
Abstract
A challenge for all toxicologists is defining what study findings are actually adverse versus non-adverse in animal toxicity studies, and which ones are relevant for generating a no observed adverse effect level (NOAEL) to assess human risk. This article presents views on this challenge presented by toxicologists, toxicologic pathologists, and regulatory reviewers at the 2019 annual meeting of the American College of Toxicology during a workshop entitled “Toxicology Paradise: Sorting Out Adverse and Non-adverse Findings.” The speakers noted that setting a NOAEL is not always straightforward, not only for small molecules but also for biopharmaceuticals, and that a “weight of evidence” approach often is more useful than a rigid threshold-setting algorithm. Regulators from the US Food and Drug Administration and European Union told how assessment of adverse nonclinical findings is undertaken to allow clinical studies to commence and drug marketing approvals to succeed, along with the process that allows successful dialogs with regulators. Nonclinical case studies of findings judged to be adverse versus non-adverse were presented in relation to the many factors that might halt or delay clinical development. The process of defining adverse findings and the NOAEL in final study reports was discussed, as well as who should be involved in the process.
Introduction
The objectives of nonclinical toxicity testing are to screen out toxic molecules, to identify adverse findings that might be of potential safety concern if induced in patients, and to provide guidance for appropriate clinical monitoring during human trials. Determining which study findings are actually adverse or non-adverse and assessing their relevance for predicting potential responses by humans impacts the generation of a no observed adverse effect level (NOAEL) for most nonclinical toxicity studies. Toxicologists and toxicologic pathologists define “adverse” as “harm” in relation to effects on a test species under the particular conditions for a given set of study conditions. Adverse findings may be identified in subreports and/or the final study report, but the NOAEL should be established at the level of the overall study report since this document integrates data from all testing categories performed throughout the study. Scientific judgement by the study director, study pathologist, and all contributing experts involved in the study interpretation should combine to provide the clearest and most concise “weight of evidence” foundation for making such decisions.
Determinations regarding whether findings are adverse or non-adverse can significantly affect a test article’s (or test item’s) development. Adverse nonclinical findings can halt or delay clinical development, and the decision regarding whether a nonclinical finding is adverse or non-adverse can lead to significant alterations in clinical development plans. Case studies can be illustrative of how these challenges might be managed from the perspectives of the company and various regulatory agencies. Interactions with regulatory agencies on this topic are critical at both the clinical trial application (CTA) stage and throughout development as new clinical or nonclinical toxicity findings are reported. Interpretation of, and agreement on, managing adversity is key to the success of clinical studies and drug marketing approvals.
During the 2019 annual meeting of the American College of Toxicology in Phoenix, Arizona, a workshop was held on defining adverse versus non-adverse findings in nonclinical toxicity studies. This paper summarizes the key messages delivered in the presentations which represented the divergent views of a variety of scientists representing toxicology, pathology, study direction, drug development, and regulatory functions. The points discussed here are the perspectives of the authors and should not be construed to embody any individual company or regulatory agency view or policy on this topic.
Handling Adverse and Non-adverse Toxicity Findings (Paul Baldrick)
Introduction
For first-in-human (FIH) clinical trials (CTs), regulatory guidance states that [t]he goals of the nonclinical safety evaluation generally include a characterization of toxic effects with respect to target organs, dose dependence, relationship to exposure, and, when appropriate, potential reversibility. This information is used to estimate an initial safe starting dose and dose range for the human trials and to identify parameters for clinical monitoring for potential adverse effects. 1 Similarly, [i]n general, the No Observed Adverse Effect Level (NOAEL) determined in nonclinical safety studies performed in the most appropriate animal species gives the most important information. 1 However, these general statements raise 2 key questions: How do we define adversity, and how should we establish a NOAEL? A number of publications have attempted to give an all-embracing definition including such options as the highest dose level that does not produce a significant increase in adverse effects in comparison to the control group 2 and [t]he highest dose/exposure that does not cause toxicologically relevant increases in the frequency or severity of effects between exposed and control groups based on careful biological and statistical analysis. While minimum toxic effects or pharmacodynamic responses may be observed at this dose, they are not considered to endanger human health or as precursors to serious events with continued duration of exposure. 3 While such definitions are useful, there is still the real-life challenge for all toxicologists in determining which nonclinical study findings are actually predictive of potential adverse or non-adverse outcomes for humans, and how to use such decisions to generate a relevant nonclinical NOAEL.
The Challenge
There are various ways of establishing an NOAEL, and it has been suggested that there are essentially three types of findings in nonclinical toxicity studies that can be used to determine it: (1) evidence of overt toxicity (eg, clinical signs, macro-, and microscopic lesions); (2) surrogate markers of toxicity (eg, serum liver enzyme activities); and (3) supraphysiologic (“exaggerated pharmacodynamics”) effects. 2 Other considerations include whether or not differences from controls are treatment related or chance deviations (eg, could be due to spurious individual values, natural variation) as well as whether treatment-related changes are adverse or not (eg, considering such factors as adaptive response, magnitude, precursor to more significant effect, secondary to general toxicity, reversibility). 4 In addition, there is the complication that [t]oxicologists from different regions have different interpretations of what constitutes an adverse event. 3 Geographic differences can also occur regarding the importance of statistics applied to the animal study findings although it has been stated that [s]tatistical significance, alone, should not be used to judge the observation as adverse. 3 A further challenge in defining whether or not changes are judged to be adverse or non-adverse is that proper interpretation of findings may not occur. Thus, the following terms are used for some findings in study reports: “expected pharmacology” or “expected immunogenicity” (ie, the findings represent effects in the study but often cannot be separated from real toxicity) and “not biologically relevant” and “not toxicologically significant” (ie, the findings may be genuine but are judged to be non-adverse with questionable relevance to assessing human safety).
Setting an NOAEL—Examples
Example 1: Measurement of cardiac troponin in a nonclinical toxicity study gives notably raised levels but no microscopic evidence of degeneration in the heart. What is the NOAEL? Obviously, other information would need to be taken into consideration to answer this question, but it highlights that isolated findings—in this case, a surrogate marker of toxicity—can have a major influence on the decision-making process.
Example 2: Emesis is seen at all drug dose levels in a dog toxicity study with inconsistent effects on body weight and food consumption. What is the NOAEL? Again, other information would be needed to assist in NOAEL setting to confirm that the findings are considered to be non-adverse due to the common physiologic (not toxic) response of dogs to test article exposure.
Example 3: A toxicity study in dogs supporting an FIH CT with a small molecule drug for an anti-inflammatory indication raised serum activity of alanine aminotransferase (ALT, a hepatocyte cytosolic enzyme released via membrane damage) by up to 4.5-fold at the mid- and high-dose levels. There were no other study findings, no visible effect on liver structure in the rat toxicity study, and no indication of the liver as a possible target based on knowledge of the mode of action. What is the NOAEL? Initial considerations include the statement that [i]n general, it is rare for a clinical pathology marker to be adverse in the absence of any other change 5 but that [a]n increase of ALT of 2-4 fold in dog and/or rat may raise concern as an indication of potential hepatic injury unless a clear alternative explanation is found. 6 A published example of altered liver enzyme activity in a short-term nonclinical toxicity study with no microscopic correlate in liver sections was interpreted to indicate “no safety issue,” but a subsequent longer duration study showed severe hepatic damage. 3 In this latter case, it was concluded that such a liver enzyme change as a predictor of potential progressive liver toxicity should be considered adverse.
