
Abstract
The inclusion of recovery animals in nonclinical safety studies that support clinical trials is undertaken with a wide diversity of approaches even while operating under harmonized regulatory guidance. While empirical evaluation of reversibility may enhance the overall nonclinical risk assessment, there are often overlooked opportunities to reduce recovery animal use by leveraging robust scientific and regulatory information. In the past, there were several attempts to benchmark recovery practices; however, recommendations have not been consistently applied across the pharmaceutical industry. A working group (WG) sponsored by the 3Rs Translational and Predictive Sciences Leadership Group of the IQ Consortium conducted a survey of current industry practice related to the evaluation of reversibility/recovery in repeat dose toxicity studies. Discussion among the WG representatives included member company strategies and case studies that highlight challenges and opportunities for continuous refinements in the use of recovery animals. The case studies presented in this paper demonstrate increasing alignment with the Society of Toxicologic Pathology recommendations (2013) towards (1) excluding recovery phase cohorts by default (include only when scientifically justified), (2) minimizing the number of recovery groups (e.g., control and one dose level), and (3) excluding controls in the recovery cohort by leveraging external and/or dosing phase data. Recovery group exclusion and decisions regarding the timing of reversibility evaluation may be driven by indication, modality, and/or other scientific or strategic factors using a weight of evidence approach. The results and recommendations discussed present opportunities to further decrease animal use without impacting the quality of human risk assessment.
Introduction
During pre-clinical drug development, reversibility of test item effects may be evaluated during a drug-free recovery period in one or more repeat dose toxicology studies. Irreversible toxicities in animals could predict permanent injury in human clinical trial participants; therefore, identification of irreversible effects is a key component of the overall risk assessment and determination of safe use conditions. In some circumstances, assessment of reversibility may not be warranted. Clarification of inclusion and exclusion criteria for evaluation of recovery animals can streamline scientific and operational processes and potentially reduce the numbers of research animals used for these studies.
Many pharmaceutical companies include recovery groups in toxicology studies based on the intended patient population or therapeutic indication. Collectively, the regulatory guidelines (International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) S6R1, S9, M3(R2)) suggest that a recovery group should be considered in those cases where prior studies indicate there is a severe adverse effect at the approximate clinical exposure, and the reversibility of that toxicity cannot be predicted.1-4
Regulatory guidance on reversibility assessment varies widely based on factors such as the indication and patient population. For example, in the ICHS9 Q&A document (Oncology), the inclusion of recovery groups is deemed not warranted for supporting advanced cancer patient populations. 5 On the other end of the spectrum, the ICH S11 document for the development of therapies in pediatric populations recommends the inclusion of a non-treatment period in juvenile animal studies in order to determine (1) whether any effects observed during treatment are reversible, persistent, or progressive and (2) whether any effects would emerge later in development because of early life exposure (i.e., delayed onset). 6 The ICH M3 (R2) Q&A document for the development of drugs in adult populations with non–life-threatening diseases indicates that demonstration of recovery is only necessary in the case where the toxicity is severe at clinically relevant exposures, is translatable to the clinic, or if the toxicity is not monitorable clinically. 7
Despite operating under harmonized regulatory guidance, the decision to include or exclude recovery animals in toxicology studies is undertaken with a diversity of approaches across the pharmaceutical industry. In some instances, greater scrutiny of findings is warranted due to novel circumstances such as a new modality, drug class, or disease setting. While the use of recovery animals may provide a valuable assessment of the reversibility of adverse toxicity (and/or delayed toxicity) in such cases, there are instances where an assessment of reversibility may not be necessary and/or may be confidently made with existing data, thus minimizing or obviating additional animal use. Consequently, using recovery animals should not be the default for every project. With the reduced availability of accepted test species such as the cynomolgus monkey 8 and a growing need to streamline development practices due to industry economic pressures, it is highly advantageous to critically evaluate recovery practices and to apply a mindset of continuous improvement.
Several previous efforts benchmarking and assessing scientific justification strategies for recovery animal practices including a global cross-company data sharing initiative have provided valuable insights.9,10 Introduction of the potential to develop and use virtual control groups (VCGs) have also initiated discussions on reducing the use of recovery control animals or control animals in general. 11 The Society of Toxicologic Pathology (STP) and an NC3R led expert working group had several recommendations regarding recovery benchmarking practices, inclusion and exclusion criteria encompassing considerations based on species, stages of drug development, modalities, etc.1,9,12 Considerable time has elapsed since these publications, and therefore, an opportunity existed to evaluate if current industry recovery practices have adhered to these recommendations. In August 2021, a Recovery Animal working group (WG) was formed by the 3Rs Translational and Predictive Sciences (TPS) Leadership Group of the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ, a consortium of pharmaceutical companies). This WG focused on understanding inconsistencies in current industry practice regarding the use of recovery animals and to identify opportunities to further decrease animal numbers without negatively impacting human risk assessment.
The goal of this manuscript is to review current reversibility assessment practices (i.e., using recovery groups on toxicology studies) amongst working group members across the pharmaceutical industry and to focus on case studies and novel strategies/opportunities that may be instructive. Additionally, we discuss regulatory outcomes of such unique submissions. The authors from several WG companies in this manuscript describe opportunities that peers can utilize to make strategically and scientifically sound decisions around the deployment of animals for assessment of reversibility that facilitate the replacement, reduction, and refinement of animal use.
