
Abstract
The European Society of Toxicologic Pathology (ESTP) organized a panel of 24 international experts from many fields of toxicologic clinical pathology (e.g., industry, academia, and regulatory) that came together in 2021 to align the use of terminology to convey the importance of clinical pathology findings in preclinical toxicity studies. An additional goal consisted of how to identify important findings in standard and nonstandard clinical pathology associated endpoints. This manuscript summarizes the information and opinions discussed and shared at the ninth ESTP International Expert Workshop, April 5 to 6, 2022. In addition to terminology usage, the workshop considered topics related to the identification and conveyance of the importance of test item-related findings. These topics included sources of variability, comparators, statistics, reporting, correlations to other study data, nonstandard biomarkers, indirect/secondary findings, and an overall weight-of-evidence approach.
Introduction
The European Society of Toxicologic Pathology (ESTP) organized an international expert panel in 2021 to help align clinical pathologists globally on the use of terminology used to convey the importance of clinical pathology findings in preclinical toxicity studies. An additional goal consisted of how to identify important differences (compared with control or pretest results) in standard and nonstandard clinical pathology-associated endpoints. This manuscript summarizes the information and opinions discussed and shared at the 9th ESTP International Expert Workshop, April 5 to 6, 2022.
Twenty-four international experts in toxicologic clinical pathology spanning the pharmaceutical and agro-chemical industries, contract research organizations, and regulatory authorities from Europe, the United States, and Canada, met for 12 preparatory teleconferences and two half-day interactive virtual workshop webinars to discuss how to identify findings in toxicologic clinical pathology and address the discrepant use of terminology describing the importance of these findings.
The workshop focused on providing recommendations on an accepted and consistent approach to data interpretation and reporting of clinical pathology findings. Recommendations exist for consistent microscopic terminology and nomenclature for use in toxicologic anatomic pathology (i.e., INHAND); however, similar recommendations for terminology of clinical pathology findings have not been a focus until more recently. The use of terms such as “biological relevance” and “toxicological relevance” to describe findings in clinical pathology data were discussed at length in the context of industry best practices. Following previous ESTP workshop efforts, this manuscript expands and builds on published literature that addresses some, but not all, commonly used clinical pathology terminology.
The preparatory teleconferences were a combination of individual presentations and group discussions. Topics covered included biologic variation, preanalytical and postanalytical variability, control (including historical) and/or pretest (also referred to as baseline or acclimation) comparisons, statistics, reporting, correlations with anatomic pathology, nonstandard biomarkers (e.g., immunophenotyping and cytokines), indirect (secondary) findings, and a weight-of-evidence approach. The overarching topic in each discussion was the identification, description, and positioning (i.e., adding context) of findings in the data. While the result was not consensus around the definitions of specific terms, there was agreement on ways to improve clarity in the interpretation and reporting of clinical pathology findings that can make a positive impact on the regulatory understanding and acceptance of submission packages. Several other key points were raised and discussed such as (1) de-emphasis of reliance on statistics based on the understanding of the limitations of common statistical approaches, (2) the inevitable contribution of sources of variability such as biological variation (BV), (3) understanding that the lack of correlative microscopic findings does not preclude the importance of a clinical pathology finding, and (4) expertise and/or formal training in clinical pathology is important to navigate data interpretation. The experts in toxicologic clinical pathology aligned on how to identify findings and address the discrepant use of select descriptive terms.
Considerations of Sources of Variability (Biological, Preanalytical, Analytical, and Postanalytical)
Sources of variability are important to consider when interpreting data because their impact could affect the overall interpretations and conclusions. Major variables that must be considered in all studies are those intrinsic to the animal being studied (biological), the conditions of the study or samples collected (preanalytical), the methodology/instrumentation used in the evaluation of the biological samples (analytical), and the reporting of data (postanalytical).
Biological variation refers to the variability in a measured analyte concentration, enzyme activity, cell count, or functional parameter (e.g., clotting time), which is unique to an individual homeostatic or physiologic range. Biological variation often exhibits daily, monthly, and/or seasonal biological rhythms. 12 Since diurnal rhythm can have an impact on the data collected in toxicological studies, the time of sample collection is an important consideration in study design. For example, steroid hormone secretion results in diurnal variation in the number and distribution of peripheral leukocytes; timing of sample collection has an impact on leukocyte counts. 5
Variation is represented by coefficients of variation (CV) calculated from serial measurements from the same animal and includes within-subject (intra) and between-subject (inter) variation (CVi and CVg, respectively). Biological variation can be determined from a cohort of approximately 10 animals/sex, whereas for historical control data (HCD), the recommendation for a reference interval is to have data from a minimum of 20 individuals but preferably more than 120 values for a robust assessment. 1 Alternatively, mean control values from a minimum of 10 studies conducted within the past five years can be tabulated and used as comparators. Recent papers provide recommendations on how to conduct and to evaluate BV studies.1,12,14
The individuality of each endpoint is determined by CVi and CVg. For an endpoint with a high index of individuality (IOI) determined by group variation over individual variation, the analyte has a marked individuality (e.g., low CVi and/or high CVg) which makes a population-based reference interval less useful for data interpretation. A low IOI indicates that the analyte has little individuality, or lower CVg, making reference intervals more useful. 12 For example, serum creatinine has a high IOI, that is most obvious when evaluating data in standard international (SI) units, and population- or group-based reference intervals are not informative compared with sodium which has a low IOI and stronger utility for population-based reference values. Intra-individual variability can be especially challenging in nonrodent species. For example, while adult nonhuman primates have low intra-individual variation for some endpoints, long-term studies starting with juvenile animals can result in maturation-related variation in an individual; similar effects can be identified in long-term studies in rats. Clinical pathology endpoints that are more sensitive to procedure- and age-related effects should be compared to age-matched concurrent controls. 4
Critical difference (CD) is another tool for assessing BV and can identify a meaningful change, regardless of statistical significance. 26 The CD (alternately known as critical change value) is the smallest difference between laboratory values that denotes a true change and it can be calculated. Calculation of the CD requires serial monitoring of an endpoint. One of the workshop experts provided personal experience with the CD for urinary albumin in male rats, which was determined to be an increase of 186% or 2.86-fold; this is notably larger than the values that would be statistically significant in a small toxicology study population and would prevent overcalling the result. While pretest and/or concurrent control data are most relevant for study interpretation, consideration of the role of BV on an endpoint or test system adds value for determination of the importance of a change.
