
Abstract
Recommendations (best practices) are provided by the Society of Toxicologic Pathology’s Adversity Working Group for making consistent interpretations of test article–related effects as “adverse” and assigning a “no observed adverse effect level” (NOAEL) in nonclinical toxicity studies. Adverse is a term indicating “harm” to the test animal, while nonadverse indicates lack of harm. Adverse findings in the study reports should be defined in relation to effects on the test species used and within the context of the given study. Test article–related effects should be described on their own merits, and decisions to consider them as adverse or nonadverse should be justified. Related effects may be discussed together; in particular, markers of toxicity that are not in and of themselves adverse ideally should be discussed in conjunction with the causal toxicity to determine adversity. Adverse findings should be identified in subreports (clinical data, pathology data, etc.) if sufficient information is available, and/or in the final study report as individual or grouped findings, but study NOAELs should be established at the level of the overall study report. Interpretations such as “not biologically relevant” or “not toxicologically important” should be avoided unless defined and supported by scientific rationale. Decisions defining adverse findings and the NOAEL in final study reports should combine the expertise of all contributing scientific disciplines. Where possible, use of NOAELs in data tables should be linked to explanatory text that places them in context. Ideally, in nonclinical summary documents, NOAELs from multiple studies are considered together in defining the most important adverse responses in the most sensitive species. These responses are then considered along with an understanding of their likely mechanisms, as well as other information such as variability in species sensitivity, comparative pathology, reversibility and progression, kinetics, and metabolism of the test substance to help assess human risk.
Introduction
The interpretation of findings in nonclinical toxicity studies as either “adverse” (i.e., harmful) or “nonadverse” and the subsequent use of the “no observed adverse effect level” (NOAEL—defined as the highest dose with no adverse effects observed within the study) have been used commonly for over 4 decades to communicate the relevance of animal toxicity data for predicting potential human outcomes to physicians and regulators. However, the absence of a universally accepted, science-based standard on how to define and extrapolate NOAEL-based decisions has made the NOAEL concept difficult to consistently apply, interpret, and use (Karbe et al. 2002). The benchmark dose (BMD), which employs data modeling to examine the dose response, is used in some settings as an alternative approach to the NOAEL (Bokkers and Slob 2005, 2007; U.S. Environmental Protection Agency [U.S. EPA] 2012). Unfortunately, a BMD cannot always serve as an alternative index of adversity as data sets submitted to regulatory agencies may not be sufficiently broad to permit the use of a model. Accordingly, the Society of Toxicologic Pathology (STP) commissioned an Adversity Working Group to develop recommendations (“best practices”) for determining, communicating, and using adverse effect data from nonclinical studies so that this information will better inform physicians and regulators about the potential human risks posed by products being developed for medical or environmental uses. Although this document is written mainly in reference to bio/pharmaceutical perspectives, many of the recommendations address fundamental views that also apply to safety assessments for environmental chemicals, food, and medical devices.
It is the ultimate responsibility of each organization to ensure that toxicology study documents and nonclinical overview regulatory documents for a given product include clear and cohesive interpretation of study findings and their relevance to humans. While it is acknowledged that a variety of approaches may be used successfully by different organizations to achieve this goal, the following recommendations represent a preferred approach to facilitate clarity in communication of adversity and human relevance in toxicology study and summary documents.
