
Abstract
Parasitic cysteine proteases are involved in parasite stage transition, invasion of host tissues, nutrient uptake, and immune evasion. The cysteine protease cathepsin F is the most abundant protein produced by fourth-stage larvae (L4) of the nematode Teladorsagia circumcincta, while its transcript is only detectable in L4 and adults. T. circumcincta cathepsin F is a recently evolved cysteine protease that does not fall clearly into either of the cathepsin L or F subfamilies. This protein exhibits characteristics of both cathepsins F and L, and its phylogenetic relationship to its closest homologs is distant, including proteins of closely related nematodes of the same subfamily.
Introduction
Nematoda are an ancient group and most terrestrial plants and larger animals are associated with at least 1 parasitic nematode. 1 Although fossilization of various nematode species has occurred throughout history, as early as 135 million years ago (mya), 2 there are no records of Teladorsagia circumcincta fossils and, as such, an estimate of their time of divergence is difficult to calculate. However, the earliest bovids appeared 20 mya 3 and since T. circumcincta is seen in sheep and goats but not cattle, and there is a closely related nematode in cattle, Ostertagia ostertagi, we can reasonably assume their divergence from related nematode species occurred within this time-frame, likely alongside the host. As such, T. circumcincta is a relatively recently evolved nematode when compared to the long history of the nematoda phylum, and some of its mechanisms of immune evasion within the host subsequently relatively recent as well. Some secretory proteins of T. circumcincta are likely to have adapted to suit the specific modern host.
Teladorsagia circumcincta is the most important parasitic nematode of sheep in cool temperate regions worldwide. 4 Eggs are passed in feces and develop into infective third-stage larvae (L3) on pasture, which are ingested by the host. L3 exsheath in the rumen, enter the lumen of the abomasal glands and molt to become fourth-stage larvae (L4) and establish within the host. L4 then molt into sexually mature adults which feed and breed on the mucosal surface of the abomasa. 5 Teladorsagiosis can result in reduced production, decreased animal welfare, parasitic gastroenteritis, poor growth performance, and weight loss. 6 Several methods are used to control T. circumcincta infection including anthelmintic treatment, nutritional supplementation, vaccination, selective breeding, and pasture management. These methods are already used with varying success,7–11 however, T. circumcincta is developing drug resistance.4,12 A vaccine against Teladorsagiosis was developed by Nisbet et al, 13 which uses larval antigens that are targets of IgA antibodies from immune sheep. IgA plays a crucial role in protection against T. circumcincta. 14 One of these antigen targets was cathepsin F, a cysteine protease.
Parasitic cysteine proteases are involved in parasite stage transition, invasion of host tissues, nutrient uptake, and immune evasion.15-18 The papain family of clan CA is the largest and most abundant family of cysteine proteases, 19 and consists of 2 major subfamilies; cathepsin B and cathepsin L. Cathepsin B is characterized by the presence of an occluding peptide loop, 20 while the cathepsin L subfamily is characterized by the presence of an ERF/WNIN motif, and comprises cathepsins L, V, K, S, W, F, and H.19,21 Cathepsin L pro-regions are about 100 residues long with 2 conserved motifs; ERF/WNIN and GNFD. 21 Cathepsin F has a longer pro-region, up to 250 residues, and, in general, is composed of 2 domains: an N-terminal cystatin-like domain and a C-terminal peptide similar to the cathepsin L pro-region. 22 However, the motifs in the C-terminal peptide are different in cathepsin F; a highly conserved ERFNAQ motif replaces the ERF/WNIN motif, and adjacent to this is an E/DxGTA motif, which was identified as a pro-region feature of cathepsins F and W, but not cathepsin L. 23 E/DxGTA has been suggested to act together with ERFNAQ as a scaffold for the pro-region, maintaining the inhibitory specificity of its α-helical structure. 19 The GNFD motif seen in cathepsin L is also present in cathepsin F and was previously identified as critical in intermolecular processing, and as the site of initial cleavage in Fasciola hepatica cathepsin L.24,25 Cathepsin L of the parasitic trematode F. hepatica has been shown to cleave and digest host IgG. 26
Cathepsin F in T. circumcincta is the most abundant protein produced by L4, and its transcript is detectable only in fourth-stage larvae (L4) and adult nematode stages, not in pre-parasitic stages.23,27 The functions of cathepsin F in T. circumcincta and its host interactions are yet to be determined, however, this protease may have a similar role to that of cathepsin L in F. hepatica, which is to digest IgG as a protective mechanism against the host immune response. 26 Secretion of cathepsin F could be digesting host IgA for protection against the host immune response or as a source of protein to aid in the development of the nematode.
This study aimed to use bioinformatic analyses to evaluate the protein sequence, structure, and gene of cathepsin F, and to find homologs with other nematode species to determine its phylogenetic history and potential functional role. This information is intended to inform the design of further functional studies.
Materials and Methods
Gene analysis and assembly
The mRNA sequence for T. circumcincta cathepsin F (GenBank accession no. DQ133568) and its translated protein sequence (Tci-CF-1, GenBank accession no. ABA01328) were obtained from The European Bioinformatics Institute (EBI). The mRNA sequence for Tci-CF-1 was used as a query for BLASTn against the draft T. circumcincta genome in WormBase ParaSite 28 (BioProject: PRJNA72569, Taxonomy ID: 45464) and against all nematode genomes in the database to identify matching genes. Predicted gene exons that matched Tci-CF-1 (>80% identity and E-value threshold of 4.5E-18) were extracted, translated into protein sequences, and aligned with Tci-CF-1 using CLC Genomics Workbench Version 9 (CLC, Qiagen) and default parameters. CLC alignments use a progressive alignment algorithm. The most closely aligned reading frame was selected and exons were re-ordered to align with Tci-CF-1.
Variants of Tci-CF-1 were constructed using PacBio RS and Illumina HiSeq 2500 sequence reads obtained from the Sequence Read Archive (BioProject PRJEB7676). 29 Sequence reads were converted to FASTq format using SRA Toolkit 2.8.2 (https://github.com/ncbi/sra-tools). The quality of reads was checked with FastQC 0.11.6 (https://github.com/s-andrews/FastQC) using default parameters. Low-quality Illumina reads were trimmed with Trimmomatic 0.32 (https://github.com/timflutre/trimmomatic). CLC was used to assemble the Illumina reads using Tci-CF-1 as the reference (variants 1 and 2). SPAdes 3.10.1 (https://github.com/ablab/spades) was used for de novo assembly of combined PacBio and Illumina reads. BLASTn of the mRNA sequence for Tci-CF-1 against the SPAdes de novo assembly in CLC resulting in contigs containing matches with >75% identity were extracted and used to assemble the Tci-CF-1 gene (variant 3).