While BV is inherent in all studies and difficult to control for, this is not true for preanalytical, analytical, and postanalytical variability. Key features of preanalytical variability in preclinical studies often include study design challenges and many can be alleviated by close scrutiny of study plans prior to study initiation. Prior to study start is the best time for many potential issues to be addressed, thus minimizing unwanted variability. Such factors that contribute to variability include, but are not limited to, animal selection, husbandry, fasting status, restraint/sedation/anesthesia, blood collection site, collection tube type, sample volume, timing of sample collection, sample collection and analysis order, and specimen processing. Preanalytical variability in toxicity studies has often a higher impact than analytical variability in routine diagnostic clinical pathology data, but analytical variability may be important for special parameters like hormones or biomarkers. Postanalytical variability also deserves mention but occurs infrequently and typically involves operator or transcription errors. These types of variability and their implications on sample analysis and clinical pathology data have been described in the literature and the reader is encouraged to refer to these manuscripts for a more in-depth review.2,3,10,22,23,38-42,47,48
Considerations for Comparators for Clinical Pathology Data
Comparators are used in toxicological clinical pathology to identify differences or effects in data sets. Appropriate comparisons are fundamental to understanding and assessing clinical pathology data in toxicologic studies and are described in several manuscripts.3,4,21,47
For rodent studies, comparisons with group means are most commonly used along with close evaluation of individual results. Rodent studies tend to have larger group sizes and animals that have lower age and genetic variability. Treated group mean comparisons are reported as higher or lower than control group means with exception when there are only a few animals per group or when describing unique changes in individual animals. In addition, comparison to concurrent control group mean can be important in studies with more than one timepoint/clinical pathology interval because of age-related effects during long-term studies. Pretest/baseline/acclimation data are often not collected in rodent studies because of the rapid growth of these animals during the study and small size (small blood volume) prior to and at the time of study start.
Comparisons of large animal data (e.g., nonhuman primate, dog, pig, and rabbit) are typically made with individual pretest data and are reported as increased or decreased from pretest. Large animals have greater genetic variability compared with rodents and typically have smaller numbers per group. In large animal studies with multiple pretest values to assess overall animal health, comparisons should be made with the value closest to the initiation of dosing. Averaging pretest values from several pretest collections is not recommended because the effects of stress (e.g., transit, early acclimation) are more pronounced earlier in the acclimation phase.4,11,42 In addition, comparison with concurrent control values in large animal studies should be used to help identify when and if there are procedure- or age-related effects (e.g., multiple blood collections, handling and restraint, anesthesia, or surgical procedures), which are more relevant in long-term studies for dogs versus nonhuman primates). Moreover, control animal results are useful to understand the dynamic range for a given endpoint in a given species, which includes the types of variability presented above, and in long-term studies, the comparison to pretest values becomes less relevant, and comparison to control becomes more appropriate.
Comparisons are often expressed using qualitative and/or quantitative descriptors with a consistent approach described in the methods section of the report. When quantitative descriptors are used, percent differences or fold change (ratios) are most commonly used but are not recommended for routine use in toxicological pathology clinical pathology reports to avoid misinterpretation. 4 Total variation of each parameter should be used for the evaluation of these comparators, and often this is based on subject-matter expert experience. When qualitative terms are used, the grades are most often similar to histopathology grades (e.g., minimal, mild, moderate, or marked). Considerations about the subjectivity and consistency of these descriptors were discussed by the group, but outlining usage guidelines was not an objective of the workshop. Additional discussion on this topic can be found in the paper by Aulbach et al. 4
Considerations for HCD and Reference Intervals
Historical control data or reference intervals are useful as a guideline for reviewing preclinical clinical pathology data but should not be used alone to interpret these study data and do not replace a systematic approach for comparing with control and/or baseline data. 4 Historical control data and reference intervals are terms commonly used interchangeably; however, it should be noted that these terms can be defined as HCD “being data from study control animals that may have received sham procedures, vehicle control article, or treatments,” whereas reference intervals “represent data from animals not on study (naïve population without procedure or treatment).” An objective approach for establishing reference intervals consists of a well-documented procedure including statistical methods for outlier detection.1,17,18,25,28,34 Historical control data can be established with values of control group individuals (e.g., for nonhuman primate or dog studies) or with control group mean or median values (for rodent studies), depending on the evaluation of the study data. For a robust and representative reference interval, the literature describes tabulating a minimum of 120 individual values (with a minimum of 20 individual values needed to create a valid reference interval) within a five-year period with regular review updates.1,17,18,25,28 Historical control data should be based on group mean data at least 10 studies (with 10 individual control data points) for establishing a robust HCD; reporting of HCD should be standardized with a set of information on metadata.6,54
Historical control data and reference intervals are considered relevant to the facility and instruments from which they were generated. They should be collected separately for each study type (e.g., regarding animal strain, study duration, administration route, blood sampling site, fasting status). Even if study design, preanalytical and analytical methods, and breeder/supplier are similar between facilities, reference intervals from other facilities are not recommended or appropriate to use or make toxicologic interpretations with. Similarly, published textbook data can be informative when reviewing data for nonstandard endpoints but cannot be used for justification of BV or test item effects within the context of interpreting toxicologic clinical pathology study data.
Recently, several groups have begun discussing study designs to decrease the number of control animals used or using virtual control groups to replace concurrent live animal controls. These discussions revolve around the 3Rs and decreasing animal numbers used in preclinical studies. Although beyond the scope of our workshop webinar, this is actively being discussed in the industry (including by several authors on this manuscript). Currently, the use of virtual control group animal data is purely ideological and will need thorough evaluation of the implications and repercussions of use in preclinical toxicology testing. At the time of the preparation of this manuscript, regulatory consideration and acceptance of derived clinical pathology data as virtual control groups is still futuristic and does not offer clear scientific benefit.
