
Abstract
The identification of adverse health effects has a central role in the development and risk/safety assessment of chemical entities and pharmaceuticals. There is currently a need for better alignment regarding how nonclinical adversity is determined and characterized. The European Society of Toxicologic Pathology (ESTP) therefore coordinated a workshop to review available definitions of adversity, weigh determining and qualifying factors of adversity based on case examples, and recommend a practical approach to define and characterize adversity in toxicology reports, to serve as a valuable prerequisite for future organ- or lesion-specific workshops planned by the ESTP.
Introduction
The concept of adversity is central to toxicological science. In a broad sense, it defines a health end point to be protected against when establishing allowable exposures to a particular substance. Identification of an effect or group of effects as adverse forms the basis of health-based guidance values used to estimate margins of exposure (MOEs) in human health risk assessment, define the entry dose of “first-in-human” studies for pharmaceuticals, and set reference levels for environmental contaminants. Interpretation of toxicological findings as “adverse” or “nonadverse” thus needs to be consistent and based on a clear scientific rationale. In current practice, however, it is often challenging to reliably determine when and why a particular biological response qualifies as an adverse effect.
Historical definitions of adversity are often vague or circular, and there has been limited practical guidance on how to evaluate adversity and to communicate the considerations underlying these decisions. This type of information is key to improved standardization in the reporting and review of toxicological findings but needs to be balanced with the large degree of case-by-case flexibility required for many adversity determinations. Considering this issue, a working group of the Society of Toxicologic Pathology (STP) recently provided an extensive review of past definitions of adversity and offered recommendations on how study pathologists and toxicologists can more clearly report adversity in nonclinical toxicology studies (Kerlin et al. 2016).
In the absence of a universal working definition of adversity, some guidelines and regulations have provided directives to categorize study findings. As an example, the European Community Classification, Labeling, and Packaging (CLP) regulation proposes classifying findings into four categories: (1) the “specific target organ toxicity” (i.e., findings characterized by a detailed description of toxic effects in humans and/or animals), (2) findings that necessitate consideration of a “weight of evidence approach,” (3) findings “for consideration,” and (4) effects that are considered “not toxicologically significant.” This regulation further subdivides these categories and provides specific examples of toxicities for each category in order to help in the identification of effects as adverse or not. This approach has the advantage of defining a classification scheme, but the complexity of many pathological effects may still lead to differences in interpretation across regulatory bodies applying the same (or other) prescriptive classification schemes, which does not promote alignment.
To complement and build on the STP effort by Kerlin et al. (2016), the European Society of Toxicological Pathology (ESTP) coordinated an international expert workshop to more clearly characterize the concept of “adversity” in current practice and to frame subsequent workshops that will be specific to particular organ or lesion types. The expert group was comprised of 21 pathologists and toxicologists from Europe, the United States, and Japan involved in industry (including Contract Research Organizations), research, and regulatory affairs for industrial chemicals, environmental and food chemicals, and pharmaceuticals. Panelists were invited by workshop chairs and the ESTP board based on their expertise in toxicology/toxicologic pathology and diverse perspectives on adversity issues.
The goals of this working group were to provide a working definition of adversity in the context of toxicologic pathology, to identify considerations for interpreting morphologic changes identified in nonclinical studies as adverse or not, and to highlight the different implications for adversity decisions across the separate fields of activity. Discussions generally focused on pathology outcomes in nonclinical guideline studies of pharmaceuticals, environmental, and food chemicals. After a 4-month preparatory phase with teleconferences and initial expert contributions, the face-to-face workshop was conducted in Alfortville, France, on June 8 and 9, 2015, in order to discuss the different aspects of adversity through case examples. This article provides a summary of these discussions, including a working definition of adversity and practical considerations for study pathologists, toxicologists, study directors, risk/safety assessors, and reviewers when attempting to classify study findings as adverse or nonadverse.
This final document has been reviewed and endorsed by major toxicologic pathology organizations including the European STP, the British STP, the Dutch STP, the French STP, the STP, the Japanese STP, the Latin American Society of Toxicologic and Experimental Pathology, the STP—India, the Chinese Pharmaceutical Association—Specialty Group of Toxicology Pathology, the Chinese Society of Toxicology—Toxicologic Pathology Specialty Section, the International Federation of Societies of Toxicologic Pathologists, and the International Academy of Toxicologic Pathology.
Past Definitions of Adversity
In a general societal context, the term adversity has been used to describe various undesirable or harmful conditions detrimental to human and animal life (natural disasters, disease, poverty, war, accidents, etc.) that can potentially have unwanted consequences for public and individual health and well-being. Any negative impact on good health can therefore be considered an adverse effect and something to be avoided if at all possible.
In the field of toxicology, the concept of adversity has historically been broadly applied but not specifically defined. For many regulatory statutes, this ambiguity is likely by design to compel scientific experts to make specific decisions on a case-by-case basis (Stansell, Marvelli, and Wiener 2005). Overly prescriptive definitions may restrict this flexibility and increase the risk of taking complex interpretive decisions out of expert hands. However, adversity decisions have often been made without clear and objective scientific explanations. This approach, which can be (or at least appear) somewhat arbitrary, can also lead to different (potentially contradictory) interpretations within or across regulatory authorities and other health organizations, lack of consistency in adversity decisions, and confusion among study pathologists and toxicologists when writing study reports.
Adverse effects have been defined by a number of different organizations and work groups, as described in the recent review by Kerlin at al. (2016). Common examples include the following: [C]hange[s] in morphology, physiology, growth, reproduction, development or lifespan of an organism which results in impairment of functional capacity or impairment of capacity to compensate for additional stress or increased susceptibility to the harmful effects of other environmental influences; … [A]ny effects which result in functional impairment and/or pathological lesions which may affect the performance of the whole organism, or which reduce an organism’s ability to respond to an additional challenge; and Changes that occur that result in impairment of functional capacity, often due to an insult that exceeds the capacity of the adaptive response to permit a return to the homeostatic state. Outcomes might include changes in morphology, development, lifespan, or growth of the organism. Although harder to define at the molecular level, potentially adverse responses might include alterations in gene expression, protein synthesis, or cell regulation. (p. 3) Adverse effect: a biochemical, morphological or physiological change (in response to a stimulus) that either singly or in combination adversely affects the performance of the whole organism or reduces the organism’s ability to respond to an additional environmental challenge. Nonadverse effect: can be defined as those biological effects that do not cause biochemical, morphological, or physiological changes that affect the general well-being, growth, development or life span of an animal. (p. 3)
Working Definition Proposed by the ESTP Working Group
While considering the existing definitions, and taking into account the consensus recommendations reached during the workshop (to be presented later in this article), the working group agreed on the following definition of an adverse effect: In the context of a nonclinical toxicity study, an adverse effect is a test item-related change in the morphology, physiology, growth, development, reproduction or life span of the animal model that likely results in an impairment of functional capacity to maintain homeostasis and/or an impairment of the capacity to respond to an additional challenge.