Phylogenetic analysis and multiple sequence alignments
Tci-CF-1 was used as a query for BLASTp analysis against EBI and the National Center for Biotechnology Information databases. The selected sequences had an e-value threshold of 1e-50. Whole sequences were extracted into CLC and duplicates were removed. One T. circumcincta papain family cysteine protease (GenBank accession no. PIO64159) matched and was excluded following further analysis which identified a mis-assembly of its corresponding gene resulting in incorrect protein sequence formation; the exons were in the wrong order (data not shown). Pairwise comparisons of the 172 total sequences were conducted, and the top 9 sequences with the highest percent identity (% ID) to Tci-CF-1 were selected for multiple sequence alignment and pairwise comparison.
A multiple sequence alignment of Tci-CF-1 and the 9 closest homologs identified in the BLASTp analysis was carried out using CLC under default parameters. An alignment between Tci-CF-1, the 3 variants identified in this study and the possible T. circumcincta cathepsin F polymorphism discussed in Nisbet et al 7 was carried out using CLC. The translated protein sequences were annotated using information from previous studies.21,23,24,30 Prediction of N-glycosylation sites was conducted via NetNGlyc 1.0 Server. 31
Phylogenetic analysis was performed using SplitsTree5 5.0.0_alpha. 32 The multiple sequence alignment of Tci-CF-1 and the 9 closest homologs was used as the original input and consisted of 8 taxa and 10 protein character sequences of length 475. The Neighbor Joining method 33 and Tree Embedder method 34 were used (default options) to obtain a rooted tree drawing, and bootstrap values calculated following 1000 replications. The Uncorrected_P method 35 was used (default options) to obtain a 10 × 10 distance matrix. The Neighbor Net method 36 and The Splits Network Algorithm method 37 were used (default options) to obtain a splits network. The Splits Network Algorithm method 37 was used (ReticulateNetwork splits transformation, default options) to obtain a reticulate splits network.
Prediction of secondary and tertiary structure
Homology modeling of Tci-CF-1, variants 1, 2, and 3, and the Nisbet et al 13 possible variant protein sequences (Figure 1), as well as the pro-regions of human cathepsins L (UniProtKB accession no. P07711) and F (UniProtKB accession no. Q9UBX1) was conducted using Protein Homology/analogY Recognition Engine V 2.0 (Phyre2, Kelley et al 38 ) under the intensive modeling mode. The predicted protein structures were analyzed using UCSF ChimeraX. 39

Multiple sequence alignment of Teladorsagia circumcincta secreted cathepsin F (Tci-CF-1, GenBank accession no. ABA01328), the polymorphism discussed in Nisbet et al 13 and variants 1, 2 and 3 identified in this study. Conserved residues indicated by a dot; stop codon by an asterisk; ERFNAQ, E/DxGTA, GxNxFxD and GCNGG motifs by square, cross, circle, and diamond, respectively; catalytic triad residues by a downward arrow; predicted N-glycosylation sites by a right-facing arrow; polymorphisms of interest in boxes.
Results
Gene analysis and assembly
Tci-CF-1 matched several exons from the draft genome assembly with exons of predicted genes TELCIR_20397, _06733, _06734, _14223, and _19209 (BioProject: PRJNA72569). Sequence _20397 has a forward orientation, while all remaining sequences are on the reverse strand. Tci-CF-1 matched at exons 2 and 3 of TELCIR_20397, matched at exons 1-3, 5 and 6 of TELCIR_06733, matched at exons 1, 2, and 4 of TELCIR_06734, matched at exons 8, 2-6, in that order, as well as some areas in the introns of TELCIR_14223, and matched at exons 1-3, as well as areas up- and down-stream, of TELCIR_19209 (Supplemental Data 1).
None of the genes in the databases encoded the complete cathepsin F sequence. Between exons and between genes were several large sections of incomplete assembly or gaps which may contain the missing regions. TELCIR_14223 was an almost-perfect match to Tci-CF-1 for the exons available. TELCIR_06733 and _06734 are adjacent in the genome. Together the separate sequences can encode the entire Tci-CF-1 sequence with 68.7% similarity, and all motifs are conserved (Supplemental Data 2). The matching of exons from different loci may be a consequence of partial sequencing of tandemly repeated genes or errors in the assembly of the genome.
The consensus sequences of the 3 variants all have high similarity to Tci-CF-1. Variant 3 gave a nucleotide and amino acid sequence similarity of 98.6% (1080/1095 nucleotides, 359/364 amino acids) to Tci-CF-1. Variants 1 and 2 gave nucleotide similarities of 98% (1073/1095 nucleotides) and 97.3% (1066/1095 nucleotides), respectively, and amino acid sequence similarities of 97.3% (354/364 amino acids) and 96.4% (351/364 amino acids), respectively. An alignment of Tci-CF-1, the 3 variants identified in this study and the Nisbet et al 13 variant show the signal peptide, catalytic triad and predicted N-glycosylation sites are conserved, as well as most of the motifs (Figure 1). The presence of a stop codon in variant 1 implies that there are at least 1 functional gene and 1 pseudogene in the T. circumcincta genome, but a sequencing error cannot be ruled out.
The gene structure of cathepsin F was determined from the PacBio and Illumina combined reads (variant 3), and has a minimum length of 9583 bp over 10 exons and 9 introns. Exons 1-10 are 119 bp, 90 bp, 70 bp, 90 bp, 145 bp, 160 bp, 85 bp, 94 bp, 178 bp, and 64 bp in length, respectively, resulting in a 364 amino acid protein, and the exon phase class is 0-2-2-0-0-1-2-0-1-2-0-1. Introns 1-9 are >937 bp, 173 bp, 1427 bp, 998 bp, 338 bp, 439 bp, 1882 bp, 705 bp, and >1589 bp in length, respectively (Figure 2).

Teladorsagia circumcincta cathepsin F variant 3 gene exon/intron structure, constructed using PacBio and Illumina reads in SPAdes and CLC. Exons indicated by solid black arrows, introns indicated by white segments, gaps in contigs indicated by a break. The positions of intron-exon junctions and phase classes are denoted by diamonds.