Considerations and Limitations of Statistical Significance Testing
The relevance of clinical pathology findings for overall study interpretation should not be based solely on the results of statistical significance testing.4,20,21 Clinical pathology data are prone to misinterpretation due to the easy accessibility of numeric data, high sensitivity to detect subtle differences, tendency for overconfidence in reported results, and overreliance on statistical significance testing without consideration for preanalytical and analytical variability, statistical/computational model limitations, and intercurrent physiologic factors that impact results. 20
Statistical significance testing is one of many tools available to assess clinical pathology data. As with all tools, statistics should be employed with an understanding of its advantages and more importantly, its limitations. For all studies, the statistical model used should be clear in the study plan, methods, tables, and reporting for each study. Statistical significance testing determines the probability (p value) that a difference between groups occurred by chance.31,44 P value cutoffs (e.g., <.05) may highlight differences that have a low probability of random occurrence, but also commonly produce type I and II errors (false positives and false negatives, respectively). In addition, they may produce incorrect conclusions when statistical model assumptions are not met. Statistical model assumptions that are commonly not met include (1) appropriate group size, (2) appropriate data type, (3) independent random sampling, (4) Gaussian distribution, and (5) homogeneity of variance.15,40 In addition, statistical significance testing as routinely applied in nonclinical toxicity studies (typically limited to a comparison of treated groups with concurrent controls) does not assess changes over time and patterns of change, which form the foundations of the weight-of-evidence approach required for interpretation. For these reasons, P value cutoffs should not be relied upon as decision thresholds for clinical pathology interpretation, and P values should be interpreted with consideration of expected signal-to-noise ratios unique to each endpoint. Statistical significance does not indicate a test item-related effect nor does a lack of statistical significance indicate a lack of test item-related effect.
In line with a weight of evidence approach, a wide variety of tools should be leveraged to assess the data in different ways. Beyond comparing values within and between individual animals and groups for one endpoint at a time, data comparisons should enable a holistic assessment of physiologically meaningful relationships within the whole data set of individual animals and the entire study groups. New statistical, visualization, and computational methods assist the clinical pathologist to help identify patterns that support prioritization of and to put the findings into context relative to background biological variability and relative to other study data. 49
Considerations for Clinical Pathology Effects Without Associated Anatomic Pathology Findings
The distinction between clinical pathology data and anatomic pathology is important because in general, clinical pathology endpoints are not limited to specific organ-based effects and are more sensitive to whole body physiologic alterations such as changes in nutrient intake (e.g. short and long-term alterations in food consumption), physical (skeletal muscle) activity, hydration status, and acid/base parameters. In addition, clinical pathology endpoints give insight into the function or injury of a system/organ while histopathology gives insight into the morphology of an organ. Effects on tissue morphology can be reflected in microscopic changes in an organ with associated clinical pathology changes, alterations in organ function might be reflected in clinical pathology changes but not associated microscopic changes in the affected organ. Associations between clinical and anatomic pathology data help to characterize the pathogenesis of test item–related effects, contribute to the weight of evidence for a causal relationship to the test item, and help to position and characterize adversity, or lack thereof, of test item-related effects in the context of the main study report. Some associations or correlations identified between clinical and anatomic pathology findings (e.g., the relationship between increased serum hepatocellular enzyme activities with hepatocellular necrosis or the relationship between reduced circulating red and/or white cells with decreased marrow cellularity) are clear and easy to identify and describe to toxicologists and regulators. However, many clinical pathology effects are not clearly associated with light microscopic observations, which do not negate the importance of a clinical pathology finding. A lack of clear or direct relationship or correlation adds an additional challenge for clinical pathologists in the interpretation and reporting of clinical pathology effects in preclinical studies and in the overall translatability and risk assessment for clinical trials. 43
The term “correlation” is often used when describing associations of clinical pathology and light microscopy (anatomic pathology) findings as part of conventional or accepted use of the terminology. It should be noted that some definitions restrict “correlate” to statistical correlation coefficients. Associated findings in study data sets are commonly described as “correlating” in preclinical toxicological pathology reports and alternate descriptions such as “concordant” findings may be used but can convey a weaker association of findings. The use of “correlate” came under some discussion and while by some definitions, “correlation” implies the use of a specific statistical method, in the context this workshop and manuscript the term “correlate” was applied to highlight interrelationships between clinical pathology and other study endpoints to allow flexibility in this terminology. If in report writing, “correlate” is used to imply “statistical correlate,” the exact statistical test(s) used should be outlined.
A higher level of concordance is expected at higher dose levels, when there is a high responder rate, when the effects are predictable and direct, and when there is homogeneity in the animal population being tested (e.g., rodents) that provides a consistent individual susceptibility. 38 A high level of concordance is often not present in toxicity studies involving outbred species (e.g., dog, nonhuman primate), when there is a multiphasic dose-response rather than a dose-proportional one and at low doses when there is a threshold effect. 38
Discordance between clinical and anatomic pathology findings occurs due to a variety of factors and biological mechanisms. The nature of clinical pathology data (e.g., numerical, systemic, sensitive, premonitory) versus anatomic pathology data (e.g., observational, tissue-specific, variables based on sampling) represents a known and somewhat predictable source of discordance encountered in routine toxicity studies. 38 In addition, the timing of clinical pathology endpoints compared with anatomic pathology endpoints can contribute to the lack of concordance. For example, transient clinical pathology differences identified during the in-life phase but not at necropsy, or clinical pathology differences that precede a microscopic lesion (e.g., increased cardiac troponin concentration in isoproterenol-treated rats can be identified as early as 1 hour post dose which precedes a light microscopic lesion in the heart). 55 Notably, routine light microscopy has inherent limitations in sensitivity as compared to ultrastructural (electron) microscopy. Therefore, differences in clinical pathology endpoints can reflect ultrastructural changes not readily identified by routine histopathological evaluation. Physiologic responses can also impact clinical pathology results but may lack corresponding microscopic findings. Examples include iatrogenic blood loss due to multiple blood draws resulting in decreased peripheral red cell mass and responsive increased reticulocyte counts, without an appreciable effect on but unremarkable marrow histology, and increased circulating peripheral neutrophil count associated with excitement and release of epinephrine found secondary to a temporary shift in marginated neutrophils rather than to an actual change in production of neutrophils or change in the tissues themselves. 22 In addition, discordance between hematology effects and microscopic findings in bone marrow may also occur because the microscopic evaluation of tissue sections is a relatively late procedure in a study (i.e., at the end of the dosing and recovery phases). Bone marrow sections are a relatively subjective and insensitive indicator of perturbed hematopoiesis when compared with the more sensitive hematology data. It is also worth considering the sensitivity of clinical pathology endpoints in the identification of minor changes in enzyme metabolism and transporter function that may not have concordance with or correlating light microscopic findings in specific organs.
An often-confounding difference during the in-life phase or at terminal clinical pathology collections is decreased transaminase activities which can at times reach statistical significance compared with controls and cause much discussion in the context of importance to the overall study conclusions. An in-depth description of the considerations of decreased transaminase activities without correlating or corresponding anatomic pathology findings is presented in Example Box 1.
In-depth example of decreased transaminase activities without associated or correlating anatomic pathology findings.