From this definition, it becomes clear that some findings will be easily recognized as inherently adverse. However, the adversity of many or perhaps most pathologic changes is dependent on specific characteristics such as severity/degree of change, distribution, effect constellations, impaired capacity to respond to additional challenges, and related lesions and is generally not simply a binary, adverse or nonadverse, decision. At the ESTP workshop, it was considered impractical to compile an exhaustive list of findings categorized as intrinsically or provisionally adverse. Rather, a primary goal of the workshop was to better clarify the factors that characterize an adverse or nonadverse conclusion.
Where and How Should Adversity Be Reported?
A tiered approach to adversity reporting was discussed by the working group for pathology subreports, integrated toxicology reports, and nonclinical overview (summary) documents. These recommendations are intended as a general working guidance, recognizing that data reporting requirements and formats will differ across sectors and organizations. The working group indicated that pathology subreports should define adversity only where possible. It was recognized that it is not always possible (or feasible) to provide a judgment on adversity in pathology subreports, particularly when the findings need to be considered within the context of other (unavailable) data in order to fully evaluate biological significance. Given this context dependence, it seems reasonable to emphasize that adversity calls be made in the integrated toxicology report at a minimum and in a pathology subreport when possible.
The integrated toxicology report should consider adversity at the study level as a basis for setting both the no observed adverse effect level (NOAEL, highest dose level at which no adverse change is observed) and lowest observed adverse effect level (LOAEL, lowest dose level at which an adverse change is observed). When adversity calls are to be made in integrated toxicology reports rather than individual subreports, it was recommended that study directors should always consult with study pathologists over questions regarding the adversity of individual findings to ensure that all relevant information is considered. It was further recommended that integrated toxicology reports should provide a thorough, descriptive, and sound scientific rationale regarding adversity (e.g., complete description of and understanding of the lesions and careful consideration of historical controls, severity, incidence, and correlations with organ weights, clinical pathology and gross observations, as well as compound characteristics and literature data). Statements supporting nonadversity calls such as “does not significantly affect organ function,” “does not significantly alter overall health,” and “represents an adaptive response” should be avoided unless clearly defined and supported by a scientific rationale.
In contrast to single study reports, nonclinical overview documents should discuss adversity at the project level and include assessment of projected relevance to human. A holistic approach, considering the implications to health status, pathophysiological process/mechanism, morphological criteria, human relevance, therapeutic indication of the molecule, pharmacology/mode of action (MOA), reversibility, patient population/population exposure, and severity of findings, should be used for human health risk assessment, particularly for findings that are more difficult to interpret. In all cases, a clear rationale for adverse or nonadverse conclusions will support consistency, transparency, and robustness in interpretation, by both sponsors and regulatory reviewers.
At Which Level of the Organism Should Adversity Be Considered?
From an interdisciplinary standpoint, adversity can be considered at the level of a molecule (e.g., DNA damage, receptor interaction, adducts), a cell (e.g., single cell necrosis, cell proliferation, hypertrophy), a tissue (e.g., full-thickness dermal necrosis, inflammation, fibrosis), or an organism (e.g., shock, death). In the context of standard guideline toxicology studies, pathologists primarily characterize adversity based on morphologic end points at the cell, tissue, or organ level, in combination with behavioral, biochemical, and clinical chemistry data. At the whole animal level, behavioral effects have also been used to identify NOAELs and LOAELs (e.g., tremors, lethargy), but functional consequences of specific morphologic findings are often difficult to assess. Further steps are needed to integrate the assessment of the study pathologist into the more integrated overview that bridges molecular events with morphological, functional, and clinical effects. Unfortunately, many target tissues do not have clear functional markers, and at this time at least, mechanistic information is typically not available for most pathologic effects, highlighting the need to focus on morphological evaluation as the current standard for nonclinical adversity calls related to histopathological changes. This does not, however, preclude the possibility of identifying adverse effects in the absence of morphologic changes (e.g., severe anemia, behavioral changes, reproductive effects, etc.).
Primary Features of Adversity (“Determinants”)
Pathological Nature of Effect
Identification of the pathological nature of a test item–related effect and its target cell/tissue/organ is a prerequisite to any adversity decision. In some “inherently adverse” lesion types, this knowledge alone is sufficient to decide on adversity. Examples include mortality, retinal degeneration, malignant neoplasms, limb deformities, and neuronal necrosis. However, for most pathologic changes other information should also be considered. Based on our working definition, the primary consideration here is whether a given change could impair cell/tissue/organ function or reserve capacity to respond to additional challenge. For example, a nasal epithelial lesion would be called adverse only if it were able to impair respiratory function (e.g., mucus in the airways), olfactory function (e.g., loss of olfactory epithelium), or ability to clear particulate matter (e.g., loss of ciliation). Conversely, if the absence of a functional correlate can be clearly demonstrated then the lesion would be considered nonadverse. In many cases, this determination will require qualifying information on severity and associated lesions, as discussed below. While there is inherent uncertainty in this type of evaluation, the work group agreed that adversity decisions ideally should be qualitative (yes/no), without hedge terms like “possibly,” “probably,” or “likely” adverse, which may further complicate interpretation.
Lesion Severity
For findings that are not inherently adverse, severity of the change can be an important factor in deciding upon adversity. Examples of such changes may include renal tubular dilatation, laryngeal metaplasia, hepatocellular lipidosis, biliary hyperplasia, and immune cell infiltrates (e.g., Kaufmann et al. 2009; Hailey et al. 2014). In some cases, dose-related increases in severity may be observed without dose-related increases in incidence due to the high-background incidence, complicating interpretation. Although dependent in part on the experience and judgment of the study pathologist, defining a threshold for adversity based on severity/incidence needs to be objective and well documented (Long and Hardisty 2012). In such cases, it becomes mandatory to provide a precise description of the grading in the pathology subreport, so that any reviewer can understand the extent of the finding and its impact on morphology (Shackelford et al. 2002). The use of severity in decision-making for adversity implies that at least for some pathologic effects, a threshold for adversity does exist. Severity (and incidence) can also qualify the intensity of effects that have been classified as adverse and, in this context, can also be recognized as an attribute of adversity (see below).