Phylogenetic analysis
The 9 complete sequences with the highest % ID on January 13, 2020 to Tci-CF-1 were Diploscapter pachys hypothetical protein WR25_25536 (GenBank accession no. PAV60527, 309 residues), Haemonchus contortus proteinase inhibitor I25 and proteinase inhibitor I29 and peptidase C1A domain-containing protein (GenBank accession no. CDJ92562, 463 residues), H. contortus proteinase inhibitor I25 and proteinase inhibitor I29 and peptidase C1A domain-containing protein (GenBank accession no. CDJ88889, 463 residues), Dictyocaulus viviparus cathepsin F1 (GenBank accession no. AFM37363, 459 residues), Ancylostoma ceylanicum papain family cysteine protease (GenBank accession no. EPB70524, 287 residues), Angiostrongylus cantonensis hypothetical protein Angca_010213 (GenBank accession no. KAE9418773, 456 residues), Strongylus vulgaris unnamed protein product (GenBank accession no. VDM81154, 264 residues), Angiostrongylus costaricensis unnamed protein product (GenBank accession no. VDM61191, 405 residues), and D. pachys hypothetical protein WR25_24125 (GenBank accession no. PAV67875, 452 residues) (Table 1).
Pairwise comparison of complete homologous protein sequences to Tci-CF-1 by sequence identity (%) and distance.
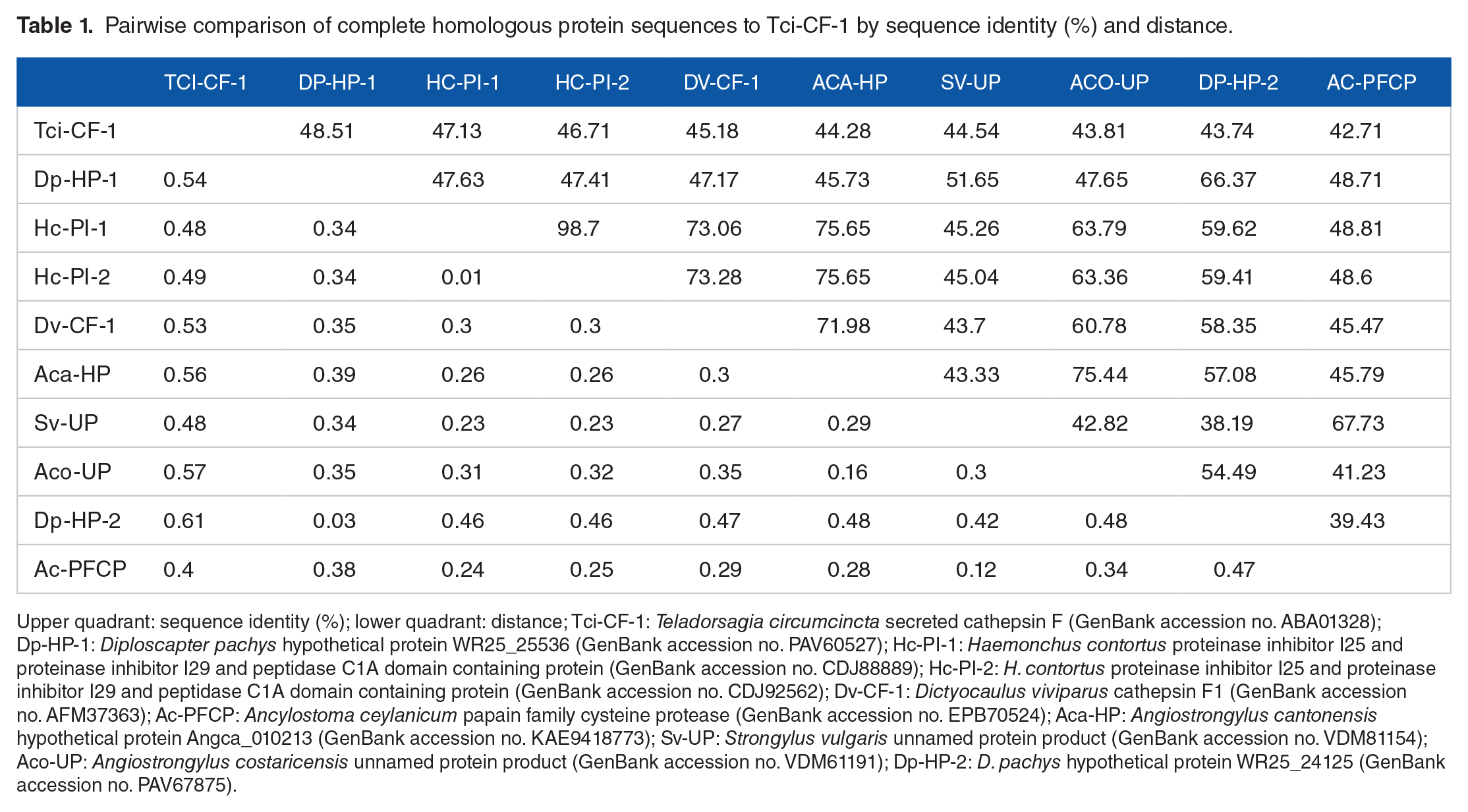
Upper quadrant: sequence identity (%); lower quadrant: distance; Tci-CF-1: Teladorsagia circumcincta secreted cathepsin F (GenBank accession no. ABA01328); Dp-HP-1: Diploscapter pachys hypothetical protein WR25_25536 (GenBank accession no. PAV60527); Hc-PI-1: Haemonchus contortus proteinase inhibitor I25 and proteinase inhibitor I29 and peptidase C1A domain containing protein (GenBank accession no. CDJ88889); Hc-PI-2: H. contortus proteinase inhibitor I25 and proteinase inhibitor I29 and peptidase C1A domain containing protein (GenBank accession no. CDJ92562); Dv-CF-1: Dictyocaulus viviparus cathepsin F1 (GenBank accession no. AFM37363); Ac-PFCP: Ancylostoma ceylanicum papain family cysteine protease (GenBank accession no. EPB70524); Aca-HP: Angiostrongylus cantonensis hypothetical protein Angca_010213 (GenBank accession no. KAE9418773); Sv-UP: Strongylus vulgaris unnamed protein product (GenBank accession no. VDM81154); Aco-UP: Angiostrongylus costaricensis unnamed protein product (GenBank accession no. VDM61191); Dp-HP-2: D. pachys hypothetical protein WR25_24125 (GenBank accession no. PAV67875).