Considerations for Assessment and Identification of Indirect Clinical Pathology Findings
Direct or primary effects of a test item are defined as an interaction between the test item (or metabolite) and the target organ or cell population, whereas an indirect or secondary effect is considered to be one which does not involve a primary test item-target cell interaction.10,35 In preclinical toxicity studies, examples of indirect or secondary test item-related effects related to stress, decreased food consumption, and/or body weight loss are frequently observed in clinical pathology data. These effects can be a consequence of body fluid loss (e.g., via emesis or diarrhea) or related to test item administration (e.g., inflammation secondary to subcutaneous administration or indwelling [intravenous] IV catheter). Another example of a secondary effect on an off-target tissue is the induction of hepatocyte drug-metabolizing enzymes resulting in increased turnover and clearance of thyroid hormone resulting in secondary thyroid hypertrophy and hyperplasia due to stimulation of the pituitary–thyroid–endocrine axis.19,56 However, it can be difficult to distinguish these effects from direct or primary effects, confounding data interpretation. In addition, primary and secondary effects contribute to the overall weight of evidence of adversity. 35
The effects of decreased food consumption or feed restriction on hematology and clinical chemistry endpoints have been extensively described in rats and less frequently in other laboratory species (e.g., beagles).24,30,32 After just 2 weeks, decreased food intake in rats is reported to be associated with bone marrow myelosuppression, as well as with effects on clinical chemistry results (e.g., increased serum urea nitrogen and bilirubin concentrations, and decreased creatinine and cholesterol concentrations). 32
Stress secondary to study-related procedures may affect total body weight, food consumption, and activity of animals which can impact multiple organ systems and are comprehensively described by Everds et al. 11 Findings in clinical pathology data that are typically attributed to a stress response include changes in circulating absolute leukocyte counts (e.g., increased neutrophil count with decreased lymphocyte and eosinophil counts). Findings in other endpoints such as glucose, acute phase proteins, or enzyme activities can also be altered secondary to a stress response.
The expert panel agreed that indirect or secondary effects could also be relevant/significant, depending on severity and downstream effects induced, and should be identified along with the clinical observations and/or findings and/or histopathological findings to which the effects are attributed. Presentation of a plausible hypothesis connecting primary and secondary effects is needed to properly interpret the data in the context of the study.
Considerations for Value and Limitations of Novel, Nonconventional, or Exploratory Biomarkers in Nonclinical Studies
In preclinical drug development, biomarkers play a critical role in successfully developing a therapeutic drug or medical device. The U.S. Food and Drug Administration (U.S. FDA) and the National Institutes of Health (NIH) as part of their joint Biomarkers, EndpointS, and other Tools (BEST) resource have defined different classes of biomarkers in the context of their respective uses in patient care, clinical research, or therapeutic development. They proposed the basic definition of a biomarker as “a defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes or responses to an exposure or intervention” which in essence incorporates all clinical pathology endpoints. 13 In the context of toxicity studies, safety biomarkers are typically being used and are defined as “a biomarker measured before and/or after an exposure to a medical intervention or environmental agent to indicate the likelihood, presence, or extent of a toxicity [. . .].” 13 The expert panel discussions focused on soluble protein biomarkers assessed by immunoassays in body fluids.
Within the category of safety biomarkers, one can distinguish further subclassifications according to the level of utilization and underlying science. Clinical pathology endpoints established over decades and now part of routine preclinical toxicology investigations are commonly referred to as traditional or conventional safety biomarkers. More recently developed biomarkers, of which only a limited number have been formally qualified for application within a specific context-of-use, are referred to as novel or nonconventional safety biomarkers. The latter are often used in an exploratory manner, meaning limited data and scientific evidence are available for their particular use and testing methods, and applications are still under development. Compared to the use of one single-safety biomarker as a standalone marker, the use of a panel of novel biomarkers reflecting various mechanisms of toxicity or physiologic responses may have the greatest promise of utility. Regardless of whether a biomarker is considered conventional or nonconventional, all data generated on Good Laboratory Practice (GLP) studies should be included in the data package according to the protocol language.
Several reasons exist to include novel safety biomarkers in preclinical studies. Early detection of undesired effects by use of novel biomarkers can aid in the mitigation and/or determining mechanism of toxicity, and aid in selection of candidate test items. Novel biomarkers add value when they are more sensitive or more specific in the detection of organ, metabolic, physiologic, or target effects or responses than conventional endpoints, and may also show additional value to assess reversibility, or for translational monitoring along the preclinical and clinical development program.
In contrast to the well-established and highly standardized conventional biomarker assays, commercially available nonconventional novel safety biomarkers often have different levels of method qualification performed by manufacturers. There are often differences across kits and/or lots in specificity, sensitivity, units, dynamic range, or different antibodies for the same or different epitopes used with different immunoassays. Together, these factors may impact the comparability of data generated and data interpretation between assays and across studies.
Biomarker panels on multiplex platforms are more common than single biomarker assays, and this adds a layer of complexity because each individual assay may not be optimized in a panel. Often the manufacturer uses either buffer or matrix samples spiked with recombinant stabilized protein to generate validation samples. The behavior of appropriate matrix samples with endogenous levels of the protein of interest can differ substantially due to the presence of matrix interference, binding proteins, variable degradation, and/or a different 3-dimensional conformation thereby influencing the accessibility of the epitope for the antibody to bind. In view of these aspects, it is considered critical for each laboratory to invest time and resources in the internal evaluation/validation of a biomarker assay using samples with adequate endogenous analyte levels whenever possible prior to study application. Once an assay or assay panel has been found suitable, it is strongly recommended to use the same method across studies for a given program to drive consistency; this may necessitate method transfer to another facility (e.g., Contract Research Organization [CRO]) as many industries outsource studies at some point in drug development. Next to the technical and analytical biomarker assay aspects, the knowledge of the biology and variability of the endogenous analytes tested as safety biomarkers and the specific context of use is key when designing a study. Therefore, the biomarker scientist/clinical pathologist should be involved in study design and is best placed to provide input on optimal sample collection timepoints, matrix, sample processing, and storage.
Published data for validation of novel biomarkers are mostly derived from studies using a tool molecule or commercial agent(s) at dose(s) which cause clear findings of degree of injury or altered physiologic response that often exceed the level of tissue injury encountered in preclinical safety studies. For example, agents which produce severe drug-induced kidney injury (e.g., cisplatin and gentamicin) have been used by many researchers.16,50,51 However, changes in novel biomarker data in nonclinical safety studies are not always as clear compared to published data. During the implementation of novel biomarkers with nonstandardized test methods in preclinical toxicity studies, it is essential that the experience with these biomarkers within a company or laboratory includes an in depth understanding of biological and analytical variables unique to that assay or test system for confidence in data interpretation. Concordance of novel biomarker changes with other study findings including traditional clinical pathology endpoints further accurate novel biomarker data interpretation.