Effect Constellations
Lesion context is one of the most important determinants of adversity for many pathologic changes. In many cases, a particular lesion may be considered adverse when it is part of a constellation of related lesions but nonadverse in isolation. Accordingly, if a group of related effects is cited as the basis for the NOAEL/LOAEL, then these should be considered as a group and not necessarily split-off and/or considered individually. For example, some chemicals activate hepatic receptors like peroxisome proliferator-activated receptor α (PPARα) and induce cytochrome P450 enzymes in the liver, which can lead to increased liver weight and hepatocellular hypertrophy. When mild and not accompanied by other histopathologic changes like necrosis or serum biochemical changes like increased alanine aminotransferase (ALT), there is general agreement that no functional impairment is present and that these changes are nonadverse (Hall et al. 2012; U.S. Environmental Protection Agency [U.S. EPA] 2002). However, when associated with degenerative changes and related responses (e.g., hepatocellular necrosis, excessive lipid accumulation, inflammation, fibrosis, and clinical pathology changes), hepatocellular hypertrophy may be considered part of a group of effects indicating adversity. Thus, a collection of changes used to set the NOAEL/LOAEL may be comprised of individual changes that may not necessarily be considered adverse when evaluated individually. A second example relates to laryngeal effects. According to Kaufmann et al. (2009), minimal to mild focal laryngeal metaplasia in the absence of other related findings likely does not impair laryngeal or respiratory function and thus should not be considered adverse. However, when accompanied by inflammation, hyperkeratinization, necrosis, and/or hyperplasia, this constellation of effects may be considered adverse. In such cases, it is the linkage of findings with “equivocal” adversity to other lesions that determines the weight of evidence (WOE).
Further Characterization of Adversity (“Characteristics”)
Several concepts are often discussed together with adversity in pathology subreports and integrated toxicology reports. The working group reviewed each of these topics separately and decided to classify them as characteristics or “modifiers” (i.e., additional explanations or factors that provide rationale and context for identifying a particular primary test item effect as adverse). They should not be used as primary determinants of adversity but may provide corroborating evidence for a difficult adversity call. The following aspects were considered by the working group as appropriate modifiers of adversity: treatment-related exacerbation of spontaneous/background findings, direct versus indirect effects, adaptive responses, reversibility of the morphological change, extrapolation of the findings to longer term or higher exposures, translatability/human relevance, and MOA/intended pharmacological action of the chemical/drug.
Exacerbation of Spontaneous/Background Findings
The working group agreed that an adversity decision should be made only after a finding is identified as test item related. This determination may be particularly challenging for exacerbated spontaneous/background lesions present in both control and treatment groups. In such cases, the initial comparison should always be made with the concurrent control group because of the same genetic background of the animal model and maintenance conditions (Keenan et al. 2009). Intergroup differences indicating a test item–related effect may be seen as changes in incidence and/or severity of a finding or an earlier/aberrant age of onset. Shifts in severity alone for higher-incidence lesions are often more difficult to interpret given the lack of statistical analysis in many cases. Conversely, extremely rare or unfamiliar findings may increase uncertainty regarding adversity and result in a higher degree of conservatism. Factors indicating that an intergroup difference is probably not a test item–related effect include lack of a clear dose–response or pairwise differences between groups, high variability or imprecision of the end point, incidence values within the normal range of biological variation, and/or lack of biological plausibility taking into account preexisting knowledge of the test item (adapted from Lewis et al. 2002).
Decisions on the adversity of findings that may also occur spontaneously raise the question of whether or not to use thresholds to record background findings. The working group recommended that the recording of spontaneous lesions in control animals should be complete. This is consistent with recommendations made for qualitative and quantitative analysis of nonneoplastic lesions in toxicology studies (Shackelford et al. 2002). Only thorough recording of background lesions will capture situations in which incidence, but not severity, is increased, show a continuity of changes allowing differentiation from physiologic changes and spurious events, and enable proper use of a historical control database. For example, thorough recording of control incidences has been recently recommended for accurate detection of test item–related increases in pulmonary alveolar macrophages in rodents (Nikula et al. 2014). Ensuring consistency of recording spontaneous pathology in the control group relies partly on threshold-setting being similar among pathologists and maintaining consistency in recording and reporting findings across studies (Long and Hardisty 2012).
Beyond the comparison with a concurrent control group, properly established historical control data can be used to support the interpretation of whether an effect was induced by the test item or is incidental. Historical data can be used to identify aberrant control data, to understand the relevance of increases in low-incidence findings, and/or to interpret minor differences from controls. Historical data are particularly important in the evaluation of proliferative rodent lesions (Keenan et al. 2009). The working group agreed that the establishment and use of historical control data should be scientifically responsible, in that only recent studies (preferably within 5 years of primary study), performed with the genetically identical animal strain of similar age and under comparable experimental and environmental conditions, should be used for reference.
Examples of test item–induced exacerbated spontaneous lesions which may pose challenging adversity decisions include vascular lesions in the Beagle dog and chronic progressive nephropathy (CPN) in the rat. In the Beagle dog, it may be extremely difficult to distinguish test item–related arterial lesions from (spontaneous) idiopathic canine polyarteritis. In a classical presentation, the characteristic histopathologic features and lesion distribution, clinical signs, and other corroborative study results would aid in the differentiation; however, there are examples in which only the dose–response curve and overall incidence of histomorphologically identical lesions are distinctive, such as treatment with benzodiazepines, endothelin receptor antagonists, vasodilators, or immunomodulators (Clemo et al. 2003). Therefore, the sensitive recording of spontaneous arterial lesions in Beagle dogs, leading to robust historical control data, is essential to interpret such lesions (Bodié and Decker 2014).
CPN is a common age-related renal disease affecting most conventional rat strains used in toxicology. This condition may be exacerbated by diverse chemicals and drugs, including those that cause α-2u-globulin nephropathy (Hard and Khan 2004). As the incidence of CPN varies largely with several environmental factors such as the protein content of the diet (Rao, Edmondson, and Elwell 1993), intergroup comparisons with the concurrent control group, severity and age of onset, and historical control data are vital to interpret whether a change is test item–related. If CPN is exacerbated to a degree in which the renal function of treated rats is impaired, it should be considered adverse.
Direct versus Indirect Effects
A direct or primary effect was considered to be one resulting from a defined interaction between the test item (or metabolite) and the target organ or cell population. Alternatively, an indirect or secondary effect was considered to be one which does not involve a primary test item–target cell interaction. The work group agreed that indirect or secondary effects could be either adverse or nonadverse. It was noted that in many cases it might be difficult to distinguish direct/primary versus indirect/secondary effects, given that many morphological findings have precursor changes. Adversity should thus not be restricted to primary effects but may be important in some cases for characterizing the stage and context of an effect in a given mechanism of action or pathway. The following examples include both adverse and nonadverse primary and secondary effects.
An example of a direct adverse effect is the inhibition of acetylcholinesterase (AChE) enzyme by cholinesterase inhibitors such as organophosphorus compounds and N-methyl carbamates. Inhibition of AChE in brain, peripheral nerves, or erythrocytes is usually judged to be adverse due to the clinical impacts of impeded neurotransmission, even though histopathological alterations are not typically detected in nervous tissues. This biomarker is generally used for setting NOAELs/LOAELs in toxicology studies of cholinesterase inhibitors.
An example of an indirect effect resulting in an adverse secondary effect is the tachycardia caused by β-2 receptor agonists used to treat asthma. Prolonged severe tachycardia may result in secondary myocardial papillary necrosis/fibrosis, which has been identified in dogs and rats and is considered to be due to local hypoxia. This indirect effect of β-2 receptor agonists depends on the dose, duration of exposure, and species differences in susceptibility.