Pairwise comparisons showed that Tci-CF-1 (GenBank accession no. DQ133568, 364 residues) has highest %ID with a Diploscapter pachys hypothetical protein at 48.51% ID, respectively. The remaining 8 closest homologs ranged from 42.71-47.13% ID (Table 1).
Phylogenetic analysis showed a rooted tree drawing with 18 nodes and 17 edges, and 4 separate clades (Figure 3). The Neighbor-Net splits network had 35 nodes and 48 edges (Figure 4A) and complements the clades in the tree. The reticulate splits network had 37 nodes and 50 edges (Figure 4B). Both split networks resulted in 20 cyclic splits. Table 1 indicates the distances between taxa calculated in the multiple sequence alignment.
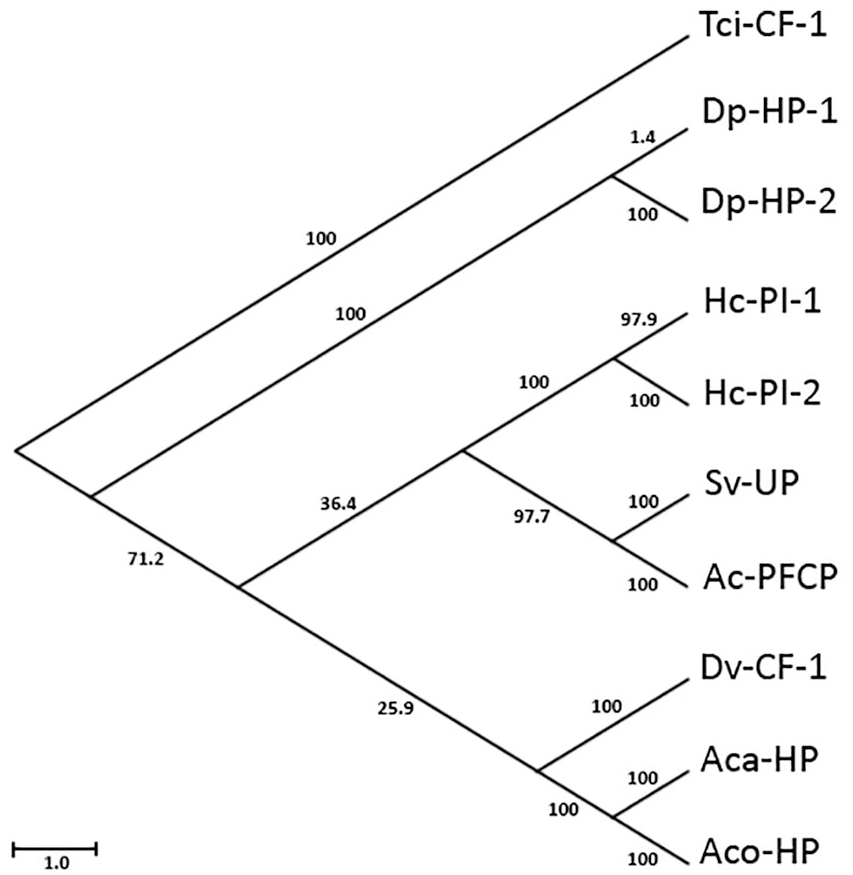
Phylogenetic tree of Tci-CF-1 and its 9 closest homologs. Tci-CF-1: Teladorsagia circumcincta secreted cathepsin F (GenBank accession no. ABA01328); Dp-HP-1: Diploscapter pachys hypothetical protein WR25_25536 (GenBank accession no. PAV60527); Hc-PI-1: Haemonchus contortus proteinase inhibitor I25 and proteinase inhibitor I29 and peptidase C1A domain-containing protein (GenBank accession no. CDJ88889); Hc-PI-2: H. contortus proteinase inhibitor I25 and proteinase inhibitor I29 and peptidase C1A domain-containing protein (GenBank accession no. CDJ92562); Dv-CF-1: Dictyocaulus viviparus cathepsin F1 (GenBank accession no. AFM37363); Ac-PFCP: Ancylostoma ceylanicum papain family cysteine protease (GenBank accession no. EPB70524); Aca-HP: Angiostrongylus cantonensis hypothetical protein Angca_010213 (GenBank accession no. KAE9418773); Sv-UP: Strongylus vulgaris unnamed protein product (GenBank accession no. VDM81154); Aco-UP: Angiostrongylus costaricensis unnamed protein product (GenBank accession no. VDM61191); Dp-HP-2: D. pachys hypothetical protein WR25_24125 (GenBank accession no. PAV67875). Scale-bar indicates branch lengths and bootstrap values are indicated following 1000 replications.