Example 4: In a nonclinical toxicity study in rats, increased incidence of hyaline droplets and/or tubular epithelial damage in the proximal kidney tubules was seen in males at all dose levels. What is the NOAEL? In theory, this situation should be easy to address, with an NOAEL at the high-dose level (if no other findings were observed that are considered adverse). Hyaline droplet nephropathy has been shown to be a progressive background lesion cuased by an impairment of α2 micro-globulin clearance specifically in kidneys of male rats; it is not seen in female rats or in either sex of other test species such as mice, dogs, and nonhuman primates. 7 However, an inexperienced study director might interpret the finding as adverse, and if the study report is not peer reviewed by knowledgeable colleagues, an inappropriate conclusion that an NOAEL was not established might be made.
Example 5: In a topical or ocular toxicity study, some local irritation findings were noted but nothing else. What is the NOAEL? It might be possible to establish two NOAELs: one on the basis of effects at the site of administration and another based on any systemic effects. In this situation, it is not clear if this dual-NOAEL approach is generally accepted, or which NOAEL might be the more relevant for predicting potential human risk.
Example 6: Immunocomplex-related study findings (secondary to antidrug antibody [ADA] formation) were seen with a monoclonal antibody in a cynomolgus monkey toxicity study. What is the NOAEL? The issue with this situation is that some toxicologists do not include such findings (ie, expected generation of an immune response following exposure of a test species to a foreign protein) in the establishment of an NOAEL, while others do. Furthermore, there can even be separate NOAELs: one based on an interpretation that the ADA finding is adverse to the monkey, and another acknowledging that immunogenicity is an expected consequence in nonhuman primates and therefore may be deemed “non-adverse” with respect to assessing risk in humans. 8 Thus, for some companies, immune complex disease is not included within the NOAEL assignment but instead is categorized as adverse in animals specifically, but not relevant for evaluating human outcomes. In such cases, predictions of human risk are built around an integrated safety assessment that is not focused mainly on an NOAEL derived from nonclinical toxicity testing. 9
Summary
Setting an NOAEL in nonclinical toxicity testing can have a number of challenges, but its use in setting safe clinical starting doses is important. 10 Challenges for toxicologists in determining whether study findings are adverse or non-adverse in order to define an NOAEL include experience (long, short, small molecule vs biopharmaceutical background, etc); scientific training (toxicology, pharmacology, another biomedical discipline); and other influences (management, regulatory agency, other constituencies). For risk assessors, incorrect NOAEL designation raises issues for values that are either incorrectly too low (findings should have been considered non-adverse) or too high (real toxicity was missed or wrongly written off). However, it is important to remember that NOAELs are just a part of the overall safety information available for a test article, and as clinical data become available such animal-derived analyses serve a progressively less important role. A final consideration is whether in the future there will be a migration away from the NOAEL paradigm. It seems that the answer is “No” for small molecules and “Maybe” for biopharmaceuticals, where toxicity is not usually a finding related to direct damage but rather represents expected but suprapharmacological and/or immunogenicity changes (with these latter effects sometimes adverse with respect to the test species but non-adverse to humans). Furthermore, for a number of years, NOAELs have not necessarily been used in toxicity testing for oncology drugs, where establishment of a severely toxic dose (STD) in rodents and highest non-STD for nonrodents is more usual as per (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidance document S9 on nonclinical evaluation of anticancer drugs. 11
An European Union Regulatory Perspective on Handling Adverse and Non-adverse Toxicity Findings (Ian Waterson)
Introduction
The NOAEL is considered by many as an important part of the nonclinical risk assessment for a new investigational medicinal product (IMP) undergoing drug development. Factors to consider when defining an NOAEL will include the exposures at which toxic effects are observed in nonclinical studies, which may be related to either on-target or off-target effects. The defined NOAEL is essentially a professional opinion. No consistent standard definitions exist for what constitutes a NOAEL. Furthermore, although NOAELs may be defined, there are examples in development history where, in isolation, NOAELs were insufficient to protect CT participants from undesirable effects produced by drug candidates.
TGN1412
TGN1412 was a first-in-class monoclonal agonistic antibody against CD28 (T cell coactivator) that was being developed as a drug for leukemia and rheumatoid arthritis. 12
An FIH (phase I) study conducted in London in 2006 led to catastrophic, systemic, multiorgan failure in 6 healthy young male volunteers. Nonclinical toxicity studies conducted previously in macaques, mice, and rats showed no adverse effects at 500 times the initial clinical dose used in the human Phase I study (50 mg/kg vs 0.1 mg/kg). 13 Studies indicated that TGN1412 was not expected to react with the receptor molecule from any species other than primates, and therefore the cynomolgus monkey was selected for toxicity studies with TGN1412. Prior nonclinical pharmacology studies in cynomolgus monkeys showed that TGN1412 had a predictable, well-defined pharmacokinetic (PK) profile following infusion of doses of 5 to 50 mg/kg body weight that maximum concentration (C max) and area under the curve (AUC) were largely proportional to dose, and that stable concentrations were observed following repeated dosing. A number of nonclinical safety studies (single- and repeat dose) demonstrated that TGN1412 was well tolerated in nonhuman primates at single doses of 2.5 mg/kg and weekly repeated doses of 5 to 50 mg/kg for 4 consecutive weeks, with no TGN1412-related signs of toxicity, hypersensitivity, or systemic immune system effects being observed. Studies in rhesus monkeys showed that after a single injection of 2.5 mg/kg TGN1412, no detectable levels of the proinflammatory cytokines interferon γ, interleukin (IL) 5, or IL-6 were found in serum, suggesting that injection of TGN1412 should not result in an acute systemic cytokine release. Using the Food and Drug Administration (FDA) guidance “Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers,” 2 a human-equivalent dose was calculated to be 16 mg/kg based on the NOAEL of 50 mg/kg established in the repeat-dose toxicity study in monkeys. Using a default safety factor of 10, the maximum recommended starting dose was defined as 1.6 mg/kg. An additional safety margin was also applied, and a proposed starting dose of 0.1 mg/kg was approved for the initial enrollees.