Hemolysis is a common example of a secondary toxicity that may be either adverse or nonadverse. Study pathologists can identify the increased turnover of red blood cells, particularly with extravascular hemolysis, as increased pigmentation (hemosiderosis) in macrophages/phagocytes in various organs/tissues. While hemolysis and the resultant pigment accumulation may be adverse, a slight increase in red cell turnover resulting in an increase in pigmentation or positive ferritin staining in the spleen, without any other adverse histological findings in hematoxylin and eosin-stained sections or alterations in hematological parameters that would impact tissue oxygenation (e.g., decreased red cell mass requiring a regenerative response), would be considered nonadverse.
A common endocrine-related example of an indirect effect is thyroid hypertrophy or hyperplasia secondary to metabolic enzyme induction in the liver. In rodents, this increase in liver metabolism can decrease thyroid hormones (T3 and T4) and lead to compensatory increase in pituitary thyroid stimulating hormone secretion, inducing thyroid follicular cell hypertrophy and growth, which will be considered adverse when considered preneoplastic in the context of a given nonclinical study and used to establish a NOAEL/LOAEL. Human thyroid is known to be much less sensitive to increased liver metabolism of T3 and T4; thus, for hazard characterization this thyroid effect would need to be considered in context of the liver effects. For chemicals, this may include a reduction in the interspecies uncertainty factor by 3-fold to account for the differences in pharmacodynamics (PDs) between rats and humans. Similar issues apply to many other endocrine effects. These examples highlight the idea that simply identifying an effect as “secondary” is not sufficient rationale on its own to consider it nonadverse.
At higher doses used to identify the maximum tolerated dose (MTD), secondary test item–related stress, decreased food consumption, and/or body weight loss are common findings. While the stress and weight loss are often associated with overt toxicity, at substantial magnitudes they are considered adverse findings. There are many findings associated with stress including decreased acinar secretion in salivary glands, lymphocytolysis, and germ cell depletion (Everds et al. 2013). Estrous cycle may also be affected by stress or body weight loss as a secondary effect, but typically severe depression of body weight (over 20% less than controls) is required to affect estrous cyclicity (Hayashi et al. 2013). The working group concluded that all of the secondary effects of overt toxicity may not be adverse in their own right and that they should be considered together with the primary finding. Overall, the working group agreed that adversity decisions should not be based on whether effects are primary or secondary in the initial assessment by the study pathologist. The subsequent integrative assessment in overview documents is more suited to discussing relevance of indirect effects.
Adaptive Responses
The working group concluded that adaptive and adverse responses are not mutually exclusive. In some cases, the term “adaptive” could be used to describe a nonadverse decision but should not be used to substantiate lack of adversity. Thus, a response should be defined as “adverse versus nonadverse” rather than “adverse versus adaptive.” Even though biological systems have a homeostatic capacity to maintain normal functions that capacity can be overwhelmed, a normal physiologic response in 1 situation could easily manifest as an adverse response following prolongation of exposure or at higher dose levels. Moreover, the argument could be made that a wide range of biological responses, from inflammation to metaplasia, are adaptive responses of the organism to tissue damage. Thus, use of the term adaptive in general should be limited to describing an event rather than defining adverse or nonadverse.
For example, many chemicals that cause only liver hypertrophy at low doses (nonadverse) will induce degeneration at higher doses. The presence of necrosis is an adverse response, and the cell replication that occurs accompanying the degenerative effects is there to replace functional deficits induced by damaged hepatocytes. Functional loss in the liver may occur, and if extensive fibrosis results, a permanent deficit of functional hepatic units may be the consequence. Under these circumstances, the constellation of changes observed (i.e., necrosis, hyperplasia, and fibrosis) is adverse. Liver lesions indicating that normal physiological responses have been overwhelmed may include zonal necrosis; increased apoptosis/single cell necrosis, excess fatty accumulation, and inflammatory cells; increased release of liver enzymes into the plasma; and decreased plasma albumin and γ-globulins. In this example, there may be a dose response with nonadverse adaptive changes such as hypertrophy, cytoplasmic vacuolization, and bile pigments at lower doses (without saturation of normal physiologic responses or development of adverse changes) and more severe adverse effects at higher doses (Maronpot et al. 2010; Boekelheide and Schuppe-Koistinen 2012; Williams and Iatropoulos 2007).
Other examples of tissue responses that are commonly characterized as adaptive (or physiologic) include laryngeal squamous metaplasia, nasal mucous cell hyperplasia/metaplasia, and increased alveolar macrophages (Burger et al. 1989). Similar to the situation described in the liver, these findings may be nonadverse in some cases when minimal or mild severity but can also be adverse with greater severity and associated changes. Adaptive does not clearly answer the question as to whether these changes impair function or the capacity to maintain function following additional challenge. Thus, the working group recommends that the term adaptive may be used as a modifier to characterize an observation but should not be used to determine if a finding is adverse or nonadverse.
Reversibility
The working group supported the position that reversibility does not automatically indicate nonadversity but that the lack of reversibility could indicate additional cause for concern and consequently increases the likelihood that a change will be considered adverse. This notion is consistent with that described by Perry et al. (2013, 1162) who concluded that “the determination of whether a finding [was] adverse [was] an independent assessment relative to reversibility.” Similarly, Lewis et al. (2002, 74) noted that “knowledge of reversibility is often used as a key part in the weight-of-evidence approach to study interpretation … [that may] influence significantly the overall interpretation and differentiation of adverse from nonadverse effects.” Guidance for Industry M3 (R2) states that evaluation of the potential for reversibility of toxicity should be provided when there is severe toxicity and that the demonstration of full reversibility is not considered essential.
There are many reversible pathological findings that should be considered adverse. Examples include axonal degeneration, osteopenia-related bone fracture, and hypoproteinemia-related edema. In contrast, an example of a reversible nonadverse finding is decreased zymogen granules in the acinar cells of the exocrine pancreas secondary to reduced food intake. This lesion is not considered adverse in isolation but should be considered in the context of other findings associated with reduced food intake. In the case of hyperplastic changes, the impact of reversibility on the determination of adversity will also depend upon its context; these changes may often be reversible after a short recovery period but in some cases may indicate early events in carcinogenic pathways. In such cases, the potential exposure scenarios may determine the impact of reversibility on interpretation. Reversibility should be used to characterize adversity in toxicology studies taking into account the duration and magnitude of exposure, the physiology of the organ/tissue affected, and the type of lesion (proliferative/nonproliferative) in scope. This assessment should provide the necessary information about the “level of risk” for subjects being irreversibly harmed following administration of a potential drug in clinical studies or exposure to an environmental chemical.