Assessments of “adversity” represent empirical measurements (i.e., objective data) integrated with well-informed subjective judgments to determine whether or not a response is considered harmful to an organism’s health. Attempts have been made to use quantitative approaches for this purpose, such as the “critical effect size,” which defines the breaking point between adverse and nonadverse changes in a toxicologic effect on an individual organism for continuous data (e.g., a 10% decrease in red blood cell count compared to the mean of the control group; Dekkers et al. 2006). However, the suitability of these quantitative strategies across species, strains, sexes, and ages is yet to be fully evaluated. The development of guidelines for standardizing the interpretation and reporting of adversity decisions using nonclinical data should retain flexibility in determining whether or not a finding is adverse or nonadverse, while increasing the likelihood that adversity interpretations will have greater consistency and transparency. The current STP best practice recommendations have been developed to provide such flexible guidance. Clarity achieved through more consistent and transparent application of the adversity concept within nonclinical reports will improve communications in regulatory submissions, benefitting both clinicians and regulators charged with protecting human health. Failure to clearly apply and justify the determination of adversity decisions with respect to test article–related effects could lead regulatory agencies to question an institution’s interpretation of nonclinical data sets in product submissions and apply a more conservative standard of adversity instead.
This publication provides a synopsis of the issues that have caused problems in defining and utilizing the concept of adversity in the past and, more importantly, offers 10 recommendations that will result in more consistent determination, communication, and use of adversity and the NOAEL by pathologists and toxicologists. The practices are based on established principles that an individual toxicity study identifies and characterizes hazards of an agent while the results from multiple studies function together to inform human risk assessment. These recommended practices will minimize misunderstandings related to evaluating the implications of nonclinical effects for assessing human risk. The ultimate objective of these recommendations is to promote more effective and efficient product development by improved communication through better reporting.
Adversity and the NOAEL as Defined in the Literature
Many technical improvements have been instituted in the conduct of nonclinical toxicity studies since the 1940s. Taken together, nonclinical toxicity studies in the 21st century generally have developed a high level of quality in all respects, including animal health and welfare, study design and analytical methodology, quality control, pathology and toxicology techniques and interpretations, scientific peer review, and detailed reporting. Despite all of these technical advances, considerable differences still remain between the spectrums of harmful effects produced by individual test articles in animals versus the adverse effects induced in people (Greaves, Williams, and Eve 2004; Olson et al. 2000). Such differences highlight the essential limitations in modern risk assessment.
One common approach to facilitate risk assessment has been the widespread adoption of the NOAEL in animal toxicity studies to predict the potential safety of novel materials to which humans will be exposed (bio/pharmaceutical agents, agricultural and commodity chemicals, food additives, etc.). As generally applied today, the NOAEL is considered to be the dose (or level of exposure) determined by empirical study at which no adverse effects (i.e.,
The following brief review showcases the range of different approaches and interpretations that have been used in the last decade to identify adversity. This background is essential because improvements in communicating adversity across institutions can be made only if prior difficulties in defining and applying the adversity concept can be minimized.
a. Adversity
Many publications written to discuss the adversity concept do not define the word (Holsapple and Wallace 2008; Hosford et al. 2004; Kimber and Dearman 2002; Muller and Milton 2012). Other publications have attempted to explain the limits of the concept (Dorato and Engelhardt 2005; IPCS 2004; Lewis et al. 2002), including at least one compilation designed to compare and contrast definitions of “adverse effects” (Table 1; Krewski et al. 2010). This compilation highlights the extent of the difficulties that can be encountered when trying to define and utilize the term adversity.
Recent Definitions of Adverse Effect.a

Adversity definitions in the recent literature (Hall et al. 2012; Keller et al. 2012) have been built up from 2 definitions published in the early 21st century. These 2 definitions acknowledge that multiple parameters may be impacted by test articles. In one case, adversity is defined as a: … biochemical, morphological or physiological change (in response to a stimulus) that either singly or in combination adversely affects the performance of the whole organism or reduces the organism’s ability to respond to an additional environmental challenge. (Lewis et al. 2002)
In the other case, adversity is defined as a: … change in the morphology, physiology, growth, development, reproduction, or life span of an organism, system, or (sub) population that results in an impairment of functional capacity, an impairment of the capacity to compensate for additional stress, or an increase in susceptibility to other influences. (IPCS 2004)
b. NOAEL
Similar to the term adversity, the meaning of NOAEL remains ambiguous until the connotation of adverse has been defined in the context in which it is to be used. Several approaches may be utilized to better define the NOAEL.