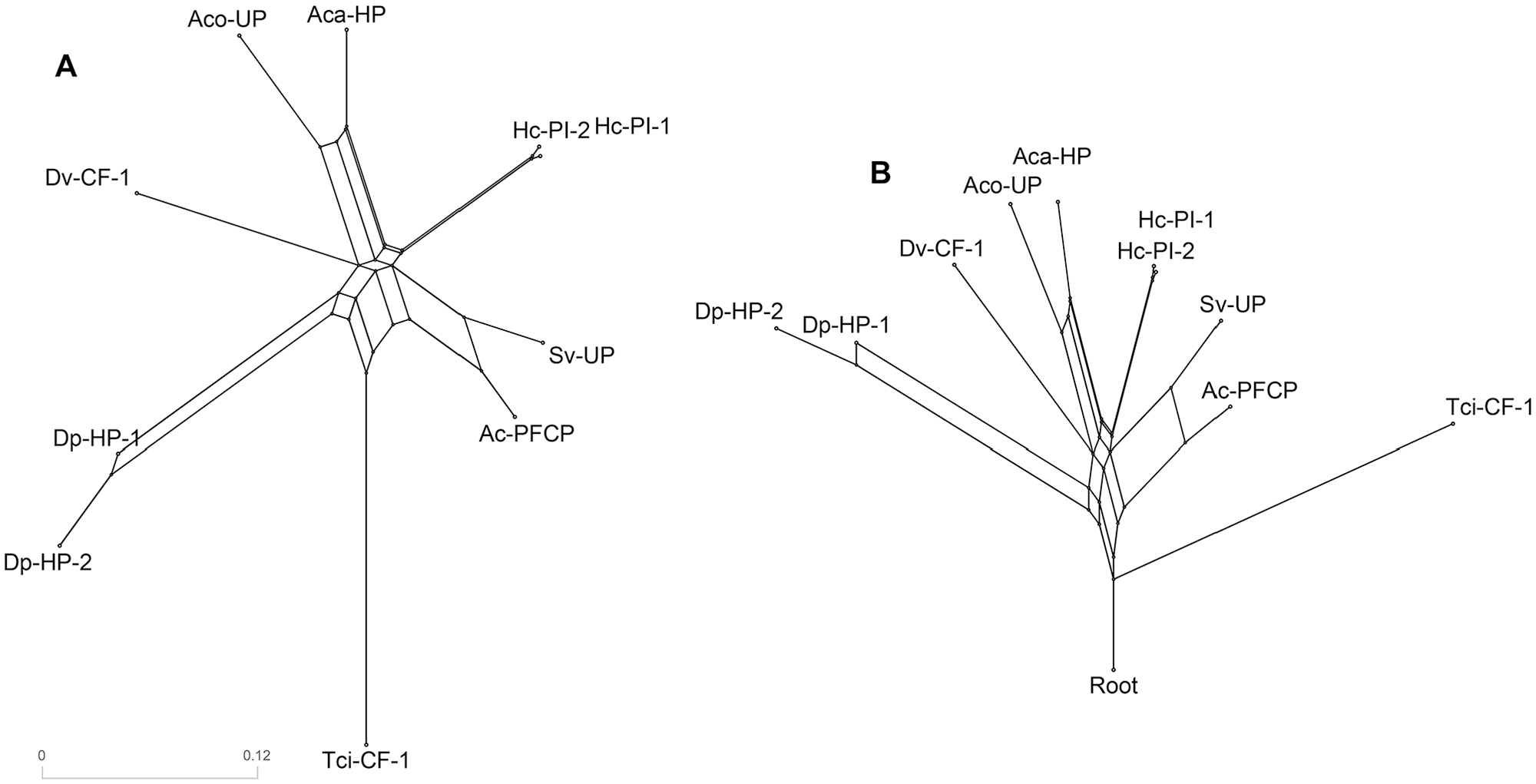
Phylogenetic networks of Tci-CF-1 and its 9 closest homologues; Tci-CF-1: Teladorsagia circumcincta secreted cathepsin F (GenBank accession no. ABA01328); Dp-HP-1: Diploscapter pachys hypothetical protein WR25_25536 (GenBank accession no. PAV60527); Hc-PI-1: Haemonchus contortus proteinase inhibitor I25 and proteinase inhibitor I29 and peptidase C1A domain-containing protein (GenBank accession no. CDJ88889); Hc-PI-2: H. contortus proteinase inhibitor I25 and proteinase inhibitor I29 and peptidase C1A domain-containing protein (GenBank accession no. CDJ92562); Dv-CF-1: Dictyocaulus viviparus cathepsin F1 (GenBank accession no. AFM37363); Ac-PFCP: Ancylostoma ceylanicum papain family cysteine protease (GenBank accession no. EPB70524); Aca-HP: Angiostrongylus cantonensis hypothetical protein Angca_010213 (GenBank accession no. KAE9418773); Sv-UP: Strongylus vulgaris unnamed protein product (GenBank accession no. VDM81154); Aco-UP: Angiostrongylus costaricensis unnamed protein product (GenBank accession no. VDM61191); Dp-HP-2: D. pachys hypothetical protein WR25_24125 (GenBank accession no. PAV67875). (A) Neighbor-Net split network. (B) Reticulate network. Scale-bar indicates the distance of the edges.
Sequence analysis
The nucleotide sequence implies a translated protein of 364 amino acids. The first 14 amino acids are the signal sequence while amino acids 15 to 150 form the pro-region. The mature protein goes from amino acids 151 (Glutamic acid) to 364 (Leucine). Protein sequence analysis of Tci-CF-1 revealed several conserved features that are typical of cathepsin F proteins. Tci-CF-1 has a hydrophobic signal sequence (residues 1-14) with a signal cleavage site between residues 14 and 15 (Figure 5). The pro-region contains cathepsin F-like motifs such as E80R84F88N91A95Q99, in which alanine (A95) has been substituted for isoleucine (I95), E/D102xG104T105A106, and G109xN111xF113xD115 (Figure 6). The predicted N-terminal of the mature processed protein is located at residue E151 (Figure 5). The mature protein contains the conserved structural motif G214C215N216G217G218 as well as the catalytic triad active site residues C177, H311, and N331. Two predicted N-glycosylation sites were found at residues N77E78S79 and N260G261S262 (Figure 6).

Annotated Teladorsagia circumcincta secreted cathepsin F sequence (GenBank accession no. DQ133568) with annotations. Signal sequence is

Homology model of Teladorsagia circumcincta secreted cathepsin F (GenBank accession no. ABA01328). (A) Ribbon structure showing alpha helices, beta sheets, catalytic triad residues (C, H, N), predicted N-glycosylation sites, ERFNAQ, E/DxGTA, GCNGG, and GxNxFxD motifs. (B) Magnified ERFNAQ, E/DxGTA, GxNxFxD motifs and pro-region N-glycosylation site. (C) Magnified catalytic triad and GCNGG motif. (D) Magnified mature domain N-glycosylation site. Sidechains of residues of interest labeled and bold.
Multiple sequence alignments
The multiple sequence alignment of the most similar sequences indicates that 6 out of the 9 sequences contain a region of amino acids in the pro-region (~97 amino acids) that is not present in the pro-region of Tci-CF-1, corresponding to a cystatin domain. The catalytic triad residues and motifs are mostly conserved amongst all homologous sequences. However, Strongylus vulgaris and Ancylostoma ceylanicum are missing the histidine and asparagine residues of the catalytic triad (Figure 7).