Extrapolation of Longer-term or Higher Exposure
Anticipation of effects in longer-term studies or of higher exposure should not influence the adversity decision in a given study, as adversity should be defined within the temporal restrictions imposed on the study design, and with the amount of information available at the time of the integrated toxicology report. In other words, adversity calls for pathologic effects should be based on observed, not hypothetical, findings. This recommendation also applies to evaluation of precursor events (e.g., PPARα activation) which may or may not lead to adverse functional effects in the target cell population. Knowledge of lesions occurring in longer-term/higher exposure studies of the same test item (e.g., pituitary carcinoma) can provide context for a shorter-term finding (e.g., pituitary hyperplasia) but should not generally dictate an adversity call for a finding that would not otherwise be considered adverse in the shorter-term study. Similarly, the lack of effects in a longer-term study (e.g., renal tubular lesions) may reduce uncertainty about a difficult adversity call in a shorter-term study (e.g., renal tubular dilatation).
Translatability/Human Relevance
While adversity should be defined only within the context of the animal model, translatability or human relevance should be considered after the primary adversity call in the nonclinical studies. This stepwise process adds clarity to whether a particular change that is adverse in the test species can be considered relevant to humans. The relevance to humans may be discussed in the pathology subreports and integrated toxicology reports when it can be supported by the literature and/or if there is a well-established view of the human relevance for a particular lesion that is generally accepted in the scientific community.
For example, a test item–related increased incidence and severity of α-2-u globulin nephropathy leading to CPN in young rats would be considered adverse in the animal model at the study level but then discussed as not human relevant in the pathology subreport and nonclinical overview documents. Where applicable, human relevance should be clearly presented in integrated toxicology study reports or subreports and emphasized in nonclinical overview documents. For drugs, the expected indication should not be taken into account for determining adversity in integrated toxicology reports, but it should rather be discussed at the level of nonclinical overview documents. Consequently, changes in the indication of the drug that may occur during its lifecycle will lead to changes in overview documents without affecting the interpretation of integrated toxicology reports. For environmental and food chemicals, exposure scenarios (and uncertainties) often differ from those associated with pharmaceuticals, and animal data are not later replaced by controlled human studies. Therefore, the point in the evaluation process where the human relevance of a given lesion is considered may differ significantly between pharmaceuticals and environmental and food chemicals. For the latter, consideration of human relevance may occur in pathology subreports (in discussion of select lesions), integrated toxicology reports (in discussion of corroborative findings), and summary/overview documents (when defining the critical end points used in quantitative risk assessment or hazard classification). In cases where adverse effects in animals are well known to lack relevance to human beings, it may be useful in the integrated toxicology report or subreport to describe these effects and to reference them with a literature citation.
In some cases, changes deemed nonadverse in test species may be potentially adverse in human populations and should be described as such. One example would be a modest test item–related decrease in spermatogenesis that could potentially decrease fertility in men but does not impact fertility in rodents, which have a greater “excess” or reserve capacity of sperm (Working 1988). Another example is methemoglobinemia, which is highly reversible in rodents and may not result in overt red blood cell defects or anemia (Marrs, Bright, and Woodman 1987). In contrast, humans have a longer half-life of methemoglobin and thus may be more susceptible to oxidant effects on erythrocyte function.
MOA—Expected/Exaggerated Pharmacology
The working group discussed the importance in distinguishing toxicological MOA (the requisite key events by which a compound perturbs normal structure and/or biological function), pharmacological MOA (the means by which a compound achieves its intended therapeutic effect or action), and exaggerated pharmacology (dose-related effects due to excessive modulation of the activity of the primary pharmacological target beyond the point necessary for efficacy). Different opinions were expressed on whether to take the pharmacological MOA into account when deciding on adversity. Such discussions reflect the range of opinions in the literature. Holsapple and Wallace (2008, 90) suggested some degree of tolerance regarding exaggerated pharmacology: “In the development of pharmaceuticals, some changes could be expected as a result of the pharmacology of the drug, and these pharmacologic changes would not be considered adverse, within certain limits, as the compound is designed to produce these changes. When the overall function and life span of an organism does not change in response to xenobiotic exposure, the changes are generally considered non-adverse.” In contrast, a more conservative approach was provided by Dorato and Engelhardt (2005, 271): “Any effect seen in a nonclinical toxicology study, whether it is broadly defined as pharmacology (on-target) or toxicology (off-target), may be considered undesirable and therefore adverse.”
As a general statement, pharmacology should not necessarily preclude a finding from being adverse, but some sort of threshold should be noted between expected pharmacology and exaggerated pharmacology. As an example, delayed gastric emptying as an intended pharmacological effect can be associated with gastric dilation. To a certain degree, this finding would not be considered adverse. However, if it is affecting the general condition of the animal (e.g., with secondary gastric volvulus), it would be called adverse. “Intended” and “exaggerated” pharmacology need to be differentiated, which may be challenging in animal models. In practice, at the study level, some pharmacologic findings might be adverse at certain doses, especially if unintended or severe, while other more minor MOA-related changes might be considered nonadverse. One example is increased mononuclear cell infiltrates evident with immunostimulants. If the infiltrates are similar to historical background but seen in more organs and/or animals, they would typically not be considered adverse. However, if the increased mononuclear cell infiltrates are associated with tissue damage or inflammation, then they may be adverse. Another example is drugs that impact clotting. Factor Xa inhibition is intended to decrease blood clotting and temporarily prolong markers of coagulation (activated partial thromboplastin time and prothrombin time). This intended pharmacology is not considered adverse. However, if the same markers of coagulation are prolonged for extensive periods of time and associated with uncontrolled hemorrhage, the MOA-related changes would be adverse. Calcineurin inhibitors intended for preventing graft-versus-host disease or treating autoimmune diseases provide another example. Atrophic changes in lymphoid organs such as a decrease in the size of thymic medulla with cortical thickening are intended pharmacological effects and not considered adverse. However, when opportunistic infection or increased tumor incidence is noted due to severe or prolonged immunosuppression at higher doses or in longer-term studies, these effects would be considered adverse.
At the summary document level, even exaggerated pharmacological effects may support continued development, depending on the benefit–risk balance for the pharmaceutical in question. In any case, a clear characterization of the pharmacological target, MOA, and pathogenesis will increase confidence in interpretation of the findings and also support consistency in subsequent adversity decisions.
Adversity and Clinical Pathology
There is limited direction in the literature and regulatory documents on how to define adversity for clinical pathology biomarkers. The World Health Organization (WHO) Core Assessment Group on Pesticide Residues (2015) has provided the most comprehensive considerations of nonadverse and “toxicologically significant” changes in hematology, clinical chemistry, and urinalysis. Examples of the latter include a 10% decrease in hemoglobin, the appearance of Heinz bodies, decreased immunoglobulin G, and the presence of cells and blood in the urine. Increased blood methemoglobin (>5% in dogs and >1.5% in rats) is the only finding specifically referred to as adverse.