Many definitions in the literature frame the NOAEL concept in terms of statistical considerations. For example, the NOAEL may be understood as … the highest dose yielding a response that is both statistically indistinguishable from the control outcome yet significantly different from the response observed at the “lowest observable adverse effect level (LOAEL). (Calabrese and Baldwin 1994) … the highest dose that does not produce a significant increase in adverse effects (i.e., responses that are statistically significant or that may be clinically significant [even if they are not statistically significant]). (U.S. Food and Drug Administration [US FDA-CDER] 2005)
A different perspective acknowledges that decisions regarding communication of adverse findings may incorporate statistical calculations but that they also need to retain flexibility to acknowledge that: … lack of statistical significance alone does not constitute a NOAEL. Rather the NOAEL is determined by the combined analysis of the biological and statistical effects. (Dorato and Engelhardt 2005) In animal studies, the highest level of drug exposure that does not lead to toxicity ([is] termed the no observable adverse effect level; NOAEL). (Muller and Milton 2012) [t]he “no observable adverse effect level” (NOAEL) is the highest dose where no effect considered deleterious to the well-being of the animal is observed. (Holsapple and Wallace 2008)
Ultimately, the difficulties in harmonizing the various definitions for the NOAEL rest in the nature of what is intended to be communicated by the concept. The problem stems from the tension between 2 diametrically opposed views regarding what may be inferred from the NOAEL: an objective value established by rigorous experimentation or a well-reasoned judgment based on both measurable and intangible factors. Thus, the concept of adversity, and the consequent assignment of a NOAEL, necessarily will merge aspects of objective and subjective analysis. By this view, a useful perspective in understanding the NOAEL and its potential utility in human risk assessment is that [i]n general use, the NOAEL … is a professional opinion about toxicologically relevant effects and interpretation of expected pharmacology, and it is the subject of iterative interpretation as the toxicology profile develops. (Dorato and Engelhardt 2005)
c. Nonadverse
Definitions of nonadverse typically are vague as they are influenced by the many potential meanings applied to adverse. The concept that a test article–linked effect may be both observable and yet not harmful is acknowledged by some definitions. For example, [c]ontrasted to adverse effects, non-adverse effects can be defined as those biological effects that do not cause biochemical, morphological, or physiological changes that affect the general well-being, growth, development or life span of an animal. (Lewis et al. 2002) [w]hen the overall function and life span of an organism does not change in response to xenobiotic exposure, the changes are generally considered non-adverse. (Holsapple and Wallace 2008) a mildly increased serum cholesterol concentration in female (Choi et al. 2011; Willson et al. 2012) and obese (Tvarijonaviciute, Tecles, and Ceron 2010) beagle dogs, which is not considered to be harmful in this species in the absence of other changes, and a slight but compensated perturbation in peroxisome function in animals such as rodents (Ammerschlaeger et al. 2004).
Interpretation of a demonstrable test article–related change as nonadverse must include the broader context explaining why the data have been interpreted in that way.
In many instances, nonadversity is associated with a list of criteria used to differentiate harmless test article–related effects from harmful (adverse) effects. An effect is less likely to be judged as adverse under the following conditions (according to Lewis et al. 2002): No alteration in the function of the test organism or affected organ/tissue is noted; the change represents an adaptive response (such as liver enlargement due to enzyme induction); the finding is transient; the severity is limited, falling below thresholds of concern (which must be clearly defined for each effect); the effect is isolated or independent (i.e., changes in other parameters that usually are associated with the effect are not observed); the effect is not a precursor lesion (i.e., part of a continuum of changes known to progress with time to yield a known adverse effect); the result is a secondary consequence indirectly caused by some other adverse effect(s); and the effect arises from some inherent biological property of the animal model.