Material and Methods
WG members designed the survey, which was then administered by an independent third-party secretariat (Drinker, Biddle, & Reath LLP) among 12 participating member IQ companies in the Recovery Animal WG. Survey links were emailed to all WG participating organizations, and participants were asked to limit the response to one per organization to limit repetition. A total of nine questions offered multiple options with instructions to choose all responses that applied, as well as a free text box to list additional responses if needed to provide further details or to present unique responses. 13 Nine questions (see Supplemental data) were posed to understand company strategies involving recovery groups including use of recovery control animals, number of recovery animals, therapeutic modality (small molecule and biotherapeutics), and regulatory feedback. Other modalities (vaccines, gene and cell therapy, or ASOs) were not included in the survey and are not discussed in this manuscript. Survey responses were compiled by the IQ secretariat and provided confidential, de-identified responses to the authors.
Results and Discussion
Key takeaways from IQ 3Rs TPS LG-recovery animal practices survey
Responses were collected to understand if companies have a formal strategy for recovery group inclusion in general GLP toxicology studies. Most respondents (8/12) reported no formal strategy on inclusion of recovery groups in general GLP toxicology studies and decided on a case-by-case, data-driven basis. Four respondents (4/12) reported that they have internal guidelines determining when general toxicity studies would include recovery groups.
When asked to identify the primary purpose of including recovery in a GLP study (Figure 1A), the majority respondents (11/12) indicated that they used the recovery group to assess reversibility of treatment-related findings. Half of the respondents (6/12) also considered recovery groups to assess delayed toxicity and/or the Pharmacokinetics/Pharmacodynamics (PK/PD) characteristics in cases of therapies with a long half-life (e.g., large molecules). A few respondents (4/12) also expressed concern that regulatory authorities may not accept studies without a recovery phase. Panel A: Y-axis represents number of responses obtained from working group survey and X-axis represents primary purpose of including recovery animal groups in a GLP study. Panel B: Histogram represents number of responses on Y-axis and major factors taken into consideration while decision making for recovery groups inclusion or exclusion NHP origin on X-axis. The shaded region and non-shaded region of histogram depicts responses during early drug development (FiH) or late drug development (Ph2/3).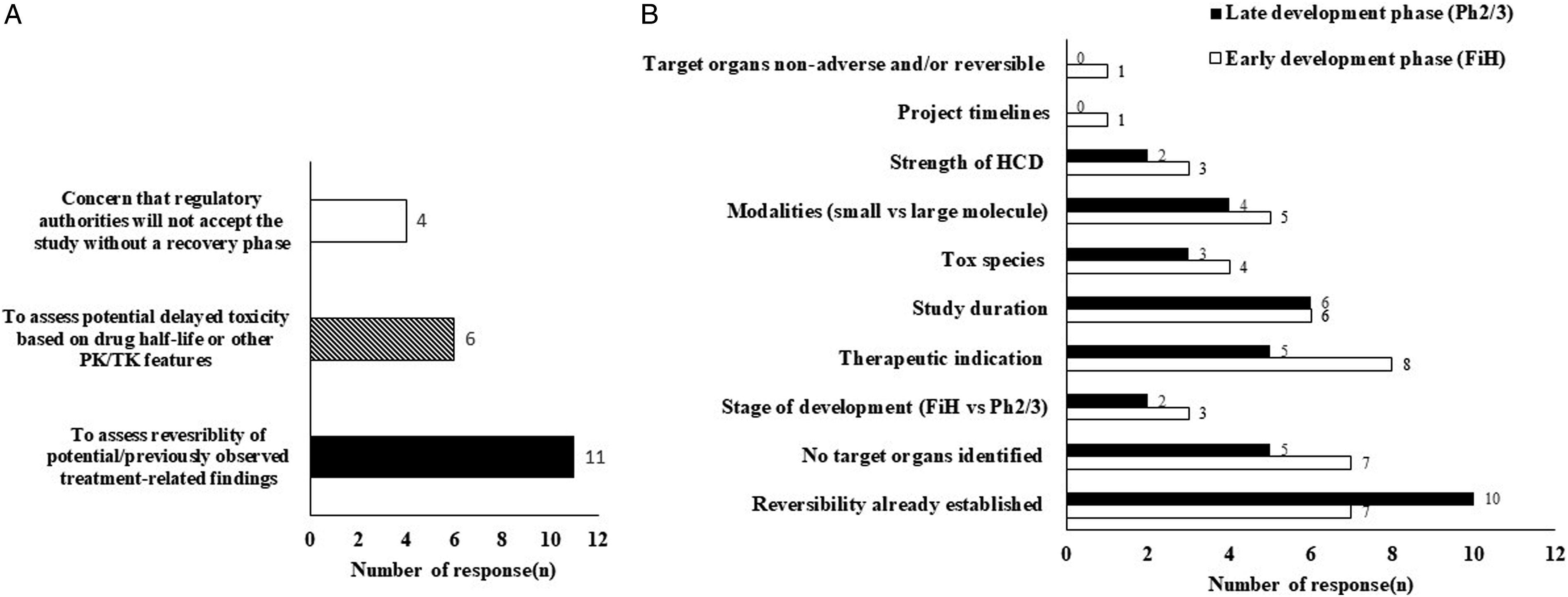
The factors influencing the inclusion of recovery groups were similar in early and late phase development and were independent of the project timeline (Figure 1B). The principal factor driving addition of recovery groups was the need to establish the reversibility of target organ findings seen in earlier studies. Therapeutic indication was also an important driver in the decision to include recovery groups (e.g., reversibility is less often an expectation in oncology/life threatening disease), whereas test species or strength of historical control data (HCD) were less influential.