Multiple sequence alignment of Tci-CF-1 with the 9 closest homologous sequences. Tci-CF-1: Teladorsagia circumcincta secreted cathepsin F (GenBank accession no. ABA01328); Dp-HP-1: Diploscapter pachys hypothetical protein WR25_25536 (GenBank accession no. PAV60527); Hc-PI-1: Haemonchus contortus proteinase inhibitor I25 and proteinase inhibitor I29 and peptidase C1A domain-containing protein (GenBank accession no. CDJ88889); Hc-PI-2: H. contortus proteinase inhibitor I25 and proteinase inhibitor I29 and peptidase C1A domain-containing protein (GenBank accession no. CDJ92562); Dv-CF-1: Dictyocaulus viviparus cathepsin F1 (GenBank accession no. AFM37363); Ac-PFCP: Ancylostoma ceylanicum papain family cysteine protease (GenBank accession no. EPB70524); Aca-HP: Angiostrongylus cantonensis hypothetical protein Angca_010213 (GenBank accession no. KAE9418773); Sv-UP: Strongylus vulgaris unnamed protein product (GenBank accession no. VDM81154); Aco-UP: Angiostrongylus costaricensis unnamed protein product (GenBank accession no. VDM61191); Dp-HP-2: D. pachys hypothetical protein WR25_24125 (GenBank accession no. PAV67875). Gaps indicated by a dash; ERFNAQ, E/DxGTA, GxNxFxD and GCNGG motif residues indicated by square, cross, circle and diamond, respectively; catalytic triad residues indicated by a downward arrow.
Secondary and tertiary structure
Homology modeling using Phyre2 revealed that the secondary structure of Tci-CF-1 comprised 37% alpha-helices, and 15% beta-strands. The tertiary structure has 100% confidence in homology to cysteine proteases, and 53% ID with cysteine protease folds from the papain-like family. The predicted structure for residues 1-60 has low confidence, while the predicted structure for residues 61-364 has high confidence (Supplemental Data 3). In the tertiary structure model, the 3 amino acids that form the catalytic triad come together to form the active site, and the inhibitor domain appears to block access to the catalytic triad (Figure 8). Polymorphisms were present on the periphery of the structure at positions 30 (E30 > D30), 56 (K56 > N56), 76 (R76 > K76), 235 (P235 > S235), and 306 (L306 > P306) (Figures 1 and 8).
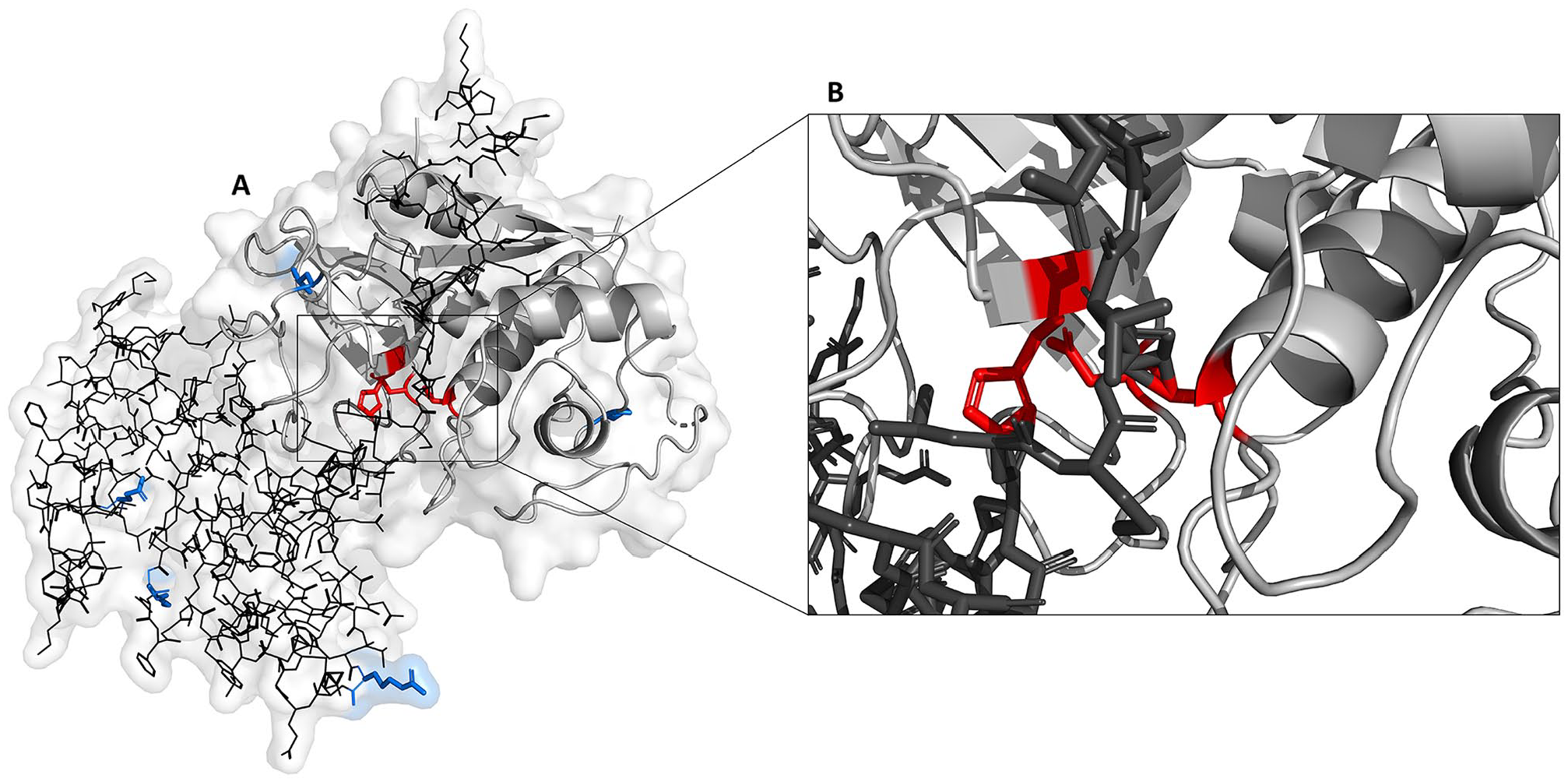
Homology model of Teladorsagia circumcincta secreted cathepsin F (GenBank accession no. ABA01328). (A) Pro-region (line residues), mature domain (ribbon), locations of polymorphisms in variants 1, 2, and 3 (bold side-chain residues), and the catalytic triad (bold side-chain residues) which is exposed following cleavage of the pro-peptide. (B) Magnified view of the active site indicating bonds between the pro-region and catalytic triad residues.