However, many authors avoid defining the meaning of nonadverse and instead permit readers to draw their own inferences for the concept from the implied meaning gained by integrating their discussion points (Dorato and Engelhardt 2005; Hosford et al. 2004; Keller et al. 2012; Kimber and Dearman 2002).
d. Other terms related to assessments of adversity
Decisions regarding the potential harm posed by a test article–related change often rest on a series of related concepts. Unfortunately, definitions for these concepts, which may be important in assisting with differentiating between adverse and nonadverse findings, are rarely offered in the “adversity-related” literature and thus can be confusing. Examples include vague definitions for critical terms such as “biological/toxicological relevance” and “biological/toxicological significance.”
Various authors have taken differing approaches to distinguish between these concepts. Some attempts are focused on “importance,” with no particular application to protecting human health. For instance,
[A] biologically significant effect [is] a response (to a stimulus) in an organism or other biological system that is considered to have [a] substantial or noteworthy effect (positive or negative) on the well-being of the biological system. The concept is to be distinguished from statistically significant effects or changes, which may or may not be meaningful to the general state of health of the system. (Lewis et al. 2002)
Toxicologically [r]elevant [denotes an] effect [that] may endanger human health. (Dorato and Engelhardt 2005)
Regulatory Perspectives on Adversity
Nonclinical data are used to evaluate the potential risk by multiple regulatory agencies that have vastly different legal statutes, scopes of influence, and regulatory requirements. The differences extend not only to distinctions in the class of materials to be regulated (e.g., chemicals by the U.S. EPA vs. medical products by the U.S. FDA) but also to various national differences (e.g., Europe vs. Japan vs. the United States). Divergence also exists in the regulatory practices of different divisions of a single agency, such as the FDA product centers engaged in assessing biologics (Center for Biologics Evaluation and Research), drugs for humans (Center for Drug Evaluation and Research [CDER]) and animals (Center for Veterinary Medicine), food (Center for Food Safety and Applied Nutrition [CFSAN]), and devices (Center for Devices and Radiological Health). Even within a single division, regulatory deliberations will contrast among offices that examine products that possess divergent safety profiles (e.g., drugs to treat juvenile diabetes vs. agents to treat cancer).
The context of exposure will dictate the risk assessment strategy for various products. For example, risk assessments for drugs and devices are based on a combination of nonclinical data from animal studies and clinical data from human trials that examine both efficacy and safety. All of this information is communicated in overview documents—both nonclinical and clinical—in order to clearly understand the implications of any adverse findings (Jorkasky 1998). In contrast, risk assessments for environmental chemicals are made chiefly from nonclinical safety studies because human epidemiological data typically are lacking and human clinical trials are not ethical (environmental chemicals) or seldom conducted (food additives). Therefore, in settings where human clinical trial data are limited or absent, adverse findings and NOAELs derived from nonclinical studies in animals carry much greater weight in the decision-making process.
At the start of the drug development process, adversity interpretations based on nonclinical data from animals play a greater role than human data in the regulatory safety assessment for a novel drug by FDA. Characterization of adversity in nonclinical studies informs the maximum starting dose to be given to human subjects during initial phase 1 clinical trials. In the relevant guideline (U.S. FDA-CDER 2005), FDA states that the true value of determining adversity, from which the NOAEL derives, is to serve as a benchmark for dose-setting, “which should be acceptable to all responsible investigators.” This same guidance also acknowledges that “the nature and extent of adverse effects can vary greatly with different types of therapeutics, and it is anticipated that in many instances experts will disagree on the characterization of effects being adverse or not….” The document concludes that “as a general rule, an adverse effect observed in nonclinical toxicology studies used to define a NOAEL for the purpose of dose-setting should be based on an effect that would be unacceptable if produced by the initial dose of a therapeutic in a phase 1 clinical trial …” (U.S. FDA-CDER 2005). Later on, the risk/benefit analysis is based much more heavily on the assembled data from multiple human clinical trials. Depending on the test system, some adverse effects may correlate well between animals and humans, while biological responses in other systems are correlated poorly (Dorato and Engelhardt 2005; Freireich et al. 1966; Owens 1962; Schein et al. 1970). As experience with human biological responses is gained during clinical trials, data derived from animals should diminish in importance as a means of evaluating an agent’s potential risk to humans, particularly when the risk perceived from animal studies can be monitored and does not occur in humans. Notable exceptions to this emphasis upon using human findings for conducting human risk assessments are for carcinogenicity or developmental toxicity end points because these outcomes cannot, or should not, be available for monitoring in clinical trials. Nonclinical data can also identify potential safety/toxicity biomarkers that might be translated from the animal studies to human clinical trials to facilitate effective monitoring for treatment-related adverse effects.