Survey results suggested that recovery animals were most frequently included in 1-month repeat dose toxicity study (8/12), followed by chronic studies (Figure 2A). Factors besides study duration that influenced inclusion of recovery animals included modality (small molecule and large molecules), toxicity profile, stage of development, and indication (2/12 respondents). Additional recovery survey findings on dose groups and the number of animals that follow general industry trends are discussed in the supplemental section. Panel A: Y-axis represents number of responses obtained from working group survey and X-axis represents study duration and factors based on which recovery groups are included. Factors other than study duration included responses suggesting the modality and evaluation based on toxicity profile, stage of development and indication were more critical than study duration in decide whether to include recovery animals. Panel B: Histogram represents number of responses on Y-axis and X-axis shows regulatory feedback on recovery arms in subsequent toxicity studies.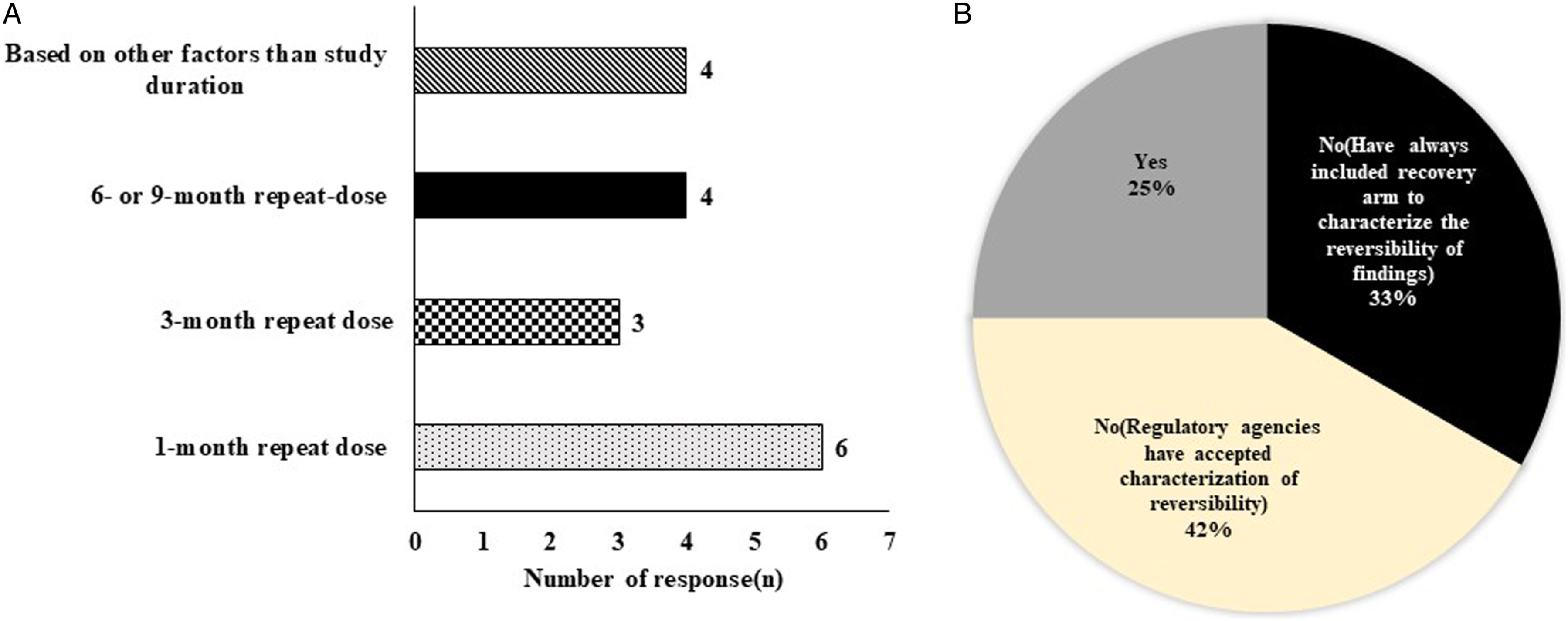
When asked whether they had received questions regarding the absence of recovery groups from regulatory authorities, four respondents stated that they had no questions as they always include recovery arms for all submissions (Figure 2B). Two respondents reported requests to include recovery in the post-IND studies after not including recovery in IND-enabling studies. Another respondent had a request to increase the duration of recovery in the next chronic toxicology study to prove recovery for a male reproductive organ finding, despite providing literature references to support reversibility. However, in nearly half of the cases (5/12), regulatory authorities have accepted characterization of reversibility based on the nature of the finding, scientific literature, HCD, and/or other rationale.
Half of the respondents (6/12) stated that they at least consider non-terminal recovery groups although they may not be actively using this option. Such an approach of using non-terminal recovery groups is considered for endpoints other than histopathology (i.e., ECG, clinical pathology, and immunophenotyping). If the endpoint(s) of interest do not include histopathology, recovery control animals may be returned to a colony at the study’s completion. A recovery phase may be deemed unnecessary due to lack of overt toxicity in prior studies. These decisions are based on the toxicity profile of the compound initially assessed under steady state drug exposure conditions.