A number of symptoms were reported by the individuals given this 0.1 mg/kg starting dose fairly soon after administration of the investigational agent. Major symptoms in most or all volunteers included headache (50-90 minutes postdose [mpd]), lumbar myalgia (rigors reported 58-120 mpd in four subjects), elevated temperature (2.5- 6.5 hours postdose [hpd]), hypotension (3.5-4.6 hpd), and tachycardia (2.5-4.6 hpd). Other symptoms reported in some individuals included nausea, vomiting, dyspnea, and bowel disturbances. As part of an investigation looking at the causes of these effects, the potential for cytokine release in humans following exposure to TGN1412 was reexamined. Findings showed that adding TGN1412 to human peripheral blood mononuclear cells (PBMCs) or diluted blood did not stimulate the release of a “cytokine storm” or stimulate lymphocyte proliferation, suggesting that the dramatic adverse effects could not be explained by a rapid induction of cytokines resulting from simple ligation of CD28. Dry-coating TGN1412 onto the walls of the wells of 96-well plates, however, resulted in significant lymphocyte proliferation in experiments using human PBMCs. One reason why it is thought that previous protocols failed to stimulate release of cytokines and lymphocyte proliferation was as a result of TGN1412 being cross-linked via its Fc region in aqueous solution. 14 Phenotypic analysis of proliferating lymphocytes identified CD4+ T-cells as the principle responding population, and all of the CD4+ cells were committed to dividing, thus leaving no resting CD4+ cells. Data from the revised in vitro assays with human cells suggested that the dose of TGN1412 given to volunteers was close to the maximum immunostimulatory dose. TGN1412 did resume clinical development as TAB08 in 2013 using doses in the new phase I trial from 0.1 µg/kg to 7 µg/kg in healthy volunteers, which were calculated using the “Minimal Anticipated Biological Effect Level” approach. None of the 30 individuals developed a cytokine release syndrome, and all cytokines remained at baseline levels. In fact, the anti-inflammatory cytokine IL-10 was detected in plasma at the higher doses, indicating that TAB08 (TGN1412) had stimulated an anti-inflammatory response.
This example demonstrates that there were clearly differences in safety and activity between the nonclinical animal models and humans, despite an NOAEL being clearly defined in nonclinical toxicity testing. This discordance, however, could have been pre-empted by a simple modification of an in vitro test, which shows that the nonclinical models selected in drug development need to be optimized in order to efficiently predict drug behavior in humans. This point is clearly stated in ICH guidance S6 (R1), 15 which acknowledges that “due to the species specificity of many biotechnology-derived pharmaceuticals, it is important to select relevant animal species for toxicity testing.” The key message from this sequence of events is that nonclinical development cannot rely on rote application (“box checking”) of a standard assay battery but instead may need to be adapted to best evaluate the potential for unusual, human-specific mechanisms of toxicity.
BIA 10-2474
BIA 10-2474 is a fatty acid amide hydrolase (FAAH) inhibitor, which potentiates the levels of anandamide and other endogenous cannabinoids by inhibiting FAAH activity. This test article was initially developed as a therapeutic modality for a variety of conditions including pain due to multiple sclerosis, cancer, anxiety, and movement disorders associated with neurodegenerative diseases. 16 BIA 10-2474 was identified as a long-acting FAAH inhibitor on the basis of nonclinical studies.
In January 2016, a healthy volunteer who received 50 mg/d BIA 10-2474 for 5 days in a phase I CT was admitted to the hospital with neurological and gait disturbances. After a dramatic and rapid worsening of neurological symptoms, the participant died. Another 5 healthy volunteers who received the same drug dose for 6 days subsequently were admitted to the hospital, four of them with similar neurological symptoms. The European Medicines Agency (EMA) coordinated 2 reviews into the incident, one concentrating on nonclinical tests and the other one addressing clinical events. In the nonclinical review, it was found that a death during the 4-week monkey study was preceded by tremors, weakness, incoordination, and a hunched posture. Similar in-life signs but not death were seen in other monkeys along with agitation and anxiety. During microscopic evaluation, axonal dystrophy of the fasciculus cuneatus in the posterior medulla oblongata, edema in the pars nervosa of the pituitary gland, and vacuolation of the submucosal (Meissner’s) plexus ganglia in all gastrointestinal segments were observed in several animals. 17 All these findings were atrributed to nerve sheath edema formation that was deemed to be a pharmacological effect of BIA 10-2474. Based on the complete or partial reversibility of the in-life and microscopic findings in monkeys 17 and rats, 18 they were not considered to be adverse by the company, even though 1 monkey had died. Wording in the protocol stated that animal toxicity studies of repeated daily dosing of BIA 10-2474 for up to 13 weeks in mice, dogs, and monkeys and up to 26 weeks in rats had been conducted and that treatment with BIA 10-2474 produced no signs of toxicity in mice, rats, dogs, and monkeys up to the NOAEL. Clearly, this assessment shows that the main focus of the risk assessment was trying to establish an NOAEL and not considering the pharmacology and its implications. It is therefore important that an investigator understand that they know what is and what is not an NOAEL, and the actual meanings and limitations of such values.
The outcome of the EMA review led to the publication of the “Guideline on strategies to identify and mitigate risks for FIH and early CTs with investigational medicinal products.” 19 This was a revision of the guideline written in response to TGN1412 and concerns the extension of the existing European Union guidance to address FIH and early-phase CTs with integrated protocols. In particular, the revised guidance provides more detailed recommendations regarding the nonclinical and emerging clinical PK to pharmacodynamic (PD), and safety data to support them while also establishing requirements that the overall clinical study design should be scientifically justified and that careful consideration should be given to the proper inclusion of each data set and the time available for integrated assessment. Moreover, it is imperative that the safety of CT subjects is not compromised in the interests of speed in acquiring nonclinical data or for logistical reasons. In summary, the revision of the guideline does not call for an increase in the amount of nonclinical data required to support FIH trials, but it does emphasize again the critical value of understanding the pharmacology profile and the mode of action when developing an IMP.
Dealing With Adverse and Non-adverse Findings in a CTA Application
An intrinsic element of uncertainty exists in assessing both the possible benefits and risks of a novel drug candidate. The nonclinical data relating to PD, PK, and toxicity and their translation to humans are an important basis for planning and conducting an FIH/early-phase CT. When planning FIH/early-phase CTs, companies and investigators should identify the potential risk factors and apply appropriate risk mitigation strategies. However, it must always be remembered that while toxicity observed in animals can be the result of suprapharmacological activity, these should not be ignored when devising safe human doses. Primary and secondary PD data can support the generation of mechanistic hypotheses regarding the nonclinical toxicities seen in vivo and help in defining the potential human relevance of these findings. An evaluation regarding whether the target organs identified during the nonclinical studies warrant particular monitoring in during CTs should be undertaken, but serious nonclinical toxicity should lead to a more cautious approach. Further follow-up studies may be required to support the CT design and/or proposed safety monitoring plan. Overall, such analyses usually are driven by exposure to the IMP.
Defining an NOAEL
As previously stated, the defined NOAEL is essentially a professional judgment, and there are no consistent standard definitions of what constitutes an NOAEL. It is probably easier to describe what an NOAEL is not than to determine what it is: A dose that only causes findings that are driven by the known pharmacology of a compound is NOT necessarily a non-adverse dose. A dose that causes findings that are shown to be reversible or partially reversible is NOT necessarily a non-adverse dose. A dose that causes clinical pathology (eg, hematology or serum chemistry) alterations that have no obvious microscopic correlates in tissue sections is NOT necessarily a non-adverse dose. A dose that causes findings which are only apparent in a few animals in a treatment group is NOT necessarily a non-adverse dose. A dose that only causes findings that are considered to be “class effects” is NOT necessarily a non-adverse dose. A dose that only causes findings in 1 test species but not in a second (or third) species is NOT necessarily a non-adverse dose.