The overarching theme of the clinical pathology discussion at the workshop was that clinical pathology changes should generally not be considered adverse in isolation but should be associated with adverse anatomic pathology findings and/or observed at critical levels with consideration for clinical adverse outcomes. Critically low levels for red blood cell mass, platelets, and neutrophils as they are related to tissue hypoxia (Ness and Kruskall 2005; Ettinger and Barrett 1995), spontaneous bleeding (Russell 2010), and increased infection (Dinauer and Coates 2005; Johnson, Thompson, and Calia 1985), respectively, were considered to be adverse examples of the latter scenario. Other changes that may be associated with adverse outcomes such as marked hypoalbuminemia and resultant edema and life-threatening alterations in serum electrolytes could also warrant the development of critical high or low values as determinants of adversity.
In many cases, however, it is not possible or reasonable to assign an adversity designation for clinical pathology biomarkers. A designation is not required if it does not assist in data interpretation. For example, an isolated change in mean hemoglobin concentration without associated changes in red cell mass should not be considered adverse, and thus a designation and specific substantiation of adversity is not specifically required. Other examples not requiring an adversity call are changes in large unstained cells because they are not an actual cell type (but a result of gating parameters on automated hematology instruments) and changes in ratios (such as the albumin:globulin ratio) because they are only as relevant as the individual changes in the specific biomarkers that are used to calculate them. Pathogenesis was considered a key factor in understanding the adversity of changes in biomarkers. Without information on associated findings, increases in serum urea and creatinine cannot be put in the proper context of dehydration versus renal disease and increases in serum liver enzyme activity cannot distinguish between damage to tissue resulting in leakage and either induction or lack of clearance.
When determining the adverse nature of a clinical pathology change, one should consider the reproducibility of the change within the study (number of animals and time points affected), control and baseline ranges, the mechanism, and the severity and the rapidity of onset of the change. Without associated clinical and anatomic pathology data, adversity should only be addressed in the subreport when it is clearly linked to a clinical outcome. However, concerns of critical values or potentially adverse findings should be clearly described in the contributor subreport. Due to the ability of clinical pathology changes to be leading premonitory biomarkers and to monitor recovery of the findings, putting every finding in the context of the overall study findings is imperative.
Discussion
There is a clear need for more unified guidance regarding the definition and interpretation of adversity in toxicology. The primary goals of this working group were to propose a working definition of adversity for evaluation of toxicologic pathology results, identify factors that influence adverse or nonadverse decisions, and provide a general workflow for communicating adversity. This workflow is presented in Figure 1, which describes the different steps leading to an adversity designation and summarizes other attributes that accompany the concept of adversity. The consensus definition of adversity from the work group should be applicable to different areas of toxicology and useful for industry as well as regulatory reviewers. Information on characterizing adversity calls should also help reduce discrepancies in the interpretation and classification of study findings and facilitate consistency in the submission and review processes. The working group shared a large number of examples from different arenas to highlight common issues in addressing adversity questions. Additional topics will be reviewed and described in future workshops targeted to specific organ or lesion types.
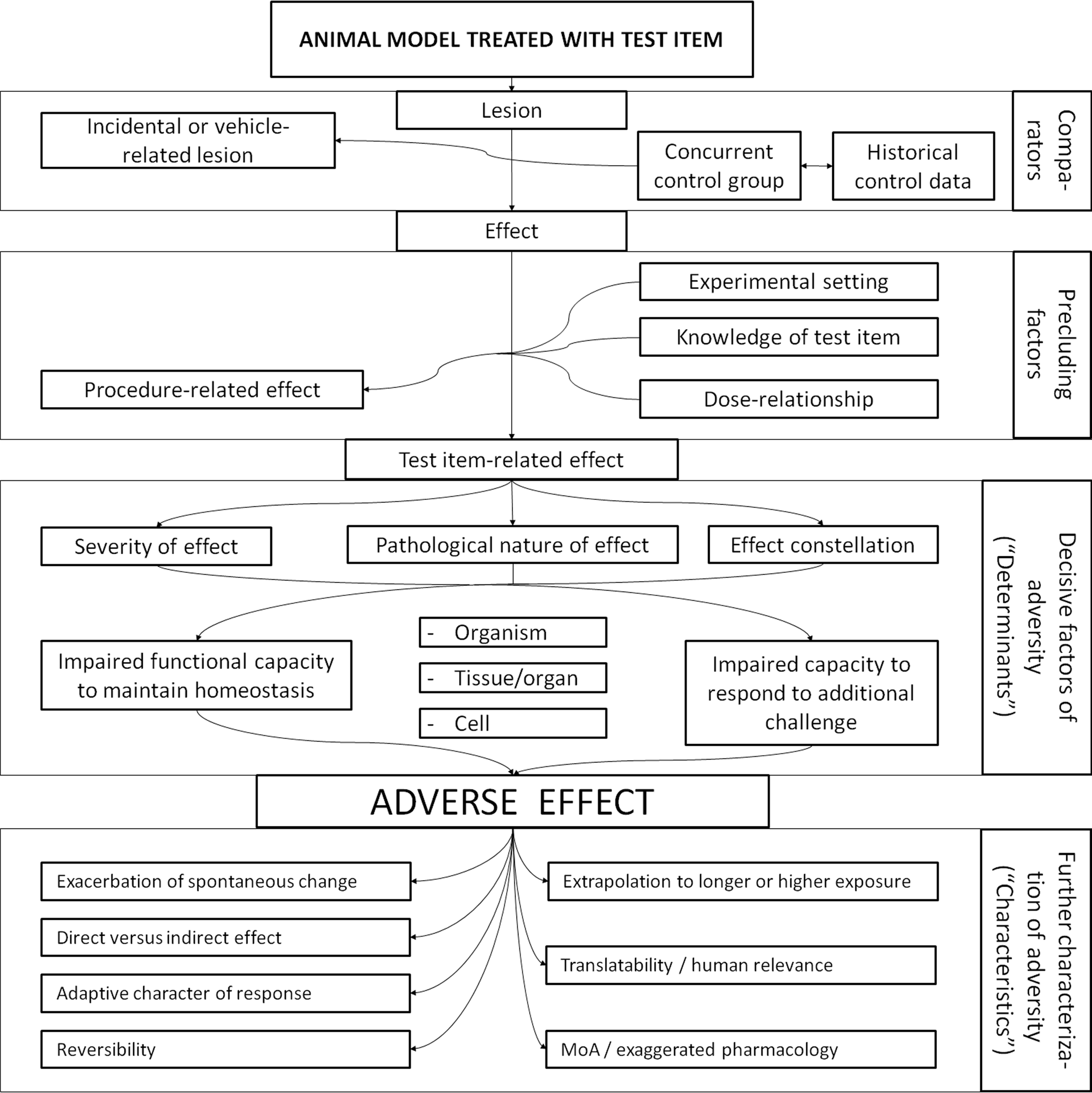
Workflow diagram illustrating the tiered approach for evaluating adversity.
Recommendations of the group generally followed an approach that would apply to interpretation of nonclinical pathology data from both environmental/food chemical and pharmaceutical studies. However, there are a number of specific factors or intrinsic differences in hazard characterization and risk/safety assessment between pharmaceutical and environmental chemicals that impact adversity decisions. Several of these considerations are discussed below.