Case Studies
The following case studies provide insights into the scientific basis for inclusion or exclusion of recovery animals in the design of toxicology studies and rationale for the optimal use of recovery arms in toxicology study design. Each case study provides rationale and considerations behind the inclusion/exclusion of recovery animals with the goal of increasing awareness of successfully progressing drugs for clinical application while also minimizing or even omitting recovery animals. These case studies and general strategies demonstrate alignment and progress from the STP recommendations towards (1) exclusion of a recovery phase in default study designs, (2) minimization of the number of recovery dose groups and/or animals/group, and (3) exclusion of recovery control groups by leveraging pre-existing control data.
Exclusion Case Study Examples
Exclusion of Control Recovery Groups
Case Study 1 (Large Molecule/Oncology/NHP)
Recovery control animals were excluded in a large molecule (recombinant fusion protein) cynomolgus monkey study for an oncology indication. Dose range-finding (DRF) monkey studies were previously not conducted for this large molecule and dose levels for the IND-enabling study were selected based on non-terminal NHP repeat dose pharmacokinetic studies with recovery phase. The 4-week IND-enabling GLP study was conducted in cynomolgus monkeys with a standard design consisting of vehicle control and three dose groups. Recovery groups for a 4-week period were limited to the high dose group only based on the following factors: (1) vehicle was well-characterized, (2) no anticipated adverse findings/target organs that may require comparison with concurrent control, (3) agreement with the project pathologist who determined that vehicle controls from the concurrent dosing phase could be used for comparison with the recovery animals in the high dose group due to the short duration of recovery, and (4) test article was not predicted to have adverse effects at clinically meaningful exposure levels based on in-vitro assays and non-GLP repeat dose pharmacokinetic studies with recovery phase. The vehicle control recovery animals were reassigned to the main study high dose group for stronger data interpretation. This exclusion of recovery NHPs was aligned with industry practices, had no impact on data interpretation, and with a successful IND submission.
Case Study 2 (Small Molecule/Oncology/Rat and Dog)
For a small molecule program in development for oncology, recovery assessment was limited to the registrational study of the most sensitive species (dog). The DRF- and IND-enabling studies (rat and dog) included a vehicle control group, three dose groups, and no recovery cohort. In these studies, dose limiting toxicity was identified at the high dose and involved the same organ system in both species.
In the rat, no anatomic pathology findings were observed at low- and mid-dose levels. In the dog, other target organ findings were identified at lower doses in addition to the dose-limiting toxicity. Subsequent 3-month registration-enabling studies were designed according to 3Rs principles. In the registrational rat study, the design was reduced to a vehicle control and 2 dose levels (low- and mid-dose levels utilized in the prior studies). For the rat, no recovery cohort was included due to lack of significant findings at the previously evaluated low- and mid-dose levels.
In the registrational dog study, the design was similarly reduced to a vehicle control and 2 dose levels (low- and mid-dose levels utilized in the prior studies). Two animals/sex (four dogs in total) were added to the high dose group to address reversibility. However, no control dogs were proposed for the recovery cohort as it was considered reasonable to use control data from the main dosing phase for comparison purposes. At the time of manuscript submission, no regulatory feedback was available for these studies.
Exclusion of Control and Treated Recovery Groups Based on Previous Pathology Readouts
Case Study 3 (Large Molecule/Oncology/NHP)
For a large multivalent molecule program in development for oncology, cynomolgus monkey was the only relevant species for nonclinical development. In the DRF/exploratory study, 2 males/group were evaluated at 5 dose levels; no vehicle control group was included in the design. Several target organ findings were noted; however, a maximum tolerated dose was not identified. The 1-month IND-enabling study included a vehicle control group and two IV dose groups, and one high dose SC group (3 animals/sex/group). Recovery animals (2/sex) were added to the high dose IV and SC groups. While two dose groups were reversed, no control animals were included in the recovery cohort as it was considered reasonable to leverage control data from the main dosing phase for comparison purposes in the cynomolgus monkey.
Exclusion of Treated Recovery Groups Based on Published Weight of Evidence and Historical Control Data (HCD)
Case Study 4 (Small Molecule/Non-oncology/Chronic/Dog)
In a 39-week oral gavage dog study with a small molecule for an auto-immune indication, test-item related increased testis weight was observed at the end of the dosing phase. Following a treatment-free period of 6 months, a minimal higher mean testis weight was still noted in high dose recovery animals compared to control recovery animals. In fact, control recovery mean testis weights were lower compared to control mean testis weights at the end of the treatment phase. HCD matching age of the dog showed that mean testis weight of the high dose recovery group was within the normal range for dogs of this age confirming reversibility of the testis weight change. Absence of a control recovery group would not have been detrimental for that study. The IND/CTA was accepted by multiple health authorities with no questions from health authorities on the toxicity study data.