The establishment of an NOAEL can act as a key tool as part of the overall data. However, some argue that researchers should move away from fixating on NOAELs and no observed effect level and, instead, concentrate on what the whole (ie, weight of evidence) of the nonclinical data are telling us. It is essential that pharmaceutical development moves away from an almost rigid “tick box” mentality of unthinkingly conforming to regulatory guidelines. It is also essential that regulators do not try to hide behind prescriptive guidelines and, instead, look to take a more flexible, scientific approach in assessing nonclinical data sets. Animal studies need to be carefully designed to provide the most useful data for deciding whether or not it is safe to progress to humans. The FIH CT protocols should be designed to have sensible starting doses as well as justifiable dose escalation steps and dose exposure caps, with the criteria for patient inclusion and exclusion as well as dose stopping also reflecting the nonclinical data.
It is the ultimate responsibility of a company to ensure that toxicity study documents and the nonclinical overview section in regulatory submissions for a given product include a clear and cohesive interpretation of study findings and their relevance to humans. The findings in a nonclinical study should be defined in relation to effects on the test species used and within the context of the experimental conditions for that particular study. Test article-related effects should be described in isolation from other factors. Decisions to consider effects as adverse or non-adverse should be discussed in overview documents rather than individual study reports where all the known information on a test article may not be known. Furthermore, decisions should be justified case by case based on a logical scientific rationale rather than some previously agreed set of global criteria that rigidly define what adverse means. No one can ever remove all the risks involved in developing new medicines. Even the growing use of in silico (computer-based) and human-derived in vitro assays (eg, cultured cells and tissues, human organs-on-a-chip) may not help to reliably identify idiosyncratic reactions. Ultimately, case-by-case decisions, a careful risk benefit analysis, and appropriate risk mitigation steps are the best that can be done.
Dealing With Adverse Findings While a CT is Ongoing
The occurrence of any new events relating to the conduct of the CT or the development of the IMP, where the new event is likely to affect the safety of the current or future subjects, means that the company and the investigator(s) are required to take appropriate urgent safety measures to protect the subjects against any immediate hazard. This requirement and its short timeline are in line with Article 10(b) of Directive 2001/20/EC. 20
It is important to realize that the company action to impose such safety measures may be taken without prior notification to the national competent authority. The competent authorities must, however, be informed forthwith or ex post of the new events, together with the measures taken, and the Regulatory Ethics Committee must be notified at the same time. In the United Kingdom, the Medicines and Healthcare Products Regulatory Agency (MHRA)’s CTs unit can be contacted to discuss the issue with a safety scientist, ideally within 24 hours of measures being taken, and a plan for further action can be mutually agreed. It is usual for any initial contact made by telephone to be followed up by traceable communications, first an e-mail and later by a written report. The company is required to inform the MHRA by email no later than three days from the date the measures are taken.
Finally, in the United Kingdom, a substantial amendment covering the changes made as part of the urgent safety measure is anticipated within approximately two weeks of notification. Any potential reason for delay to submission of the substantial amendment should be discussed and agreed with the MHRA at the time of initial notification or through a follow-up call. The submission of the substantial amendment should not be delayed by additional changes outside of those taken and required as an urgent safety measure. Unrelated and unacceptable changes can result in rejection of the amendment. The annual submission of the development safety update reports should also take into account all new available safety information received during the reporting period. Scientific advice may be sought via the MHRA and/or the EMA’s committee responsible for human medicines, the Committee for Medicinal Products for Human Use.
A US FDA Perspective on Handling Adverse and Non-Adverse Toxicity Findings (Tessie Alapatt)
Introduction
Distinguishing between adverse and adaptive effects is vital for pharmacology/toxicology reviewers in regulatory agencies, including the US FDA, in order to make informed decisions regarding doses and durations of drug exposures, preferably before they progress into the clinic. Nonclinical toxicity studies, as per ICH and US FDA guidance documents, usually are recommended by the agency to gather data on the safety of a drug, including its adverse and non-adverse effects, as well as the doses at which these effects occur. Typically, a NOAEL generally is an accepted benchmark for safety when derived from appropriate animal studies and can serve as the starting point for determining a reasonably safe starting dose of a new therapeutic candidate in healthy (or asymptomatic) human volunteers. The NOAEL is the highest dose level that does not produce a significant increase in adverse effects in comparison to the control group in repeat-dose animal toxicity studies. In this context, adverse effects that are biologically significant, even if not statistically significant, should be considered in the determination of the NOAEL. 2 Collective data, including clinical observations and macroscopic and microscopic analyses obtained from a battery of nonclinical studies of the test article, may be used to assess potential risks in the clinic. However, distinguishing an adverse effect to be a direct effect of the drug vs a possible adaptive effect of the organ system can be a challenge for nonclinical reviewers. The following sections will summarize some of the aspects and issues that pharmacology/toxicology reviewers face during review of nonclinical studies of test articles, escpecially for new molecular entities (NME) that are yet to be introduced in the clinic. However, this does not reflect the views of the US FDA per se, as perspectives may vary to some extent among various Centers, offices, and divisions, depending on the test article, indication, and duration of treatment in the clinic.
Considerations of NOAEL
While the indiscriminate use of an NOAEL derived from animal experimentation may be an oversimplified estimate of potential human risk, pharmacology/toxicology reviewers of nonclincial studies appreciate its utility in making a judgment call about a test article. 21 A dose–response relationship exists if a change in the dose of a test article causes a change in the effect observed, often calculated as a change in the quantifiable endpoint parameter being measured. However, determination of an NOAEL largely depends on correlated dose–response changes occurring at various tissue levels in the species tested. For example, changes in clinical pathology parameters (hematology, serum chemistry, urinalysis, etc) alone are difficult to classify when assessing whether or not an effect is adverse or non-adverse; therefore, clinical pathology alterations alone may not be sufficient to establish an NOAEL. To elaborate further, Pandiri et al 5 point out that clinical pathology parameters are rarely adverse by themselves and typically do not determine NOAELs, but they support the determination of the NOAEL based on morphologic lesions or clinical signs. Therefore, an overall determination of adversity and consequently an NOAEL should be pathogenesis-based rather than focused on individual clinical pathology parameters. Nonetheless, this is a broad statement and may not definitively cover all instances. As an example, consider a 28-day toxicity study in animals, in which serum chemistry analytes measured in the middle and at the end of the study period showed markedly elevated liver enzyme levels (50-100 times the levels in control animals), while liver specimens showed no necrosis in tissue sections, either due to clearance of single necrotic cells by macrophages by the end of the study period or due to the absence of single-cell necrotic foci in poorly-sectioned liver specimens. Here, altered serum chemistry (ie, elevated liver enzyme levels) would likely be indicative of liver injury and could be regarded as a signal for monitoring liver function in the clinic.
Another consideration is that the NOAEL established in a study is specific to the particular species tested under the conditions of that study. While risk assessment for a test article should be inclusive of an evaluation of findings observed across multiple species, findings in a single species may be considered species specific. For example, an NME was shown to cause severe vacuolation in major endocrine organs, including the ovaries and adrenal glands, along with a dramatic increase in serum cholesterol concentrations after 28 days of treatment in rats, but not in dogs. However, the single ascending dose CT with the NME was considered safe to proceed with the FIH dose adjusted to have a 10-fold or greater safety margin over rat NOAEL.