Considerations for Assessment of Immunophenotyping and Cytokine Data
Immunophenotyping refers to the identification and characterization (e.g., activation state, functional endpoints) of immune cells using labeled antibodies (Abs) directed against specific markers on the surface or interior of specific cells. In a typical clinical pathology setting, the techniques employed are flow cytometry and immunocytochemistry. The expert panel focused on immunophenotyping via flow cytometry.
There are certain limitations inherent in analyzing and interpreting immunophenotyping data derived during toxicity studies. Typically, measurements are conducted using peripheral blood. However, peripheral blood findings may not accurately reflect the immune status. Factors which influence circulating immune cells include circadian rhythm, stress response, and redistribution due to homing mechanisms.8,22,36 Furthermore, not all immune cell phenotypes circulate in large enough numbers to enable measurement in peripheral blood and many findings may not have cross-species relevance. In addition, the number of circulating immune cells does not necessarily imply their functionality. The proportion of lymphocytes, lymphocyte subsets, and other leukocytes varies between laboratory animal species and humans. In addition, the response to a given stimulus may vary between species. For example, in peripheral blood, neutrophils predominate in humans, nonhuman primates, and dogs, whereas lymphocytes predominate in rodents. An inflammatory response in a neutrophil-rich species can often present as an increased absolute neutrophil count, whereas inflammation in a lymphocyte-rich species (e.g., rodents) typically manifests as an increased absolute lymphocyte count. 16
Immunophenotyping should not be a routine assessment for every toxicology study or program. The International Conference on Harmonization S8 regulatory guidance (ICH S8) describes standard endpoints in toxicity studies that should be employed to identify unintended immunotoxicity and additional assays only conducted if there is cause for concern. 7 The assays selected should be based on a specific concern and may include immunophenotyping. Triggers for the conduct of additional studies include the mechanism of action of the drug, known biology of the target or structural alerts, the intended patient population and/or drug indication, and findings in toxicity studies or clinical trials which suggest an impact on the immune system. 7
A typical panel would include T cells (often subdivided into CD4+ and CD8+ T cells), B cells, and natural killer (NK) cells. When driven by specific questions, other (sub)populations may be evaluated (e.g., monocyte subsets, plasma cells, NK cell subsets, double positive T cells). Moreover, cell activation or other functional states may be evaluated under certain circumstances, but the absolute numbers of cells in peripheral blood expressing the markers (e.g., CD69 in activated T cells) may be quite low. In this case, the T cells expressing CD69 should be evaluated as a percentage of total T cells and not total lymphocytes.
To deliver valid data, panels of labeled antibodies should be qualified as fit-for-purpose for the species and the instrument used. This qualification should include binding of reagents, choice of “stains” (organic fluorophores and synthetic dyes), presence of the marker on the cell of interest, optimum concentrations, inter/intra-assay repeatability, sensitivity, and sample and reagent stability.
Cytokines are mediators of immune effects produced by many cell types; they induce effects by binding to specific receptors on effector/suppressor cells. Most of their release and activity is local in the tissue and may not be reflected in peripheral blood. However, drugs acting on immune cells can trigger systemic release with potentially life-threatening consequences.9,45
Cytokines may be measured by enzyme-linked immunosorbent assay (ELISA) or multiplex methods. There are commercial kits available, but due to species differences in cytokine structure, and in kit methods and reagents even for the same species, kits are not directly interchangeable. Cytokine assessment should not be performed routinely. It can be added as endpoint of a toxicity study if systemic cytokine response is part of expected pharmacology to determine the relevance of the toxicology species and to assess activity and provide potential biomarkers for activity. If an in vitro cytokine release assay is positive, measuring blood cytokine levels in toxicity studies may confirm this potential safety liability.
Depending on the expected target(s) and mechanism of action of a given test item, the general recommendation is to measure a panel of cytokines (e.g., IFNɣ, interleukin [IL]-2, IL-10, tumor necrosis factor alpha [TNFα], and IL-6), rather than a single cytokine. Collecting multiple timepoints within a 72-hour period after dosing may be critical to capture the full-time course of events; when cytokine release occurs, cytokines themselves stimulate release of other cytokines which enhance and/or dampen the effects. A single timepoint in the course of a cytokine response will provide an incomplete and potentially misleading picture. It is important to note that, due to the dynamic nature of cytokine release and short half-lives in circulation, cytokine data should not be expected to necessarily correlate with pharmacokinetic data or histopathology findings.
Specific considerations are needed for study design, interpretation, and reporting of immunophenotyping and cytokine data. When there are endpoints added to monitor specific immune function or immunotoxicity, clinical pathologists and immunologists and/or immunotoxicologists should work together closely on study design, particularly endpoint selection and blood collection timepoints, and when interpreting study results. Ideally, the immunophenotyping and cytokine report(s) should be incorporated into or integrated with the clinical pathology report. Usually, the data are used for determining proof of concept, mechanism of action, or extent of exaggerated pharmacology, rather than unanticipated toxicity. It is very important to have enough animals to identify “real” effects, as most of these endpoints have high interindividual variability in values, and there is little data in the literature regarding dynamic ranges or the prevalence of more uncommon immunophenotyping markers in mice or humans, rats, dogs, nonhuman primates, or minipigs. Pretest cytokine concentrations in peripheral blood are low or even below the limit of detection, but non-drug-related factors such as stress may result in measurable levels. For both immunophenotyping and cytokine assessments, it is important to obtain pretest data (ideally two or more samplings) or at a minimum concurrent control data, and to determine normal variability within individual animals, particularly if the group size is small. In the case of more than one pretest sampling, the result closest to the initiation of dosing is recommended to be used as the comparator and for calculations (as noted previously, averaging pretest results is not recommended).
The data should be interpreted in light of the complete blood cell count (CBC) results and other study data (e.g., potential for stress responses, clinical signs of infection or for cytokine release and histopathological findings); there should also be an understanding of sample quality, which can greatly affect results, particularly in mice. For immunophenotyping, absolute numbers (e.g., not percentages of total lymphocytes) should be used to interpret and express the data for lymphocyte subtypes. However, within a given major lymphocyte subtype, the use of percentages of that subtype to describe activation or other markers which may seem insignificant when described in absolute numbers is recommended. For example, the percentage of activated CD4+ cells or the percentage of a cell subtype expressing a marker for apoptosis is more meaningful than the absolute value of cells expressing activation or apoptosis markers. Reference to specific cell subtypes should be as clear as possible and may include a description of the presence and/or absence of the marker (e.g., CD4+ and CD8+ T cells) rather than using older terminology (e.g. “helper” or “killer” T cells). If descriptive terms are used, they should be described in detail in the study plan and report.