Exclusion of Treated Recovery Groups Based on Weight of Evidence Approach (target biology and internal experience)
Case Study 5 (Small Molecule/Oncology/4-Week/Rat/NHP)
The decision not to include recovery groups in the GLP toxicity studies for the two examples provided by a company was made considering the target biology, prior experience with molecules targeting this pathway, results from DRF toxicity studies, and was consistent with the 3Rs principles.2,4,5
A small molecule selective kinase inhibitor for use in oncology patients was similar (i.e., same chemical series) to another selective kinase inhibitor in Phase 2 for a non-oncology indication. Since a sub-chronic toxicity study was completed with the other molecule and familiarity with the target, the study designs leveraged the previous knowledge to design one month rat and NHP toxicity studies. The vehicle was a well characterized methylcellulose suspension. Recovery groups were not included in either study. The IND was filed and allowed to proceed with no comments on the nonclinical package.
Case Study 6 (Small Molecule/Non-oncology/4-Week/Rat/Dog)
For a small molecule inhibitor of human acyltransferase for treatment of an auto-immune disease, 1-month GLP toxicity studies were conducted in rats and beagle dogs. The rat study (10 animals/sex/group) and dog study (3 animals/sex/group) were administered the test item (3 groups) by oral gavage for 4 weeks. There were no recovery animals used in either the rat or dog study as this was the typical approach for this company for First in Human (FiH) enabling toxicity studies. The CTA was submitted to the MHRA to support FiH studies and could proceed without comments on the non-clinical package.
Case Study 7 (Large Molecule/Oncology/4-Week/NHP)
A large molecule (immune-stimulating antibody) was being developed for oncology with cynomolgus monkeys as the only relevant nonclinical species. Histopathology findings from DRF studies were consistent with immune stimulation and the pharmacology of the target. These findings were expected to be reversible based on previous experience molecules in the same class. Therefore, no recovery arm was included in the IND-enabling 4-week repeat-dose toxicity study. Histopathology findings from the 4-week toxicity study included expected changes due to immune stimulation but also revealed an unexpected toxicity not consistent with the known pharmacology of the target. Although recovery animals had not been included in the study, the pathologist concluded that the finding was both irreversible and non-monitorable due to the target tissue and severity of the changes. Since this adverse finding occurred at all doses, no safety margin could be established for the adverse toxicity noted so the decision was made to terminate the program. The absence of recovery animals in the study had no impact on the program’s termination decision.
Case Study 8 (Large Molecule/Non-oncology/4-Week/NHP)
A large molecule (antibody) was developed for a non-oncology indication with cynomolgus monkeys as the only relevant nonclinical species. No adverse findings were identified in single dose non-GLP studies at doses that were shown to fully saturate the target and provided a robust safety margin over predicted human efficacious doses. Therefore, no recovery animals were included in the 13-week IND-enabling repeat-dose toxicity study and no safety findings were observed at any dose in this GLP study. No questions were received from regulatory authorities regarding the absence of recovery animals during IND/CTA filing and no recovery animals were included in the chronic 26-week study to support longer duration clinical trials.
Case Study 9 (Large Molecule/Non-oncology/13-Week/NHP)
The pharmacology of target inhibition of a large molecule (antibody) for a non-oncology indication was well characterized preclinically and clinically. Vascular toxicities were anticipated but highly tolerable and reversible; therefore, recovery groups were excluded. Studies were carried out by IV administration to rats (2x/week for 4 weeks) and cynomolgus monkeys (1x/week for 13 weeks). The highest dose was the NOAEL in the rat study. In the monkey study, 1 monkey in the low dose group was euthanized at week 11 due to moribundity. Localized clinical signs in this monkey included swelling, exudate, and/or discharge at cephalic vein dose sites and were related to immunogenicity/hypersensitivity and ADA formation. The weight of evidence indicated that the immunogenicity/hypersensitivity response was not considered to have direct relevance to the safety of Phase I patients and the high dose in the monkey study was set as the NOAEL. Experimental recovery demonstration was not considered required in the monkey study, a rationale accepted by the FDA, EMA, and other agencies.
Inclusion of Recovery Groups in Preparation for Request by Health Authorities
Case Study 10 (Small Molecule/Non-oncology/13-Week Rat and Dog)
A small molecule was being developed for a novel target for a chronic indication with no clinical precedent and dose-limiting tolerability in rats and dogs in 1-week DRF studies. A prior related compound demonstrated no dose-limiting findings in the 4-week studies but unanticipated findings in the 13-week studies in both species. Therefore, studies for the new compound were carried out by daily oral dosing to rats and dogs for 13-weeks with a 10-week recovery. Findings in the rat included adverse microscopic findings in the epididymides and testes of male rats which demonstrated reversibility. Findings in the dog included adverse, but monitorable and reversible findings in the liver. The mid-dose for each study was considered the NOAEL. Recovery groups were included in these studies to be prepared to demonstrate the reversibility of unanticipated findings. Findings in each species required demonstrations of reversibility for acceptance of Phase I study protocols by the EMA. Although doses for the planned chronic toxicology studies will be lower than those at which findings were observed in the 13-week studies, recovery groups will be included to demonstrate reversibility of unforeseen chronic toxicity.
General Strategies
General Recovery Practices From Four Working Group Member Companies Based on Modalities (Small and Large Molecule) and Oncology Indication.