The NOAEL serves as only one element in evaluating the potential for human risk and is primarily used to get the test article into the clinic. Once the test article begins to gain clinical experience, the relevance of an NOAEL wanes and at that point, the complete analysis of human risk encompasses likely systemic exposure, any available clinical information, and other available information (eg, published class effects) that might be accessible to individuals performing the assessment. The amount of nonclinical and clinical data, as well as other information, increases as the development program progresses.
Adverse Effects
Typically, in the case of an NME, initial nonclinical testing involves the treatment of 2 mammalian species, often rodents and nonrodents such as dogs, but in some cases nonhuman primates, with the proposed NME. Administration of the test article to the animal via the proposed clinical route of exposure may cause one or more changes in gross and microscopic structure, clinical pathology, and in-life functionality. Among all the test article-related changes observed, adversity may be applied appropriately only to those changes in which the animal is harmed to some degree. As mentioned above, the final determination of whether a finding is adverse or non-adverse should be done in the context of the particular study under review. To further elaborate this point, an adverse effect should not be extrapolated on the assumption that the NME might prove harmful at higher doses or after a longer duration of exposure in the clinic if those conditions were not tested in the nonclinical study under review. Another aspect of this assessment is that when toxicity is interpreted as being specific to the test species and lacking relevance to humans, the effect may still be an adverse response for the animals and therefore cannot be ignored.
All observations and changes seen during the nonclinical study typically should be compiled into the final study report and submitted to the FDA for a comprehensive review of the safety profile of the test article prior to initiation of a CT. The toxicity study report should typically strive to unambiguously communicate all effects so that pharmacology/toxicology reviewers may understand the rationale for adverse versus non-adverse designations when making informed decisions regarding potential human risk. Compiling all correlated adverse findings with a clear relationship to test article exposure would reduce the complexity of making a reasonably accurate judgment about the fate of the test article in the clinic. Similarly, minor findings or non-adverse findings should be addressed clearly, and provide justification as to why any modest test article-related finding was not considered adverse. For example, mild test article-related emesis in dogs is not uncommon as they are especially responsive to emetic stimuli; intermittent emesis in this species may not cause body weight loss or even alter food consumption. Therefore, emesis should not be considered adverse in the context of a dog toxicity study unless body weight and/or food and water consumption is affected substantially. As another example a test article that interacts with the metabolism of bilirubin and may cause a transient increase in serum bilirubin levels, which may be erroneously regarded as liver toxicity. However, in the absence of altered liver enzymes i.e., alanine (ALT) and aspartate (AST) aminotransferase activities and adverse microscopic findings in liver sections, increased bilirubin may be justified as non-adverse, specifically if bilirubin levels are normalized at the end of the recovery phase.
Drug development often advances over time, leading to a test article's evaluation for several diseases and indications. However, toxicity that would be permitted as allowable for one indication could be considered an inappropriate risk for another indication. 5 For example, a drug that causes severe fetal malformations and hence is contraindicated in women of child-bearing potential and pregnant women for one indication might still be used in the clinic for women who are surgically sterile, menopausal, or postmenopausal for the same or another indication. Another example would be a drug originally intended for cancer in older patients that may be repurposed as a topical antibiotic in children. However, in both cases, the same nonclinical study report may be used—if written appropriately—to support both indications. Thus, all adverse and non-adverse findings are recommended to be reported in unambiguous, descriptive terms only to support different indications. Repurposing an approved drug for a different indication may be driven by the urgent need for an effective therapy in the clinic. In this case, toxicologists may find themselves precariously balancing the risk–benefit scale to advise clinicians about the safety, as well as the potential efficacy, of the repurposed drugs in the clinic.
Intermingling of descriptions with interpretations and mechanistic speculations at the study report level may prevent the report from being used to repurpose the test article for new indications. In general, interpretations and speculations should be communicated in a nonclinical overview document as an integrated message assembled from multiple study reports, for the same test article. Even within a single regulatory center, decisions will differ among offices and divisions tasked with examining products that have diverse safety profiles when used for various indications and administered via divergent routes, eg, a drug delivered intravenously for treatment of cancer and topically for treatment of inflammation in children.
Reversibility
Reversibility can be an important factor in the holistic interpretation of nonclinical toxicity studies. 4 In assessing the level of concern assigned to a given biological effect, a change that is readily and completely reversible on cessation of treatment is considered to indicate a lower level of concern. 4 Typically, the recovery phase of a nonclinical study is based on the half-life of the test article (ie, the recovery arm should span 5 half-lives of the test article), which reasonably ensures systemic clearance of the test article. Regarding reversibility, the guidance states that evaluation of the potential for reversibility of toxicity or return to the original or normal condition, should be provided when there is severe toxicity in a nonclinical study with potential adverse clinical impact. The evaluation can be based on a study of reversibility of effects, using one or more recovery time points within a study. Alternatively, reversibility may be based on a scientific assessment that includes the extent and severity of any structural lesions, the regenerative capacity of the organ or tissue showing the effect, and knowledge of other materials that can cause the effect. Thus, recovery arms are not always critical in concluding whether or not an adverse effect is reversible, and demonstration of full reversibility is not considered essential for completing the risk assessment. A trend toward reversibility, eg, decrease in incidence and/or severity, and a scientific assessment that this trend would eventually progress to full reversibility are generally considered to be sufficient. If full reversibility is not anticipated in the test species, this should be considered in the clinical risk assessment.
While reversibility of a finding may be enough to justify it as non-adverse, reversibility per se is insufficient to establish lack of adversity. For example, a test article may cause necrosis in the stroma, causing transient organ atrophy (determined by reduced organ weight) without damaging infrastructure of the whole organ. Although, normal organ weight at the end of the recovery phase indicates that the organ likely is restored to normal; the initial finding should be considered adverse, specifically while establishing an NOAEL. Language in the guidance, ICH M3(R2), 1 pertaining to reversibility is noteworthy in this respect: [t]he goals of the nonclinical safety evaluation generally include a characterization of toxic effects with respect to target organs, dose dependence, relationship to exposure, and, when appropriate, potential reversibility.
Adaptive Effects
An adaptive response may not cause any measurable alteration in function. Instead, adaptive changes are those that the organism makes to allow continued normal function in the face of a persistent stimulus. Cellular changes that do not lead to cell death or death of the animal may be called “adaptive,” and they can be considered either adverse or non-adverse depending on the nature of the change. Some cellular adaptations involve metabolic or functional alterations that leading to increases in the number and/or volume of cellular organelles as well as intracellular accumulations of a variety of endogenous and exogenous substances; either of these allow the affected cells and the animal to survive and often live normally. 22 Adaptations involving a test article-related response with no deleterious effects on the test species should be considered non-adverse within the context of the study. Designation of a specific response as adaptive in a given case does not necessarily carry over to all scenarios.