When values for cytokines or specific cell subtypes are below the limit of detection in pretest and controls, it is not possible to express treatment group data as percent change or ratios compared with pretest or controls. When this is the case, the presence of these cytokines or cells is described as the finding. Furthermore, it is not advisable to compare results across studies due to differences between instruments, platforms, and reagents. In general, immunophenotyping and cytokine data should not be used to assign adversity because there are no established criteria to do so. Exceptions would be complete depletion of a very well characterized and essential cell type or part of a weight of evidence review with corroborative data (e.g. infection, clinical evidence of cytokine release syndrome or cytokine storm, histopathologic findings).
In summary, immunophenotyping and cytokine analysis are not part of routine toxicity testing but should be considered based on biology, pharmacology, and/or previous findings. The need for immunophenotyping and cytokine measurements should be determined in advance to ensure appropriate endpoints are selected and sample collection and processing guidelines are optimal. Study design must account for stability of cells and ensure appropriate validation of reagents and methods for the given species. The study design strategy should be tailored to the molecule and the scientific objectives. While cytokine measurement can be done retrospectively using samples stored frozen, the data are more useful when planned prospectively and appropriate collection timepoints are incorporated into the study design. It is extremely important to understand species differences in terms of biology, dynamic range, and relevance of animal species for desired pharmacology and/or safety, reagent applicability, data interpretation, and human risk assessment.
Considerations for Reporting and Positioning Test Item-Related Clinical Pathology Findings
Format and style of clinical pathology reports can contribute considerably to clearly conveying the importance of clinical pathology findings. However, the approaches to clinical pathology reporting are variable amongst the different individuals and organizations due to different training and certification requirements and internal company policies. Clinical pathology report style is described in the literature and regardless of format, the report should prioritize clarity, accuracy, and consistent wording. 4 Ideally, a trained clinical pathologist should write or review all GLP and non-GLP clinical pathology contributions. The most common approach for GLP studies is an independent clinical pathology report authored by a trained clinical pathologist appended to the main study report. Non-GLP studies can use an independent report (similar to a GLP report) or use a clinical pathology report summary that is integrated into the main study report or combined with the anatomic pathology report.
Aspects discussed by the expert panel members agree with current literature recommendations for clinical pathology reporting.3,4,38,47
Preference is for a scientist with formal education and qualification in clinical pathology to interpret the data and write the report or the corresponding summary section in the study report.
Standard vocabulary/terminology should be used, avoiding diagnostic terms (i.e., decreased red cell mass instead of anemia, higher absolute neutrophil count instead of neutrophilia).
Stand-alone reports should follow a common format (e.g., table of contents, objectives, summary, material and methods, results, and discussion/conclusion).
Clinical pathology findings should be correlated with relevant study data, including in-life and microscopic findings, when possible, to provide context.
Critical and pertinent background information should be available to the clinical pathologist (e.g., test item type and indication, results of any previous studies, in life findings, cause of any early deaths, toxicokinetic data, and anatomic pathology). Active communication between the clinical and anatomic pathologists assigned to the study is important for an integrated interpretation of the study clinical pathology data for inclusion in the individual contributor report and/or the main toxicology report.
Clinical pathology reports provided by CROs should undergo a comprehensive review by the sponsor, similar to that done for other contributor reports (e.g., anatomic pathology report), but this is not a regulatory requirement.
Considerations of Terminology and How to Convey the Importance of Test Item-Related Clinical Pathology Findings
The overarching intent of the expert panel discussions and workshop was to add structure to the process of conveying the importance of clinical pathology findings in toxicology studies using accepted and understood terminology. During the discussions, it was quickly identified that no one formula or method can be applied to all descriptive clinical pathology data interpretations, and an informal or formal rubric may be used by each individual depending on training and experience. Alternative descriptive approaches were discussed, especially the use of the terms “toxicological relevance” or “biological relevance.” The use of these terms was variable and inconsistent amongst the expert panel. Attempts were made through several in-depth discussions to align their use, but the expert panel did not achieve consensus on definitions of these terms and therefore did not endorse their use.
An initial poll of the 24-member expert panel indicated approximately 1/3 of the group used “biological relevance” and approximately 2/3 used “toxicological relevance” when describing a wide range of differences in clinical pathology data. In the companies that use these terms, perceived alignment on the use was only noted in about 1/2 of the group. It was determined that the terms are inconsistently used in reports and variably included in the summary and/or conclusion statements (statements which are often included in regulatory submission documents). In some instances, these terms were used to describe or infer perceived adversity. Although several expert panel members reported using these terms in the positive context, they were more typically used in the negative context (i.e., “not biologically relevant” or “not toxicologically relevant”). This inconsistency within the expert panel underscored the importance of the workshop and highlighted the need for more consistent and clear descriptive terminology to illustrate the importance and interrelationships of clinical pathology findings.
To better understand the use of these terms in the larger clinical pathology community, a survey of the webinar participants was conducted at the beginning of the workshop webinar and a similar survey was conducted at the end of the webinar to assess impact of the discussions (Figure 1). Consistent with the expert group, the results indicated that many of the webinar participants use “toxicological relevance” and “biological relevance,” yet when asked to align on defining the terms, consensus was not achieved. A wide variety of opinions were expressed, and close to 50% of participants preworkshop would consider using the terms if better defined and a similar percent were undecided on their use postworkshop. Within the workshop discussions, it was identified that similar to the expert panel, most of the participants use the terms more commonly in the negative context (e.g., “not biologically relevant” or “not toxicologically relevant”). The suggestion that “toxicological relevance” implied adversity of a clinical pathology finding was discounted during the discussion because a clinical pathology finding is most often not adverse in isolation and in most cases, adversity should be discussed in the main study report in the context of all relevant study data or in the regulatory filing in the context of all relevant data across studies. 39 Shifts were noted in individual acceptance or avoidance of these terms over the course of the workshop webinar (Figure 1). These terms generated much discussion, and several possible algorithms were developed; one such approach was used as an illustrative decision tree example to aid in the interpretation of toxicologic clinical pathology data (Figure 2). Even the example algorithm presented in Figure 2 sparked lively discussion amongst the panel and participants. There was consensus that any use of the terms “toxicological relevance” or “biological relevance” would need additional description(s) or context in the narrative of the report. When these terms are used in isolation, they lack clear meaning because use is based on individual perception of meaning and a unified definition was not achieved.