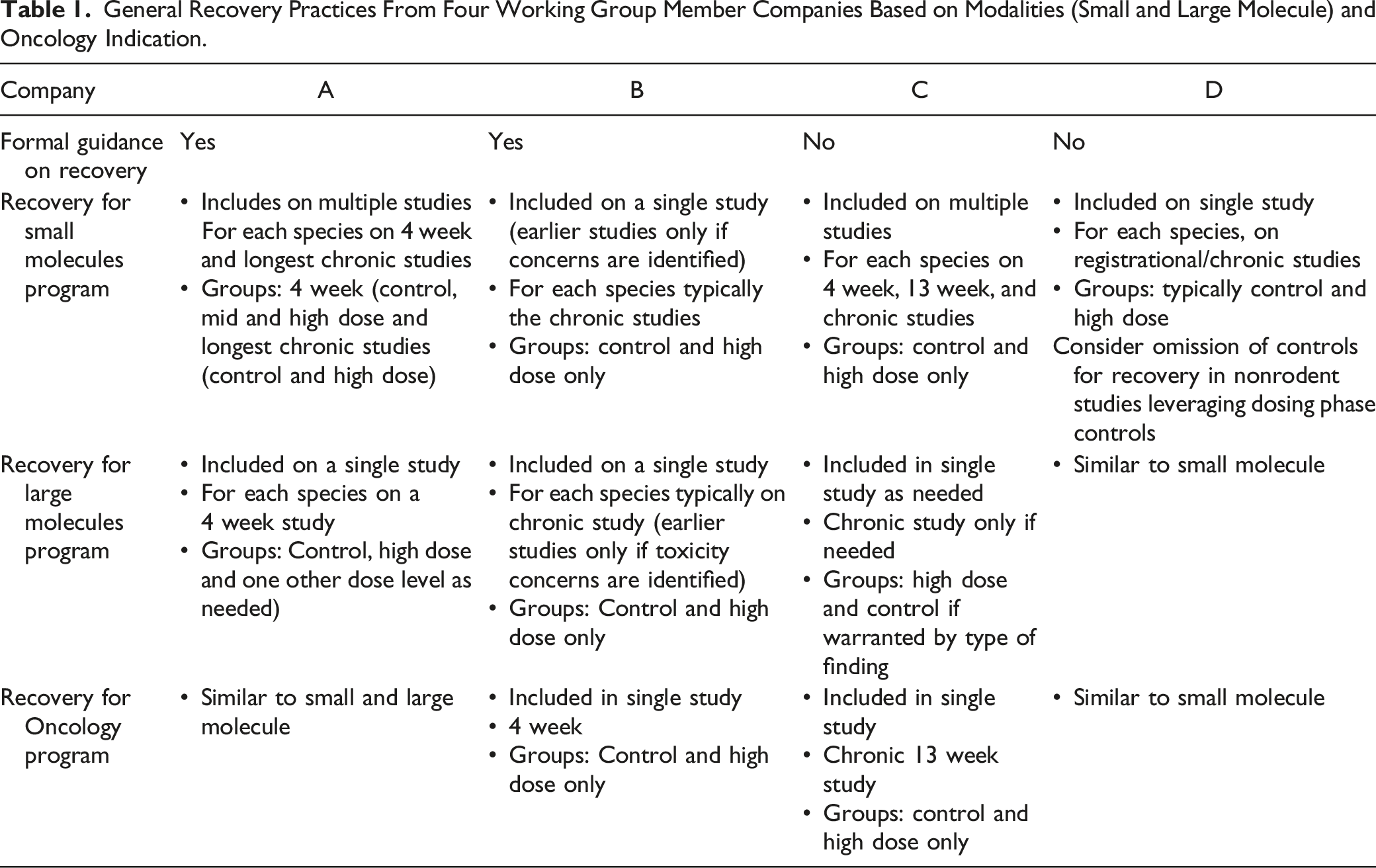
Since recovery investigations seldom impact safe starting dose calculations and are usually focused on dose multiples well above clinically relevant exposures, some organizations consider them unnecessary to support early clinical development. Omission of recovery cohorts from IND-enabling studies has the potential to reduce animal use across the industry and to reduce time to clinical trial initiation. Evaluating recovery on only the registrational study (study of longest duration) ensures that reversibility of any finding emerging late in a program is addressed and is a strategy that can effectively reduce redundant reversibility assessments in shorter studies and reduce recovery animal usage in those programs that fail in earlier in development.
