeate the blood-brain barrier can induce an adverse drug reaction in the brain.
Peonidin 3-O-rutinoside is shown (Table 3) to be a substrate of permeability glycoprotein. Also, none of the reported compounds is an inhibitor of the Cytochrome P450 isoforms engaged in the study. Contrarily, Gefitinib is predicted to be an inhibitor of 2C19, 2C9, 2D6, and 3A4 isoforms. Inhibiting any of these crucial enzymes can induce a drug-drug reaction and disrupt the metabolism of the administered drug. All the reported Anthocyanins are not inhibitors of any of the isoforms and would not induce drug-drug reactions. Additionally, the bioavailability score of all the reported Anthocyanins is 0.17 while Gefitinib returned a value of 0.55. Bioavailability scores measure the tendency of a compound to be an oral drug candidate. Finally, the toxicity profiles of the compounds showed better prospects than Gefitinib (Table 4). All the anthocyanins are predicted to be non-hepatotoxic, non-carcinogenic, and non-mutagenic. However, Gefitinib was predicted to be hepatotoxic.
Conclusions
The differences in the pathophysiology of breast cancer cases have led to the advent of individualized breast cancer treatment. In this study, 5 compounds namely; Pelargonin, Delphinidin 3-O-rutinoside, Malvin, Cyanidin-3-(6-acetylglucoside), and Peonidin 3-O-rutinoside were identified. These compounds have the potential to inhibit the progression of a wide range of breast cancer types due to their robust inhibitory molecular interactions with the associated receptors. The MM-GBSA-based binding energies calculated returned impressive results and further pharmacokinetic screening also showed favorable results. They are therefore recommended for further analyses as they can be employed in treating ER+, HER2+, and EGFR+ breast cancer.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
PAA and SOO conceptualized the study, performed the research experiment and analysed the research result. AOA and TAN wrote the original draft of the article. SAS supervised and validated the experiment. All authors read and approved the final manuscript.