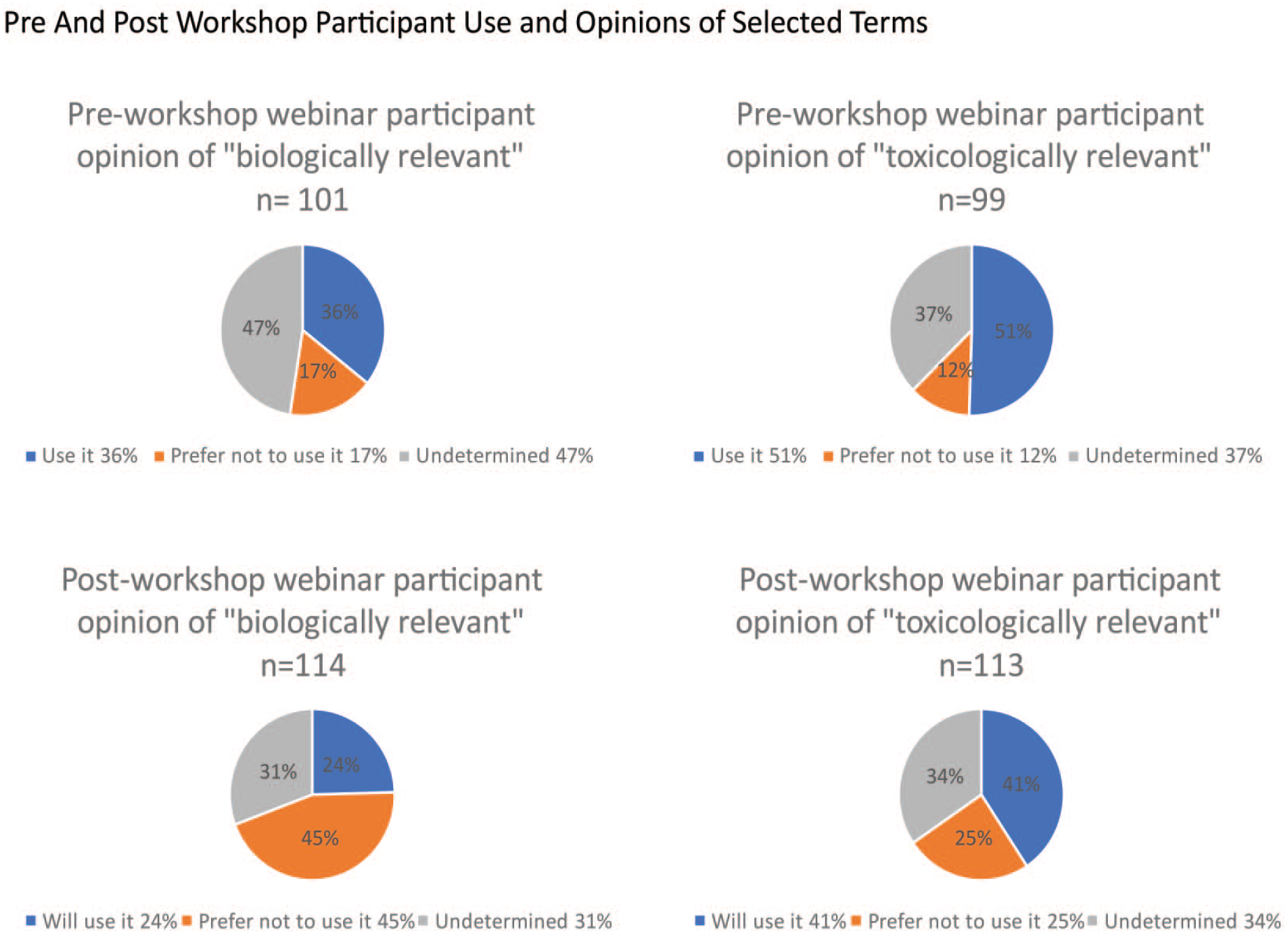
Preworkshop and postworkshop participant use and opinions of selected terms.
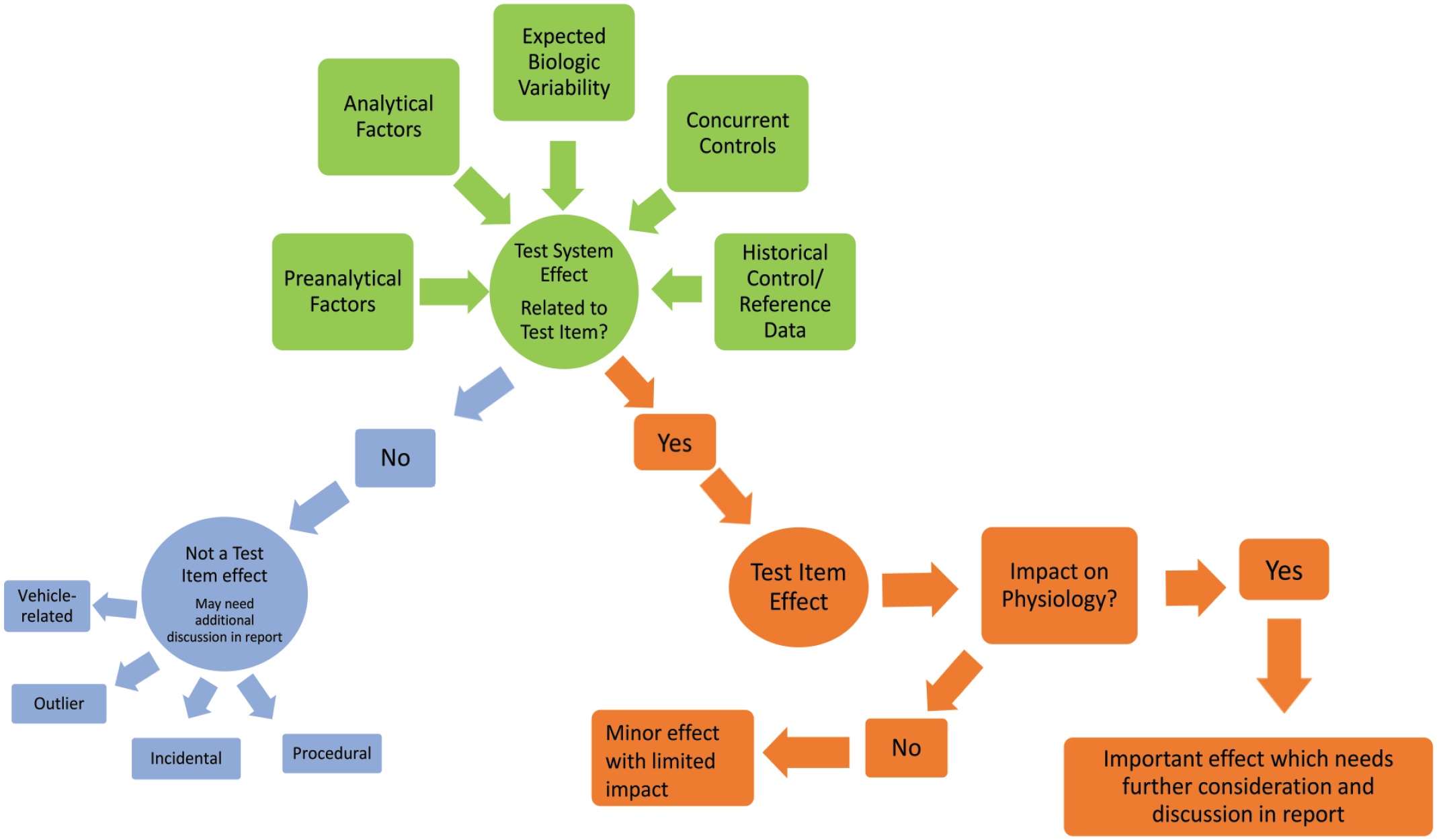
A possible approach to determine the importance of differences in clinical pathology data (consensus was not reached on this approach or definitions).
