
Abstract
Triacylglycerol lipases have been thoroughly characterized in mammals and microorganisms. By contrast, very little is known about plant lipases. In this investigation, a homology model of
Introduction
Lipases are glycerol ester hydrolases that act on triacylglycerols to release fatty acids and glycerol.
1
A major part of lipases used in today's industrial processes stems from microbial or animal sources. The oretically speaking, plant enzymes may have an advantage over animal or microbial enzymes due to their availability from natural sources, lower cost and apparent purification ease.
2
Lipases widely exist in microbe and plant species, but their structure is poorly characterized. Plant lipases are generally considered to be involved in particular in the regulation of plant growth and development. They are mainly located in seeds where triglycerides are stored in intracellular structures called oil bodies. Triglycerides are hydrolyzed by lipases to glycerol and fatty acids that provide energy needed for seed germination and seedling development.
3
Plant lipases can be classified into three major groups. The first group consists of the triacylglycerol hydrolases that are primarily present in seeds. Their study is of economic importance since they are largely responsible for seed alteration during storage. Lipases of the second group, called acylhydrolases, are present in various plant tissues. These enzymes exhibit little specificity for their substrate and are unable to hydrolyze triglycerides but can catalyze some trans-esterification reactions.
4
The main acylhydrolases are phospholipases A and B, glycolipases, sulfolipases and monoglyceride lipases. The third group consists of phosphorlipases C and D. Lipases are also involved in plant metabolism, rearrangement and degradation of chlorophyll during leaf growth and senescence as well as in fruit ripening process.
5
Recently, researches in this field became more and more attractive. Although several candidates from
An updated and revised classification of family I bacterial “true” lipases mainly based on a comparison of their amino acid sequences and some fundamental physicochemical and biological properties, identified 11 subfamilies. 10 Although lipases belong to many different protein families, they have the same architecture, the a β-hydrolase fold as defined by Ollis et al. 11 Their activities rely mainly on a catalytic triad usually formed by Ser, His and Asp residues. 12 In amino acid sequences of α/β hydrolases, the three residues follow the order Ser-Asp-His. Also, lipases share a consensus sequence of Gly-Xaa-Ser-Xaa-Gly. 13 where X may be any amino acid residue.
Three-dimensional structure of proteins gives valuable insights into the molecular organization, function, docking simulations and also effective drug designing experiments. In the absence of an experimentally-determined crystal structure, homology modeling could provide a rational opportunity to obtain a reasonable 3D model. It is generally recognized that homology modeling of proteins is currently the most accurate method for 3D structure prediction, yielding models suitable for a wide spectrum of application, such as structure based molecular design and mechanism investigation.
30
This approach is able to provide a reasonable structure model with related template sharing more than 25% sequence identity. The present study attempts to propose a model of
Methods
Target and template proteins
With the aim of finding an adequate template for homology modeling of
General, physicochemical and structural data of

Homology modeling
Homology model was constructed using Swiss-PDB Viewer version 4.0.1.
18
This application provides a user friendly interface that allows analyzing several proteins at the same time. Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. Three different types of modeling requests can be made in the Swiss-PDB Viewer version 4.0.1 program. They are automated mode, alignment mode and project mode. The automated mode is suited for cases where the target-template similarity is sufficiently high to allow for fully automated modeling. The alignment mode allows the user to test several alternative alignments and evaluate the quality of resulting models in order to achieve an optimal result. In difficult modeling situations where the correct alignment between target and template cannot be clearly determined by sequence-based methods, visual inspection and manual manipulation of the alignment can significantly help improve the quality of the resulting model. We used the project mode for obtaining the modeled structure of
Model validation
To obtain an accurate homology model, it is very important that appropriate steps are built into the process to assess the quality of the model. 19 Therefore, accuracy of the predicted models were subjected through a series of tests. Stereochemical quality were evaluated using Ramachandran plots obtained from the RAMPAGE server 20 and amino acid environment was assessed using Verify 3D 21 and Errat 22 from the UCLA-DOE server. 23
Docking
The lipase natural substrate was
Results and Discussion
Homology models and validation
Studies of Yang et al
27
demonstrated that a sequence identity higher than 25% between two proteins is indicative of similar three-dimensional structures. In our case, target and template protein share 32% of sequence identity. Based on this alignment, a model of
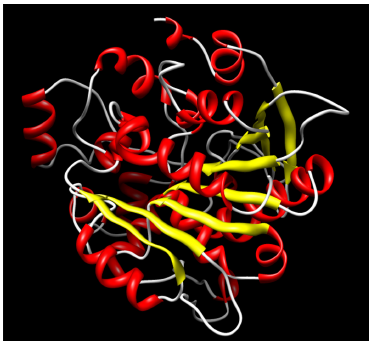
Ribbon representation of a three dimensional structure of

Ramachandran plot values showing number of residues in favoured, allowed and outlier region.
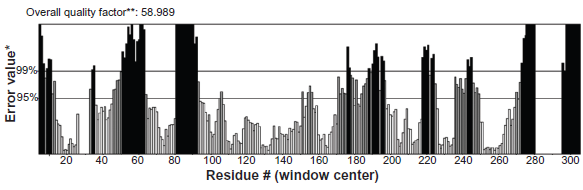
Errat plot for

The verify score diagram validate the
Docking
For docking, the ligand structures were obtained from the PubChem database.
33
In order to investigate the substrates binding with the enzyme, we attempted to dock A) tributyrin, B) trioctanoin and C) triolen to the lipase enzyme model. The top docking solutions of 3000 interaction results for each ligand were selected. The total binding energies are respectively: –201.09 (kcal/mol), –191.11 (kcal/mol) and –272.83 (kcal/mol). This result confirms that the most preferred substrate for
Figure 5 shows that triolein does not bind the lipase at the same site as tributyrin and trioctanoin. In addition, the catalytic active site, serine S166, is not involved in the binding site, this result confirms that the lid domain which covers the active site is not accessible to solvent.
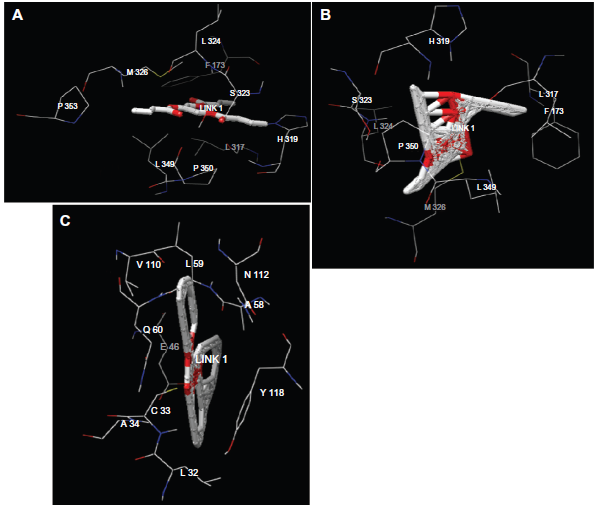
Three-dimensional representation of substrates (A), tributyrin (B), trioctanoin and (C) triolen in the
Conclusion
In summary, the constructed homology model of
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.