Hepatocyte hypertrophy (Figure 1) is the most common finding identified as adaptive in nonclinical toxicity studies. Hypertrophy is not considered a direct toxic effect when the severity is low and it is not accompanied by hepatocyte degeneration or cytotoxicity. This adaptive response is consistent with test article-related microsomal enzyme induction to promote increased hepatic clearance of the test article. However, if hepatocyte hypertrophy is present in combination with a pattern of effects that together would be considered adverse, such as increased serum activities of hepatocyte leakage enzymes (ALT, AST), increased hepatocellular lipid or increased plasma cholesterol levels, and scattered single-cell hepatocyte necrosis, then this entire constellation of changes—including hepatocellular hypertrophy—may be considered as an adverse rather than an adaptive event. Additional data may be requested by FDA to support the designation of a finding as an adaptive response. In addition, information regarding the reversibility of findings and evaluation of cytochrome P450 induction in animal/human hepatic microsomal preparations, along with careful monitoring of serum activities of liver enzymes in the clinic, may be helpful.
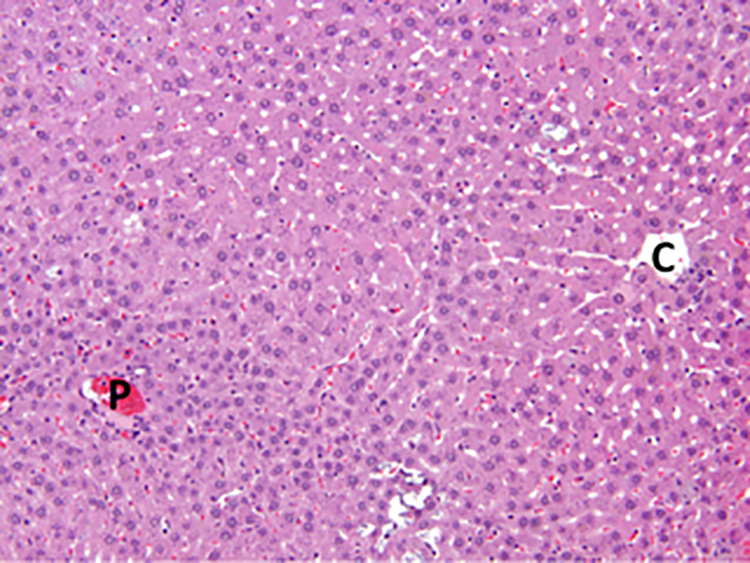
Hepatocellular hypertrophy—compare enlarged hepatocytes near the central vein (C) with the smaller hepatocytes near the portal region (P)—is a frequent liver response after extended exposure to test articles that induce cytosolic metabolic enzymes (especially cytochromes P450). Adult rat. hematoxylin and eosin.
Generating, Interpreting, and Communicating Adverse and Non-adverse Pathology Data From Animal Toxicity Studies (Brad Bolon)
Introduction
Interpretation of findings in animal toxicity studies as either adverse or non-adverse has been used for decades to predict potential human risk of test article exposure. Adversity assessments integrate empirical data with informed judgments to define whether or not a response is considered to be harmful to an organism’s health. Nonetheless, the kinds of adverse effects induced by test articles in animals diverge to a variable degree from the adverse effects produced in people exposed to the same agent. 21,23,24 Such differences highlight the essential limitations in modern risk assessment.
The principle of “adversity” has been approached in many ways in the scientific literature. Some papers have assessed the concept without offering a detailed definition, 25 -28 others have outlined the potential limits and relevance of adversity and the NOAEL as useful predictive parameters, 3,4,29 -31 and 1 paper compares and contrasts possible definitions of adversity. 32 The multiplicity of options resulted in a recent decision by the Society of Toxicologic Pathology (STP, based in North America) to convene a working group to devise recommendations (“best practices”) for effectively determining, communicating, and using adverse effect data in nonclinical studies. 33 The resulting 10 recommendations were designed to offer clarity and consistency in establishing adversity decisions while improving understanding of information included in regulatory submissions. A concurrent expert workshop undertaken by the European Society of Toxicologic Pathology, which included some overlapping membership with the STP working group, provided additional insight on important factors to consider in weighing such adversity decisions. 34
Recommendations for Generating Adversity Versus Non-adversity Data
The first four STP recommendations were devoted to determining whether or not findings should be interpreted as adverse: No. 1 is that adversity is a term denoting harm, which is applicable to any sort of observed change (biochemical, functional, and/or structural). The implication of this statement is that only a fraction of changes seen in subjects during animal toxicity studies will be capable of detrimentally impacting an animal’s performance or life span either under normal conditions or if challenged; the corollary is that test article-related findings that are not harmful by definition are interpreted to be non-adverse. No. 2 is that decisions regarding whether or not test article-related changes are judged as adverse or non-adverse should be unambiguously stated and explained in the pathology subreport and/or full study report. This recommendation is critical as adversity decisions related to pathology data sets are informed expert interpretations based on an objective exploration of the study materials—founded in the shared training of pathologists and a burgeoning menu of harmonized diagnostic terminology (STP, 2020 35 )—and an individual professional judgment of its meaning based on the toxicologic pathologist’s unique experiences. No. 3 is that adversity as described in a study report should be applied only to the specific test species and under the particular conditions (dose, duration, route, etc) used in a given animal toxicity study. The presence, incidence, and severity of findings seen in 1 animal species in a given context cannot be extrapolated to other species (animal or human) nor would effects seen in short-term, high-dose studies be deemed automatically to predict the likelihood of related adverse findings in longer-term, lower-dose studies. This accommodation is required because some test article-related effects in animals are known to be exacerbations of incidental species-specific changes with no known human counterpart, which even if adverse in the animals may have no relevance for human risk assessment. 33 Finally, No. 4 (which is an extension of No. 3) is that effects observed in cells, tissues, organs, or systems within the test animal should be assessed on their own merit; in other words, interpretations should not incorporate conjectures unsupported by actual experimental data regarding possible mechanisms that might produce the effect or the putative equivalence of animal and human responses. Taken together, recommendations (3) and (4) state that adversity decisions should be based on observations and not speculation on either a possible pathogenesis of a particular finding or its presumed relevance in predicting risk to humans.
Recommendations for Interpreting Adversity Versus Non-adversity Data
The next four STP recommendations addressed several means for optimizing communication of adversity decisions. No. 5 is that communication of adversity decisions should be made in the pathology subreport and reiterated in the full study report, while assignment of an NOAEL or equivalent threshold value for the study should be done only in the overall study report and not the various subreports (eg, pathology report) used in compiling the study report. A critical consideration in this regard is to avoid utilizing ambiguous statements (eg, not biologically relevant or “not toxicologically important”) unless a detailed explanation is included to support such contentions. No. 6 is that communication of adverse findings (and the NOAEL) should include direct interaction among all staff from different scientific disciplines who are involved in performing a nonclinical study. The rationale for this recommendation is that a single toxic event may manifest in different ways to scientists with diverse backgrounds, which implies that the full picture of a test article-related effect requires discussion of these various views when defining the integrated message that will be communicated in the final study report. No. 7 is that the NOAEL for a test article should be stated in an overview document based on data from multiple studies (eg, investigator’s brochure, investigational new drug application, CTs application, new drug application, biologics licensing applications). Integration is necessary because selection of the NOAEL in the most sensitive species is made using data from many studies in at least 2 species, and an NOAEL identified in 1 animal toxicity study may be overridden based on data from another toxicity study. No. 8 is that the use of NOAELs in data tables within reports should be linked to appropriate explanatory text (eg, a cross-reference to the narrative and/or a footnote of the table) in order to place them in proper context. This recommendation acknowledges that nuances explained at length in the report narrative often provide critical insight regarding assignment of the NOAEL.