Conclusions
While the initial purpose of this working group expert panel was to find alignment on terminology used to convey importance of clinical pathology findings, clarity on other aspects affecting data interpretation were also deemed important outcomes of the workshop. There were expert presentations and discussions on the interpretation of clinical pathology data with consideration of the following aspects: sources of variability, appropriate comparators, statistics, reporting, correlations with anatomic pathology, nonstandard biomarkers, indirect (secondary) findings, and a weight-of-evidence approach.
An understanding of influencing factors and sources of variability (biological, preanalytical, analytical, and postanalytical) was considered critical. Even though the numerical assessment of biological variability is not routinely performed, awareness aids in the understanding of analytes and performance of assays, especially when using a new analyte or new test system. With regards to comparators, the concurrent control group and/or pretest values are deemed the most relevant depending on species (small vs large animal species) and endpoint. The use of HCD may be used as additional information for evaluation of clinical pathology data. Identification of indirect/secondary changes is beneficial because these effects are typically important to provide the appropriate context of the data. The underlying pathophysiology of these effects should be included when known or likely, and supporting literature for such is beneficial. For discordant findings that have no apparent relationship with other study results, an integrated approach to data interpretation with all available study data is imperative to understand test item relationship and importance to the study. The absence of expected pharmacology may be important to the study and can be noted in the report.
While statistics are a standard tool in clinical pathology, the limitations need to be understood to optimize data interpretation. Statistical results are best used to highlight differences between groups and cannot be used to definitively identify or exclude test item-related effects. The use of a wide variety of complementary statistical, computational and visualization tools is recommended to better assess patterns, correlations and trends in the data while minimizing the limitations of each tool.
For reporting of preclinical toxicology clinical pathology report (GLP and non-GLP), it is preferred that a formally trained veterinary clinical pathologist performs the data interpretation and report writing, or at a minimum reviews the data and report. For clarity, the clinical pathology reports should use standard and consistent vocabulary/terminology and avoid diagnostic terms. An integrated assessment of clinical pathology data with relevant study data (e.g., in-life, exposure, microscopic findings) should be made when possible. For nonstandard clinical pathology evaluation, it is strongly recommended that trained scientists familiar with clinical pathology and immunosafety and/or the assayed biomarkers collaborate to interpret the data and write the report. Other considerations for the evaluation of immunosafety endpoints were to use standard vocabulary/terminology, consider the whole panel evaluated (not just a single endpoint), and to consider weight of evidence and concordance with other study data (e.g., hematology data) to provide an integrated assessment of all relevant data.27,29 In addition, novel biomarkers were considered more challenging to interpret without relevant context due to limited (or lack of) experience and understanding of timing of change or dynamic range within the context of the study conditions. For cytokines, the influence of stress and handling was considered highly relevant for interpretation. Comparison to concurrent controls was recommended to rule out procedure-related findings.
In closing, the preparation and conduct of this ESTP expert workshop resulted in and endorsed important recommendations for the evaluation of preclinical toxicologic pathology clinical pathology data. A more robust understanding of BV and other sources of variability and the limitations of statistics should be considered to prevent overcalling small differences or changes in data. The discussions underscored the importance of a scientist trained in clinical pathology interpretation, particularly the value of a formally trained clinical pathologist in toxicology, to make sound interpretations in the context of a weight of evidence approach considering all relevant study data. In addition, and perhaps more critical, is the importance of using consistent and clear terminology and providing distinct descriptions if less unified terms are used.
Footnotes
Acknowledgements
The authors would like to thank Anne Provencher, CRL (retired); Luann McKinney U.S. FDA; Lynda Lanning U.S. FDA (retired); Joanna Harding, AstraZeneca; and Amy Narewski, ESTP for their valuable contributions to the preparatory sessions and workshop and/or for their critical and impactful review comments on this manuscript.
Authors’ Note
The opinions expressed in this document are those of the authors and do not reflect views or policies of the employing institutions, including the U.S. FDA. Mention of trade names or commercial products does not constitute endorsement or recommendation for use. This expert workshop article is the product of an International Expert Workshop commissioned by the European Society of Toxicologic Pathology. This final document has been reviewed and endorsed by the European Society of Toxicologic Pathology (ESTP), the British Society of Toxicologic Pathology (BSTP), and the Society of Toxicologic Pathology (STP), but it does not represent a formal Best Practice recommendation of the Society; rather, it is intended to highlight expert perspective on emerging toxicologic pathology issues that are relevant to the development of appropriate industry practices and good regulatory policies. The points expressed in this document are those of the authors and do not reflect views or policies of the employing institutions. Readers of Toxicologic Pathology are encouraged to send their thoughts on this article to the Editor.
Author Contributions
All authors (TA, MK, OB, LB, FC, DE, EE, AF, JH, SJ, CFK, PM, PO, LR, IR, VS, PV, DBW, MW, GPE, LT) contributed to the conception and/or design; data acquisition, analysis, or interpretation; drafting this manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigate and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This workshop was coordinated and supported by ESTP and hosted by Boehringer Ingelheim (Germany).