Pharmaceuticals
For clinical pharmacologists, there is a risk that integrated toxicology reports only present an overview of adverse effects and neglect or exclude other (nonadverse) effects, whereas a broader view, captured under the concept of “safety signals,” is considered more appropriate to support first-in-human studies. Among the different existing definitions, one of them defines a safety signal as a report or reports of an event with an unknown causal relationship to treatment that is recognized as worthy of further exploration and continued surveillance (Council for International Organizations of Medical Sciences VI [CIOMS] 2005).
Consequently, the most important issue for clinicians involved in early clinical development of drugs is not necessarily whether a nonclinical finding is adverse but whether it is relevant in humans, and the potential of early detection and efficient minimization. In order to cap the dose level in clinical studies for nononcologic drugs with healthy volunteers, the NOAEL is the most important determinant since, if properly applied, it should help define the tolerability limits of the potential therapeutic agent. The intended pharmacological action may not be adverse for the relevant patients but might be harmful for healthy volunteers (e.g., weight loss for lean subjects, sleepiness for workers). Therefore, the NOAEL as well as the no observed effect level (NOEL) and pharmacological active dose (PAD) should be considered in determining the safe starting dose in the first-in-human study with healthy volunteers. The NOAEL is critical to determine the permissible daily exposure/acceptable daily exposure and occupational exposure limits, but the NOEL and/or PAD should also be taken into consideration (U.S. FDA 2005; European Medicines Agency [EMA] 2007; Ministry of Health, Labour and Welfare [MHLW] 2012; Nielsen et al. 2008).
From a U.S. pharmaceutical regulatory perspective, the required package to open an investigational new drug (IND) usually includes data from 2 animal species (rodent and nonrodent), and the lowest NOAEL out of these 2 species is used to identify a safe starting dose for clinical trials. Consequently, reviewers must decide how adverse effects in animals affect both the starting dose and the maximum dose to which clinical subject exposures can be escalated. The difficulty is that a given set of toxicity studies on a compound may need to support markedly different patient profiles (e.g., oncology vs. immunology). Workshop attendees felt that in cases that lack information or have unsubstantiated data in integrated toxicological reports, the regulatory reviewers might be forced to make their own interpretation, possibly without all the relevant results in hand.
In Japan, from the collective experience of the authors, there are no substantial differences in the interpretation of adversity between the Pharmaceuticals and Medical Devices Agency (PMDA) and other international regulatory agencies. However, a unique regulatory process in Japan is that IND/Investigational Medicinal Product Dossier (IMPD) packages do not include the final integrated toxicology reports. Assessment by the PMDA is made based only on the investigator’s brochure (IB). The final reports are required upon submission of the new drug application following completion of the clinical trials. This unique situation should change following the implementation of the standard for exchange of nonclinical data initiative. Finally, the PMDA requests not only an explanation of the findings determining the NOAEL but also those findings used for determining the MTD as per the “poisonous and deleterious substances control law.”
There is no specific guidance in Europe relating to the definition of adversity for determination of a LOAEL. The package of nonclinical studies required to support a clinical trial application for an IMP in Europe is generally based on similar guidance (e.g., International Conference on Harmonization [ICH] nonclinical guidance documents) as for the United States and Japan. However, there are differences in the process for data review and approval of clinical trial applications compared to the United States and Japan. A major difference is that data review and approval are based on summarized nonclinical data (e.g., the IB or similar level summaries in the nonclinical section of the IMPD) rather than the actual nonclinical study reports, although full data from the studies and copies of the references should be made available on request. These nonclinical summaries should provide a critical analysis of the available nonclinical data, including justification for deviations and omissions from the detailed guidance and an assessment of the safety of the product in the context of the proposed clinical trial rather than a mere factual summary of the studies conducted.
In the absence of actual study reports, European nonclinical assessors do not have access to data to reinterpret study-specific NOELs, NOAELS, LOAELs, and MTDs. Thus, it is important that there is an overreliance not only on the NOAELs derived from study reports but also on the assessment of safety in the context of the proposed clinical trial, including a discussion of dose/concentration relationships for pharmacological and toxicological effects. An example of the importance of the context of the clinical trial is provided by the ICH S9 guidance that toxicology studies to determine a NOAEL or NOEL are not considered essential to support clinical use of an anticancer pharmaceutical; in such an advanced cancer patient population, use of the highest nonseverely toxic dose may be more appropriate. For IMPs considered “high risk” (EMA 2007), both the NOAEL and the minimum acceptable biological effect level (MABEL) can be of value to guide first dose selection for a clinical trial. MABEL is a pharmacologically based approach like the PAD approach and utilizes all in vitro and in vivo information from pharmacokinetic/PDs data.
Food and Environmental Chemicals
Risk assessment approaches vary widely across different types of chemicals. These differences may be driven by the size of the available database, potential exposure scenarios, and other regulatory protocols and considerations. In the case of pesticides, an extensive toxicological database is required as part of the registration process to assess the potential for adverse outcomes. Laboratory animal data are requested for different species, treatment durations, and routes (oral, inhalation, dermal), and follow well-established guidelines and good laboratory practice (GLP) processes as for pharmaceuticals. These studies are used to identify potential adverse effects to assess potential human risks for different durations (e.g., acute, short/intermediate term, chronic) and routes (oral, dermal, inhalation) of exposure to various populations (e.g., adults, pregnant women, children). Assessments may include evaluations of worker or consumer exposures or dietary (food or drinking water) exposures in the general population. Selected NOAELs or benchmark dose levels for the most sensitive critical effects form the basis of the safety evaluations or risk assessments used in chemical risk management.
The risk assessment process for chemicals that become environmental contaminants most often requires the development of standards for exposures that are anticipated to be without increased levels of risk. For many chemicals, this determination is often based on results from non-GLP studies. In some cases, data submitted for registration purposes may not be available to other assessors evaluating human health risks of individuals who are exposed to the same chemicals through environmental contact. This information gap means that dose–response data, often from animal studies, are extrapolated to identify human exposures that are deemed acceptable. These acceptable levels are then compared to exposures to estimate the likelihood of adverse effects occurring. Differences in the variability of exposures forces some added considerations. While MOEs might be anticipated for some specific human population groups with some appreciable degree of accuracy, this is not the case with the general population, which results in substantially higher uncertainty surrounding MOE values.
As for pharmaceuticals, the quality of documentation and its transparency and consistency will have an important impact on interpretation. The integration of data should follow a WOE approach. In particular, the judgments and choices made, difficult or debated issues, and inherent uncertainties in some of the data should be clearly explained. Consideration should be given to findings observed in more than one study, more than one species, both sexes, or at different treatment durations. Moreover, the time dependence of severity, the toxicity profile in relation to structurally similar chemicals, the response at or near MTD, steepness of dose–response slope, and dose spacing between NOAEL and LOAEL all need to be discussed at the level of summary documents for pharmaceuticals or in the overall assessment for chemicals.