Recommendations for Communicating Adversity Versus Non-adversity Data
The final two STP recommendations address the use of nonclinical adversity interpretations and NOAEL assignments in assessing human risk. No. 9 is that all scientists (toxicologists, toxicologic pathologists, and other contributing subject matter experts) who acquire and interpret animal toxicity data should actively assist in assessing and communicating human risk. This recommendation acknowledges that individuals who generate specific subreports during the course of an animal toxicity study are best equipped to explain the data in their own statements (especially its interpretation). No. 10 is that all available data from all animal studies for a given test article must be assessed together in defining potential toxicities and predicting human risk. This point acknowledges that elements not evaluated in conventional Good Laboratory Practice (GLP)-compliant nonclinical toxicity studies (eg, lesion pathogenesis, molecular mechanisms) but more commonly assessed in non-GLP discovery and/or nonclinical efficacy or nonclinical combined efficacy/toxicity studies also have relevance in predicting potential human outcomes.
Final Considerations for Adversity Versus Non-adversity Decisions
Two additional considerations are important in determining, communicating, and using adversity in animal toxicity studies. First, the presence of premonitory biomarkers for an adverse finding typically should be associated with the adverse finding in establishing adversity in a final study report rather than being interpreted independently in subreports. An example of collation for liver, a common target organ for many test articles, would be to construct a final study report that integrates the composite meaning of test article-related changes in organ weights, serum enzyme activities (ALT and AST), and microscopic findings that were originally described in individual anatomic pathology and clinical pathology subreports. Such integration has been instrumental in differentiating genuine test article-induced liver damage (usually associated with substantial elevation in ALT and/or AST activities with microscopic evidence of hepatocyte necrosis) from adaptive changes associated with test article-related enhancement of liver metabolic capacity (shown by increased liver weight due to hepatocyte hypertrophy without elevated serum activities of hepatocyte leakage enzyme activities or microscopic lesions in liver). 36 The second factor is that reversibility of an otherwise harmful finding is not a sufficient reason to interpret a change as non-adverse. As noted above (STP recommendation No. 3), adversity decisions are made based on the specific observations made during the course of a study and not on any possible recovery that might take place in the future.
An Industry Perspective on Adverse and Non-adverse Toxicity Findings in Product Development (Melissa Rhodes)
Introduction
In order to consider how adverse and non-adverse nonclinical toxicity findings may affect product development, it is important to establish consensus on the definitions of these terms. An adverse finding is one that indicates harm to the animal within the constraints of the study design. 5 This is both species- and study specific. Secondary toxicity refers to adverse findings that occur as a result of a primary toxicity potentially in a different organ. 24 Effect constellations are findings considered to be adverse when part of an assemblage of related lesions, even though each is potentially non-adverse when considered in isolation. 34 Adverse findings due to enhanced efficacy based on the compound’s mechanism of action are referred to as exaggerated or suprapharmacology. 5 Non-adverse findings are test article-related effects that do not cause biochemical, morphological, or physiological changes that affect the general well-being, growth, development, or life span of an animal. 5
Risk assessment consists of the identification of hazards and the analysis and evaluation of risks associated with exposure to those hazards. 37 In product development, the risk assessment depends on many factors. The treatment indication is paramount in determining and balancing potential benefits and risks. For example, pharmacological interventions for patients with late-stage cancer may be associated with more risks of adverse but not immediately life-threatening effects than treatments for such chronic conditions as insulin-resistant (type 2) diabetes or asthma because the oncology therapy may still be deemed to have a positive risk: benefit ratio relative to the alternative strategy of no therapy for a dying cancer patient. The exposure margin between the identified hazard (toxicity) and the therapeutic effect should also be considered. Larger margins between efficacy and toxicity are more desirable, even for therapeutic candidates destined for use in terminal diseases. The ability to monitor for adverse events is also a critical component of the risk assessment. Test article-related changes that are easily detected by standard minimally-invasive laboratory tests (eg, hematology or serum chemistry analysis) or self-reported symptoms (eg, headache and nausea) can be easily monitored as opposed to severe or unpredictable events (eg, seizures) or lesions that only can be detected microscopically. Risk assessment requires scientific judgment by qualified scientists and clinicians.
While risk assessment may contribute to decisions about product development, there are separate and distinct considerations for product development that may not be altered by a positive or negative risk assessment. In risk assessment, we consider if the potential benefits of treatment outweigh the potential risks; in product development, there are a number of business and logistical considerations that are also evaluated including feasibility and cost of the development path, timeline to market and anticipated competitive landscape at launch, ability to differentiate the new product from competitors, and patent life. Thus, findings that do not negatively impact the risk assessment nevertheless can have substantial effects on product development.
Adverse Findings Affecting Product Development
A compound in development for muscle wasting secondary to chronic heart failure underwent a standard battery of nonclinical toxicity studies to enable clinical progression. Treatment-related myocardial injury was observed at all doses in a 13-week oral gavage toxicity study in rats. A NOAEL for cardiac effects was not identified. The AUC exposure at the lowest dose in the study was approximately the same as the anticipated efficacious clinical exposure. A risk of cardiotoxicity could not be tolerated in this patient population. Therefore, product development was terminated.
Secondary toxicity can also impact product development. Nonhuman primates that experience daily emesis and diarrhea may deteriorate in condition leading to morbidity or mortality secondary to the treatment-related effects on the digestive tract. In this case, it may be possible to proceed with clinical studies, even with a relatively small therapeutic index, provided that gastrointestinal effects are closely monitored in patients.
Effect constellations may be adverse or non-adverse, depending on the dose, and can impact product development. For example, clinical signs of widespread, progressive skin dryness with various associated discolorations occurred in all treated groups in a 39-week monkey toxicity study for a test article being developed for a serious neurodegenerative disease. Additional secondary skin effects including scaling, scabbed areas, abrasions, cracking of the dermal tissue resulting in bleeding, and swelling around the eyes were also noted. The skin effects correlated with an increased incidence of microscopic findings in the skin of treated animals including epidermal hyperplasia, subacute/chronic inflammation, and alopecia/hypotrichosis. The findings were considered non-adverse at the low dose but adversely affected the overall health of animals at higher doses. Because of this constellation of effects, there was a hypothetical concern for potential of the compound to cause test article-related toxic epidermal necrolysis (Stevens-Johnson syndrome), although the risk: benefit was still favorable for continuation of development in the face of this for a serious life-threatening condition. In order to monitor and potentially mitigate such serious effects, dermal changes were listed as an adverse event of special interest, and a dermatologist was required to review and document any potential dermal changes in the clinical studies.