An alternative approach used in another company is to include recovery groups (when appropriate) in IND-enabling repeat-dose toxicity studies, but not include in subsequent sub-chronic or chronic studies unless warranted based on the findings in the IND-enabling studies. This approach can be taken regardless of species, indication, or modality. Additionally, recovery animals would only be included in the IND-enabling studies if warranted based on findings from the non-GLP pilot studies or historical knowledge of the mechanism of action. This approach may also be most appropriate for certain target organs findings that are more straight forward to evaluate in younger animals, for example, the reproduction system changes in female rats which begin to show spontaneous age-related estrus cycle irregularities as early as 5 months of age. 15
Non-terminal Control
For scientific reasons if recovery control groups are included, other novel approaches such as not euthanizing the recovery control animals can and should be considered. A recent survey across 4 major organizations found that in non-rodent nonclinical toxicology studies where a recovery vehicle control group was included and euthanized at the end of the study, euthanasia of vehicle control recovery animals was not necessary for the interpretation of findings in ∼95% (116/122 studies) dog and ∼91% (109/120 studies) NHP studies. 14 In ∼5% dog and ∼9% of NHP studies where euthanasia of vehicle control recovery animals was considered necessary for study interpretation, it was considered to have no impact on the study/program outcome except where historical control data (HCD) were not robust. The survey results further indicate that not euthanizing the vehicle control animals could have saved around 460 dogs and 428 NHPs over 14 years without compromising the overall conclusions. What can we do with the animals that are not euthanized in the study? This question needs thoughtful consideration to have a stronger impact on our 3Rs strategies. Naive non-euthanized animals can be used in training protocols, non-GLP studies, and pharmacokinetic/pharmacodynamic studies. In addition, dogs can be moved to in-house dog adoption programs. While these are some of the common scenarios, the scientific community can devise other novel strategies for utilization of non-euthanized control animals
Working Group Recommendations and Considerations for Recovery Groups
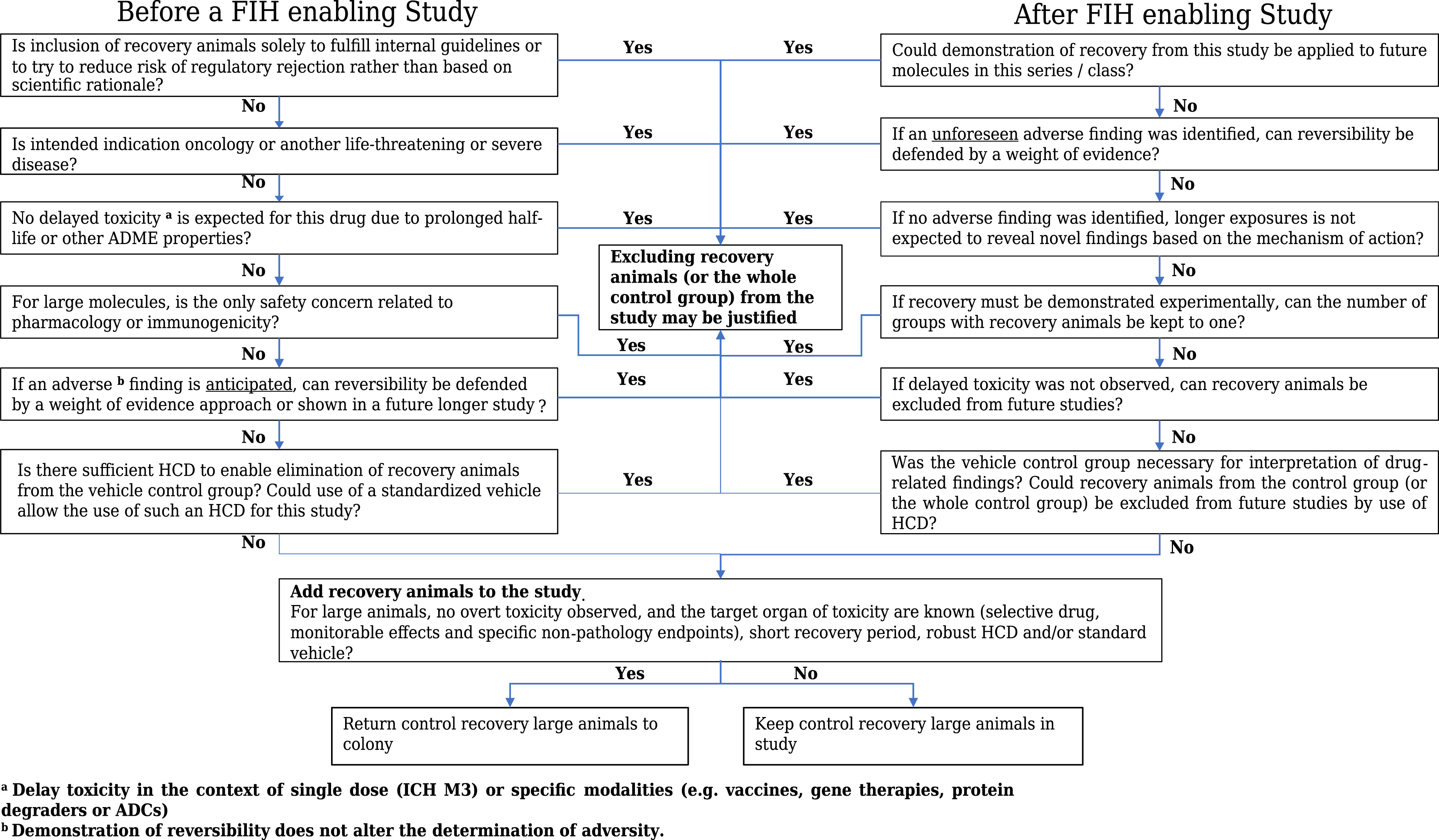
c. When reversibility investigation is justified in non-rodent studies, an emerging approach that is gaining acceptance incorporates 2 animals/sex in a recovery dosing cohort but omits the 2 animals/sex in the recovery control cohort (reduction of 4 dogs or non-human primates per study), instead relying on a combination of pre-study, main phase, HCD, and literature to interpret the limited recovery dataset.