Differences between Pharmaceuticals and Chemicals in the Concept of Adversity
The amount of available information for environmental chemicals can vary from data limited to data rich. For many or most environmental chemicals, there are little to no data on potential toxicity, while for others like food-use chemicals there is often a large nonclinical database. Information gaps for a particular chemical (and thus lack of contextual data) may influence the degree of conservatism in the interpretation of a finding that may represent a potential human health risk. Moreover, for environmental chemicals, there are no human clinical trials to confirm or negate findings in nonclinical studies, and exposure estimates and target populations are often less defined, which can impact human relevance evaluation of adverse effects. Finally, the incorporation of potential health, economic, or other societal benefits into assessments varies across and within regulatory organizations for environmental chemicals. In many cases, there may be no presumed benefit taken into account (by directive), which is different from pharmaceuticals and may influence evaluation of potential risk signals. Many of these considerations occur beyond the individual study level but may nonetheless influence adversity and human relevance decisions about particular findings.
The European CLP regulation mentions that adverse health effects include consistent and identifiable toxic effects in humans or, in experimental animals, toxicologically significant changes which have affected the function or morphology of a tissue/organ or have produced serious changes to the biochemistry or hematology of the organism and that these changes are relevant for human health (CLP guidelines; European Chemical Agency 2015). The ability to establish whether a finding is human relevant (or not) based on human clinical trial data does not exist for chemicals. Moreover, for chemicals, the default assumption is that an adverse effect in experimental animals is relevant for humans. Some regulatory agencies may not use findings that have been conclusively demonstrated to have an MOA that is unlikely to occur in human populations, preferably by provision of literature references (e.g., α-2-u globulin nephropathy in male rats). Others may reduce the uncertainty factor for these MOAs to account for the PD differences between humans and the animal species. This evaluation includes qualitative factors (e.g., target receptor is not expressed in humans) and quantitative factors (e.g., toxicokinetic differences are so marked that it is certain that the effect will not be expressed in humans; Boobis et al. 2006; Boobis et al. 2008).
Future Trends
There is a long-standing recognition of the value of mechanistic information in understanding toxicological effects and the need to apply this knowledge in ways that make the current assessment paradigm more predictive and efficient. Over the past 15 years, the MOA framework has provided an important scaffold for organizing pathway-based toxicity data (Boobis et al. 2006; Meek et al. 2014). To the extent possible, MOA knowledge should be taken into account in a WOE of adversity. For pharmaceuticals, the MOA may inform whether a particular change is related to the intended molecular target, as discussed above. For chemicals, the MOA may inform dose response, susceptibility, cumulative risk, and human relevance, as outlined in previous WOE approaches (using Bradford Hill-like considerations) developed by the WHO International Program on Chemical Safety (Meek et al. 2014; Boobis et al. 2006; Boobis et al. 2008). In many cases, however, this MOA information is not available for pathologic end points routinely encountered in nonclinical studies.
A number of efforts are ongoing to develop new types of tests that would enable a shift toward a more mechanism- or pathway-based approach. One such effort under the auspices of the Organization for Economic Cooperation and Development (OECD) aims to develop a library of adverse outcome pathways (AOPs; OECD 2013). An AOP is defined as an analytical construct that describes a sequential chain of causally linked events at different levels of biological organization that lead to an adverse human health or ecotoxicological effect. AOPs are the central element of a toxicological knowledge framework being built to support chemical risk assessment based on mechanistic reasoning (OECD 2013). The AOP framework is conceptually similar to the MOA but intended to be more prospective in application and relevant to both health and ecological outcomes (Ankley et al. 2010; Becker et al. 2015). Early molecular effects from bioactivity assays, in vitro models, and genomic biomarkers are an important focus of the AOP framework, which is designed to enable predictive models that integrate molecular data streams with more traditional “apical” outcomes. Although the AOP and MOA concepts provide a useful analytical construct to organize, evaluate, and integrate data, the interpretation of whether a molecular perturbation is adverse or not will be challenging. Greater understanding of linkages and dose–response relationships between lower and higher levels of biological organization will be necessary. At least in the short term, pathology will serve as an important bridge in interpreting the adversity of responses at lower levels of biological organization.
Conversely, as more pathway-based mechanistic data become available, this information should have a greater role in characterizing adversity of specific pathological outcomes and their potential relevance to human health. Currently, the vast majority of nonneoplastic pathologic effects used for NOAEL/LOAEL determinations do not have corresponding mechanistic data or functional assays. The MOA and AOP frameworks have been developed to help facilitate the generation, organization, and application of newer data streams. In the future, these constructs are intended to help shift the classical concept of adverse effects based predominantly on morphologic outcomes to more integrated assessments that include newer molecular data streams (Keller et al. 2012).
Conclusions
The ultimate goal of this workshop was to increase consistency in the characterization and interpretation of adversity based on pathology findings in nonclinical studies. While considerations discussed here should add structure to this process, an important conclusion of this workshop was that there is no one formula or method that can be applied to all adversity decisions. The consensus definition of adversity by the work group was based on impairment of functional capacity to maintain homeostasis or respond to an additional challenge. However, the participants acknowledged that adversity decisions for many pathologic end points should remain context dependent and be conducted on a case-by-case basis using a WOE approach. In addition to histopathology, this evidence may include associated findings, pathophysiological processes, and study design considerations (e.g., number of animals, time course, and mode of administration). Where possible, adverse effects should be further characterized by parameters such as reversibility, relationship to physiologic or adaptive responses, and MOA, which should be used to accompany and justify adversity statements for questionable lesions. General steps to be followed include (1) evaluation of whether a finding is test item related, (2) determination of adversity (in the test model), and (3) further characterization of adversity including assessment of human relevance. This information should provide study pathologists, toxicologists, and reviewers with guidance regarding when, where, and how adversity should be determined. Future steps will consist of organ- or lesion-specific workshops to help clarify problematic or uncertain adversity issues in toxicologic pathology, including the use of standard thresholds for some findings.
Footnotes
Acknowledgments
The authors would like to thank Inger Thorup (Novo Nordisk A/S, Denmark) and David Jacobson-Kram (ToxRox Consulting, USA) for their valuable contribution to some of the presentations at the workshop. We are grateful to Jennifer Sims (Integrated Biologix GmbH, Switzerland) and reviewers from the ESTP and U.S. EPA for their review comments on this article.
Author Contributions
All authors (XP, JB, HC, VD, PF, JF, SF, PG, SG, TH, JH, KI, WK, BL, HN, GP, AS, MS, LT, CW, MY) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Authors’ Note
The opinions expressed in this document are those of the authors and do not reflect views or policies of the employing institutions including the U.S. FDA. This article has been reviewed by the U.S. EPA and approved for publication. Approval does not signify that the contents necessarily reflect the views or the policies of the Agency. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This workshop was coordinated and supported by ESTP and hosted by Sanofi (Centre de Recherche de Vitry-Alfortville, France).