Adverse Findings With No Negative Effect on Product Development
An oral gavage toxicity study in rats revealed treatment-related mortality at the highest doses administered in two animals on study. Microscopic findings were observed in the nasal cavity and sinuses of these rats and included degeneration/regeneration of olfactory and respiratory epithelium, inflammation, and exudates containing foreign material (likely to be the test article). These findings are consistent with gastric reflux of gavage material into the nasal cavity. This is clearly an adverse finding to the rats in this study. However, the route of administration in humans was oral, not oral gavage, and consequently these findings were not judged to be clinically relevant. Therefore, there was no effect on product development as a result of these adverse findings.
Suprapharmacologic findings may be adverse but not negatively affect product development. While it is inadequate to refer to these types of findings as exaggerated pharmacology that was “anticipated” and therefore “acceptable,” these heightened responses may be very useful in development. This is the situation for case study BIO-1 described by Brennan et al 9 in which a monoclonal antibody was being developed to treat autoimmune diseases. The expected pharmacology included inhibition of proinflammatory activities of macrophages. In toxicity studies, it was not surprising that foamy macrophages and adverse buildup of foreign material, cholesterol clefts, and granulomatous inflammation due to reduced macrophage function were observed. These adverse findings informed human dose modeling and therapeutic index margin predictions which were used to identify a safe starting dose and dose escalation schemes for the phase 1 clinical study.
Non-adverse Findings Affecting Product Development
Phospholipidosis (PLD), or excess accumulation of phospholipids in tissues, is a fairly common finding in product development. In this example, PLD was observed in multiple organs in rat and monkey toxicity studies of two-weeks duration through chronic administration. By routine microscopy, this finding was characterized by foam cell infiltration, vacuolation, and increased tingible body macrophages (ie, phagocytic cells in lymphoid organ germinal centers that contain ingested fragments [“tingible bodies”] of apoptotic cells). Phospholipidosis alone is generally not considered a manifestation of toxicity but rather an adaptive response, with >50 marketed drugs containing a cationic amphiphilic structure and reported to induce PLD in vivo and/or ex vivo. 38 However, there are steps that may be taken to determine if patients in CTs are experiencing PLD, including testing for visual acuity and conducting a thorough cardiovascular assessment (QT) study which examines the interval of time from the beginning of the QRS complex to the end of the T wave on a standard electrocardiogram. These activities were added to the product development plan for this test article.
Multiple compounds across indications are associated with non-adverse liver findings. These include clinical pathology alterations such as increased serum activities of ALT and alkaline phosphatase, serum bilirubin and cholesterol concentrations, and/or prothrombin time (PT) increases (ie, coagulation abnormalities) with or without microscopic correlates such as centrilobular hepatocyte hypertrophy (Figure 1). Organ weight increases (liver/body weight ratio) may also be observed. Collectively, these liver findings are evidence of a test article-related physiological adaptive response to an increased metabolic workload but not necessarily indicative of a toxic response. These types of findings may progress to adversity in longer toxicity studies (eg, where prolonged hepatocellular hypertrophy reduces blood flow in sinusoids and thus induces hypoxia and eventually necrosis of hepatocytes), but there are many examples where these findings do not reach the level of toxic adversity as defined previously (ie, they do not indicate harm to the animal within the constraints of the study design).
The bile salt export pump (BSEP) is an ATP-dependent transporter expressed on hepatocytes that functions to mediate bile flow. This transporter has important safety and risk assessment implications that should be considered in product development. Many compounds that inhibit BSEP have been associated with drug-induced liver injury (DILI). Troglitazone is a hallmark example of a BSEP-inhibiting drug removed from the market for liver injury. Patients reported elevations in serum ALT activity, but the onset was typically delayed, with only 1 patient having an elevation during the first month of therapy. In most patients, the peak ALT values occurred between the third and seventh months (mean 147 days; range, 1-287) after initial treatment. 39 Interestingly, an analysis of 85 drugs showed no clear correlation between BSEP inhibition (IC50), unbound C max, and DILI. However, all 17 drugs with BSEP IC50 values <100 mM and unbound C max values >0.002 mM caused DILI. 40 Toxicity studies are not often strong predictors of this delayed potential for DILI because animal models have been poor predictors of liver injury associated with BSEP blockers. For test articles that potently inhibit BSEP, this pattern must be transparently communicated to investigators, and implementation of additional safety monitoring during the CTs should be considered.
Similar to non-adverse liver changes, many test articles in nonclinical development have been associated with non-adverse effects on coagulation parameters, such as moderate alterations in activated partial thromboplastin time. In such situations, nonclinical testing also should investigate whether any correlated alterations (eg, increased PT, abnormal platelet numbers, or microscopic findings demonstrating hemorrhage or increased hematopoiesis) are suggestive of an increased tendency for bleeding. Nonclinical testing should be performed to determine if these parameters return to normal levels following a recovery/off-treatment period. Coagulation markers are not necessarily standard for collection in clinical studies. Therefore, the toxicologist should discuss these nonclinical findings with team clinicians to determine if adding these easily measurable end points to clinical studies is warranted.
Summary
Determination of findings as adverse or non-adverse can significantly affect whether or not a test article can be developed successfully. Adverse nonclinical findings can halt or delay clinical development, but in many instances adverse nonclinical findings can be safely monitored in clinical studies. Non-adverse nonclinical findings can lead to significant alterations in clinical development plans and should not be considered inconsequential simply because they do not adversely affect the health of the animals in the toxicity study. Secondary toxicity findings and suprapharmacologic effects in nonclinical studies should be discussed in the risk assessment of a test article and may alter the product development pathway. Some non-adverse toxicities may be viewed in combination as adverse effect constellations, and these may have a potentially large impact on the development pathway. All classifications of nonclinical toxicity findings may require in-depth scientific justifications, scientific judgment, and careful consideration of the patient population as clinical studies are designed.
Workshop Summary
Decisions regarding whether findings in animal toxicity studies are adverse or non-adverse, determinations of NOAEL or similar threshold values and the relevance of such information with respect to assessing human risk are complex endeavors typically tackled using a weight of evidence approach. As shown by the current workshop synopses, scientists with differing backgrounds and professional roles have divergent but overlapping views of adversity versus non-adversity decisions and NOAEL calculations. Such decisions may be simpler for small molecules relative to biologics (since human-derived protein test articles often elicit an immunogenic response in animals, which may be harmful to the animal but is not a genuine manifestation of direct target-mediated toxicity and thus is of no relevance to human risk assessment). Findings linked to suprapharmacology (exaggerated efficacy) or that exhibit reversibility may be non-adverse but must not be assumed to be interpreted as such. Regulatory scientists welcome insights from product development teams regarding the interpretation and justification for both adverse and non-adverse findings. Taken together, the speakers agreed that adversity decisions and establishing threshold values such as NOAEL must be handled on a case by case basis, and that the scientific basis of such professional judgments must be supported as needed to build a logical scaffold on which regulatory determinations can be based.
Footnotes
Authors’ Note
This publication reflects the individual views of the authors and should not be construed to represent the policies or positions of their respective institutions. The authors thank Joanne Berger, FDA Library, for manuscript editing assistance.
Author Contributions
All authors contributed to the conception, design, drafting, and critical revision of the manuscript for important intellectual content. All authors gave final approval and agree to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.