Homology modeling of Tci-CF-1 illustrates structural similarity to x-ray crystallographic human cathepsin L and F structures.40,41 The mature domains appear structurally similar (Figure 9) despite the aminoacid sequences being quite different between proteins, and the pro-region of human cathepsin L is more structurally similar to Tci-CF-1 (Figure 9B and C) than human cathepsin F (Figure 9A).

Structural comparison of human and Teladorsagia circumcincta cathepsin proteins. X-ray crystallography of mature domains of human cathepsins L 40 and F, 41 and homology modeling of T. circumcincta cathepsin F (GenBank accession no. ABA01328) and the pro-regions of human cathepsins L (UniProtKB accession no. P07711) and F (UniProtKB accession no. Q9UBX1). (A) Human cathepsin F; (B) T. circumcincta secreted cathepsin F; (C) Human cathepsin L; pro-region, mature domain and cystatin-like domain highlighted in different colors.
Discussion
Bioinformatic analysis of Cathepsin F of T. circumcincta has assembled a putative gene encoding the protein, described the gene structure, discovered several potential polymorphisms, failed to identify any close homologs and predicted the structure of the protein. Most cysteine proteases are synthesized as an inactive precursor. Cathepsins, a family within the cysteine proteases, maintain typical features including an N-terminal hydrophobic signal peptide sequence, a pro-peptide domain, and a mature domain containing the active site. 23 The active site consists of a catalytic triad of cysteine, histidine, and asparagine residues.42,43 Cathepsin pro-peptide inhibitor domains contain an α-helical structure that prevents access to the active site and proteolytic cleavage is needed to remove the inhibitor and activate the zymogen 44 (Figure 8). Conserved motifs of cysteine proteases are critical for characterization of the different sub-families.
When cathepsin F of T. circumcincta was first identified, its closest homologues were hypothetical proteins of Caenorhabditis briggsae and Caenorhabditis elegans. 23 The highest % identity in the databases on April 2020 is with a D. pachys hypothetical protein and a H. contortus proteinase inhibitor I25 and proteinase inhibitor I29 and peptidase C1A domain-containing protein at 48.51% and 47.13%. The similarity between these proteins is not particularly high, and molecules from species other than T. circumcincta appear more closely related to one another, ranging from 38.19% to 98.7% identity (Table 1). Closely related species are expected to have more similar proteins. For example, tropomyosin from T. circumcincta and Trichostrongylus colubriformis has been shown to differ by only 1 amino acid, 45 both nematodes belong to the order Strongylida and are in the same clade V. Cathepsin L of Fasciola hepatica has been shown to digest IgG. 26 If Tci-CF-1 has a similar role, it could suppress the immune response given the important role that antibody responses play in resistance to this parasite. 46 The absence of close homologs suggests that this protein has emerged relatively recently in evolutionary history. Since there are no fossil records of T. circumcincta specifically, an estimate of its divergence from relatives must be made. The earliest bovids appeared 20 mya, 3 and since T. circumcincta is seen in sheep and goats but not cattle, we can safely assume their divergence from related nematode species occurred following or alongside bovid divergence. Chilton et al47–49 have shown the evolutionary relationships between T. circumcincta (sheep and goats), O. ostertagi (cattle), and H. contortus (sheep and goats), and that they are very closely related. There was no similar protein detected in this study between T. circumcincta and O. ostertagi, the more closely related of these 3 species, but there was a protein detected from H. contortus (Table 1), a slightly more distant species but still within the same Trichostrongylidae clade and with the same host species. Whether the gene for cathepsin F in T. circumcincta was inherited from an ancestor or developed later is unknown, but it has diverged enough from relatives to be a distinct protein.
One of the methods in which a new gene arises is by exon shuffling, however, there was no evidence of ancestral exons, as there is no 1-1 class phase observed in the cathepsin F gene (Figure 2). 50 Gene assembly demonstrated that the Tci-CF-1 gene is composed of 10 exons spanning a minimum length of 9.5 kbp. The SNPs with transcribed substitutions, resulting in the 5 polymorphisms identified in this study, are located on the periphery of the protein structure (Figure 8A). Of particular interest are the 2 polymorphisms located in the mature protein (proline (P235) to serine (S235), and leucine (L306) to P306), as these are likely to affect mature protein function and/or recognition. The substitution of P235 to S235 is conservative as proline and serine are both small amino acids and serine is commonly present within tight turns on protein surfaces because its hydroxyl oxygen can form a hydrogen bond with the protein backbone and mimic proline. 51 The location of the L306 in Tci-CF-1 indicates that it may be influencing the catalytic triad in some way. Leucine is a hydrophobic amino acid and its position on the outside of the protein structure suggests that it may be involved in substrate recognition. 51 The substitution of leucine to proline is interesting due to the ability of proline to introduce kinks into the sequence. The substitution of leucine to proline may change the way the catalytic triad interacts with host molecules.
The pro-region of Tci-CF-1 does not have typical cathepsin F characteristics. A typical cathepsin F has a long pro-region (up to 250 residues) and in general, the pro-peptide of cathepsin F is composed of 2 domains; an N-terminal cystatin-like domain and a C-terminal peptide similar to the cathepsin L pro-region. 22 In Tci-CF-1, homology modeling indicates that the cystatin-like domain is missing (Figures 7 and 9), resulting in a much shorter pro-region, and it is more similar to a typical cathepsin L pro-region. Clonorchis sinensis cathepsin F (GenBank accession no. AF093243) is also missing the cystatin-like domain in the pro-region of its cathepsin F protein. 19 Typical cathepsin F pro-regions also contain a highly conserved ERFNAQ motif, in place of the cathepsin L ERF/WNIN motif . 43 Adjacent to the ERFNAQ motif is an E/DxGTA motif, which was identified as a pro-region feature of cathepsins F and W, but not cathepsin L. 23 These 2 motifs together are thought to act as a scaffold of the pro-region and maintain the inhibitory function of the α-helical structures of cathepsins F and W. 19 Redmond et al 23 demonstrated that Schistosoma mansoni cathepsin L (GenBank accession no. AAC46485) contains the ERFNAQ motif, and not the expected ERF/WNIN motif, which is consistent with the Tci-CF-1 in the current study.43,52 In addition, S. mansoni cathepsin L does not contain a cystatin-like domain, which is consistent with Cathepsin L, but also with Tci-CF-1 in the current study (Figure 9). Despite the conservation of the cathepsin F/W E/DxGTA motif in Tci-CF-1, the lack of a cystatin-like domain in Tci-CF-1 coupled with the presence of the ERFNAQ motif in S. mansoni cathepsin L, illustrates that the distinctions between cathepsins F and L may not be as clear as often assumed.