17
d. HCD at the Contract Research Organization (CRO), matching sex, age, origin/strain, and/or study duration and type, can provide organ weight and clinical pathology reference ranges or incidence/severity of a microscopic finding in untreated control animals. In the literature, global review publications provide a general overview on incidence and severity of usual background findings in large animal species.18-22 Multiple additional publications offer a deeper review of findings for a specific tissue/organ system (i.e., Keenan and Vidal 2006 for heart findings of dogs and NHP), vehicle, and/or study type.23,24 Omitting control recovery of non-rodent animals may be a reasonable approach particularly when biomarker(s), HCD, or literature are available to identify and track reversibility of a change documented during the dosing phase. Use of literature and/or HCD is also accepted by HA to characterize and support reversibility. Control recovery groups would only be necessary for unusual vehicles or unique studies such as juvenile or chronic studies (in aged animals) which have little/no HCD and/or literature and where an age drift would be anticipated. Decision tree for determining when to add recovery (or control recovery groups) in a toxicology study.
Conclusion
This manuscript’s purpose has been to review and present opportunities for continuous improvement regarding toxicology study design focused on the inclusion or exclusion of recovery group animals. Although a relatively small subset of companies represented in the IQ Working Group and survey (n = 12 for the survey), the learnings from these examples are valid from a study design and strategy approach. Case study discussions and general strategies can add further value in internal study design discussions and also for sponsors in future by input from contract research organizations who perform a majority of the work and ethical review and have collected the HCD that would be used as part of the determination of whether using animals in recovery groups. The individual companies are accountable for influencing and accepting the risk assessment strategy for the individual projects. Through this information sharing and examples of successful regulatory outcomes with toxicology study designs, this WG intends that we reduce or effectively refine our current recovery practices. Limiting recovery groups to fewer dose levels, use of non-terminal recovery control animals, conduct of first in human enabling studies with no recovery groups, and/or using a weight of evidence and historical control data are approaches currently being explored to assess reversibility with reduced animal usage. The hope and intention are to encourage data driven decisions leading to alternative toxicology study designs. Implementation of 3R in toxicology relies on fewer animals while producing interpretable and acceptable nonclinical safety data that finally supports the safe progression of candidate medicines in the clinic.
Supplemental Material
Supplemental Material - Recovery Animals in Toxicology Studies: An Innovation and Quality Consortium Perspective on Best Practices With Case Study Examples
Supplemental Material for Recovery Animals in Toxicology Studies: An Innovation and Quality Consortium Perspective on Best Practices With Case Study Examples by Smita Salian-Mehta, James D. Smith, Thierry D. Flandre, Amy L. Lambert, Joan H. Lane, Alan H. Stokes, Kathy Orsted, Natalie A. Bratcher-Petersen, Kyathanahalli S. Janardhan, and Elizabeth G. Tonkin in International Journal of Toxicology
Supplemental Material
Supplemental Material - Recovery Animals in Toxicology Studies: An Innovation and Quality Consortium Perspective on Best Practices With Case Study Examples
Supplemental Material for Recovery Animals in Toxicology Studies: An Innovation and Quality Consortium Perspective on Best Practices With Case Study Examples by Smita Salian-Mehta, James D. Smith, Thierry D. Flandre, Amy L. Lambert, Joan H. Lane, Alan H. Stokes, Kathy Orsted, Natalie A. Bratcher-Petersen, Kyathanahalli S. Janardhan, and Elizabeth G. Tonkin in International Journal of Toxicology
Footnotes
Acknowledgements
The author(s) would like to acknowledge the contributions of the following individuals from the 3R recovery WG during the case study discussion, survey drafting and analysis process: Noel Dybdal (Genentech), Marc DeCristofaro (Daiichi Sankyo), Renee Hukkanen (previously employed at Eisai), Binod Jacob and Christine Lynn Lanning (Merck), Stephan Kopytek (Bristol Myers Squibb), Mike Boyle (previously employed at Amgen), Radhakrishna Sura, Fatima Arjmand and Megan Wilichinsky (Gilead Sciences), and Rachel Goldsmith (previously employed at Jansen). Authors would also like to extend thanks to Fatou Sarr (IQ Secretariat) and Alexis Myers (IQ 3R TPS LG) for all their support for anonymizing and compiling survey results and their support during review of this manuscript.
Author Contributions
Salian-Mehta, S. contributed to conception and design and contributed to acquisition, analysis, and interpretation; Smith, J. contributed to conception and design and contributed to analysis and interpretation; Flandre, T. contributed to conception and design and contributed to analysis and interpretation; Lambert, A. contributed to conception and design and contributed to analysis and interpretation; Lane, J. contributed to conception and design and contributed to analysis and interpretation; Stokes, A. contributed to conception and design and contributed to analysis and interpretation; Orsted, K. contributed to conception and design and contributed to analysis and interpretation; Bratcher-Petersen, N. contributed to conception and design and contributed to analysis and interpretation; Janardhan, K. contributed to design and contributed to analysis and interpretation; Tonkin, E. contributed to conception and design and contributed to analysis and interpretation. All authors drafted manuscript, critically revised manuscript, gave final approval, and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: This publication was developed with the support of the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ, ![]() ). IQ is a not-for-profit organization of pharmaceutical and biotechnology companies with a mission of advancing science and technology to augment the capability of member companies to develop transformational solutions that benefit patients, regulators and the broader research and development community.
). IQ is a not-for-profit organization of pharmaceutical and biotechnology companies with a mission of advancing science and technology to augment the capability of member companies to develop transformational solutions that benefit patients, regulators and the broader research and development community.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.