Substitutions at specific residues between cathepsins F and L may provide insights into Tci-CF-1. The substitution of alanine (A) to isoleucine (I) in the ERFNAQ motif of Tci-CF-1 places this motif halfway between ERFNAQ (cathepsin F) and ERF/WNIN (cathepsin L). Both alanine and isoleucine can be readily substituted for one another due to their small size, and non-reactive sidechains. The ERF/WNIN and ERFNAQ motifs are known to form α-helical structures, 51 however, isoleucine is known to be restricted in its conformations, and finds it difficult to form an α-helical structure. Similarly, asparagine (N) can be readily substituted for glutamine (Q) as both these residues can be substituted by polar amino acids, are similar in structure, and are frequently involved in protein active or binding sites, of which the ERFNAQ and ERFNIN motifs inhibit. 51 Whether Tci-CF-1 has evolved from ERFNAQ or ERFNIN to ERFNIQ remains unknown. Phylogenetic analysis of our top 10 similar sequences does not provide insights either.
Tci-CF-1 is quite isolated from the rest of the proteins as seen in Figures 2 and 3 and illustrates that although these homologous proteins are closest by sequence %ID, they are closer to one-another than Tci-CF-1. Phylogenetic networks show the possible relationships in a dataset. Taxa are represented by nodes and their evolutionary relationships are represented by edges. 32 Recombination, hybridization, gene conversion and gene transfer all lead to phylogenetic relationships that cannot be adequately modeled by a single tree. Even when the underlying history is treelike, sampling error and parallel evolution may make it difficult to establish a single, accurate phylogenetic tree. 32 Parallel edges are used to represent the splits of the taxa, instead of single branches of a tree. 36 Split networks often contain nodes that do not represent ancestral species and, therefore, can only provide a suggestive representation of evolutionary history. Two split network methods were applied in this study to portray the relationship between Tci-CF-1 and its 9 closest homologs: The Neighbor-Net split network and the Reticulate Network.
Neighbor-Net split networks use multiple sequence alignment distance calculations to construct a circular collection of weighted splits and can represent conflicting signals in the data, whether they arise from sampling error or genuine recombinations 36 and show the uncertainty of the phylogenetic history. The more tree-like the network, the more confidence that the tree constructed is an accurate representation of the phylogenetic history with the data used. Figure 4A shows that the groupings observed in the network are complementary to the clades in the phylogenetic tree (Figure 3). Tci-CF-1 is distinctly isolated from the other groupings. The Neighbor-Net gives confidence that although there are several alternative phylogenetic tree outputs possible, they will follow roughly the same paths, with the different clades consistently grouping together (Figure 4A).
Reticulate Networks illustrate evolutionary histories, and their splits are a result of reticulate events such as hybridization, horizontal gene transfer, or recombination. The internal nodes represent hypothetical ancestral species, and nodes with 2 or more parents correspond to reticulate events such as hybridization or recombination. In this study, the reticulate network in Figure 4B shows that when Tci-CF-1 is selected as the outgroup, the homologous proteins are quite evolutionarily distant because many parent nodes correspond to many possible reticulate events between them. The long edge lengths are indicative of the weight of the associated split and are analogous to the length of a branch in a phylogenetic tree. 36 The distance between Tci-CF-1 and its closest homologs is relatively large compared to the distances between the homologs themselves and this complements the %ID between species in Table 1. The increased number of parent nodes illustrates there are many hypothetical ancestral species between these proteins, highlighting how divergent they are.
In summary, T. circumcincta cathepsin F has no close homologs even in closely related species such as H. contortus which is a member of the same subfamily. 53 The absence of close homologs indicates Tci-CF-1 may have evolved relatively recently, whether because of host immune pressure or other factors leading to rapid change. Cathepsin F has characteristics of both cathepsins F and L. The Tci-CF-1 pro-region contains motifs characteristic of both cathepsins F and L; however, homology modeling indicates that it lacks a cystatin-like domain, making it structurally more similar to cathepsin L. The bioinformatic investigation of Tci-CF-1 provides insights into the presence of amino acid changes/substitutions, and how these may influence the function of cathepsin F. This information can be used to design powerful functional studies to improve our understanding of the role of cathepsin F in the immunogenicity of T. circumcincta.
Supplemental Material
200115_Supplementary-Data-1_xyz42578e15bab79 – Supplemental material for Cathepsin F of Teladorsagia circumcincta is a recently evolved cysteine protease
Supplemental material, 200115_Supplementary-Data-1_xyz42578e15bab79 for Cathepsin F of Teladorsagia circumcincta is a recently evolved cysteine protease by Sarah Sloan, Caitlin Jenvey, Callum Cairns and Michael Stear in Evolutionary Bioinformatics
Supplemental Material
200115_Supplementary-Data-2_xyz42578dce9de09 – Supplemental material for Cathepsin F of Teladorsagia circumcincta is a recently evolved cysteine protease
Supplemental material, 200115_Supplementary-Data-2_xyz42578dce9de09 for Cathepsin F of Teladorsagia circumcincta is a recently evolved cysteine protease by Sarah Sloan, Caitlin Jenvey, Callum Cairns and Michael Stear in Evolutionary Bioinformatics
Supplemental Material
200725_Supplementary-Data-3_xyz4600490785289 – Supplemental material for Cathepsin F of Teladorsagia circumcincta is a recently evolved cysteine protease
Supplemental material, 200725_Supplementary-Data-3_xyz4600490785289 for Cathepsin F of Teladorsagia circumcincta is a recently evolved cysteine protease by Sarah Sloan, Caitlin Jenvey, Callum Cairns and Michael Stear in Evolutionary Bioinformatics
Footnotes
Acknowledgements
We would like to thank the La Trobe University High Performance Computing cluster team for access to their systems and support. Molecular graphics and analyses performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from the National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by a grant from La Trobe University.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ Contributions
SS, CC, and MS conceived of the presented idea. SS developed the theory and performed the computations, research, and wrote the manuscript. CJ and MS verified the analytical methods and supervised the findings of this work. All authors discussed the results and contributed to the final manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.