In contrast, the risk assessments made by FDA-CFSAN incorporate nonclinical data to a much greater degree as compounds to be added to food are rarely tested in human clinical trials. Indeed, food safety assessments generally are made exclusively based on toxicity data derived from subacute and long-term animal toxicity studies (U.S. FDA CFSAN Redbook 2000). In contrast to drug candidates, the statute giving the FDA regulatory responsibilities related to food ingredients mandates consideration of a compound’s safety profile while expressly prohibiting consideration of any of its inherent benefits (FDA Code of Federal Regulations [CFR]—Title 21, Part 170—food additives). The safety threshold is “proof of reasonable certainty that no harm will result from the proposed use of an additive.” Although harm is not specifically defined in the legislation, “the legislative history reflects that an effect is harmful if it affects health, not if it is simply an undesirable or unexpected effect that has no adverse health consequences” (Mattia 2011). Similarly, with regard to food additives, the Delaney Clause in the Food, Drug and Cosmetics Act (FDCA of 1938 and its 1958, 1960, 1997 amendments 1938) indicates that with regard to carcinogenic effects, the risk assessment pertains not only to neoplasms observed in humans but also to those observed in animals: No additive shall be deemed to be safe if it is found to induce cancer when ingested by man or animal or if it is found after tests which are appropriate for the evaluation of the safety of food additives, to induce cancer in man or animal….
The target audience needs to be considered when study findings are being reported by pathologists and toxicologists. The ultimate target audiences of pathology data presented in regulatory submissions are global and national regulatory agency reviewers and clinicians, who, together, are responsible for approving and overseeing human clinical trials, respectively. These documents will have additional audiences such as project and management team members charged with reviewing nonclinical study reports before they are issued. Consequently, the report authors should take great care to communicate all study findings in a clear, complete, and sufficient manner to meet the needs of the target audience(s). Specifically, it is recommended to make certain that the report includes an appropriate rationale why test article effects within the study are considered to be either adverse or nonadverse.
Recommended (“Best”) Practices for Determining, Communicating, and Using Adversity in Nonclinical Studies
Ten recommendations are presented below in the sequential order that a toxicologist and/or toxicologic pathologist might encounter them when interpreting toxicity study data. Recommendations 1 through 4 address critical points related to determining adversity and a NOAEL. Recommendations 5 through 8 focus on communicating adversity and the NOAEL. Recommendations 9 and 10 consider using adversity and NOAEL in assessing human risk. These 10 recommendations specifically differentiate between communication of adversity in subreports, and/or final study reports from interpretation of their implications for human risk as conveyed in nonclinical overview documents. For clarity, the principal components of each recommendation are in italics at the beginning with the main points underlined, followed by explanatory discussion of each recommendation.
When using these recommendations, authors must recall that a critical aspect in using the adversity concept in nonclinical studies is the decision regarding “when” to use it. The notion of defining a test article effect as adverse or nonadverse should be limited to studies that are to be used to design human clinical trials and limit human exposure (intentional with regard to volunteers or patients given drugs and consumers eating food additives, and inadvertent for people exposed to environmental or industrial chemicals). In contrast, while they may be included at the discretion of the company generating the data, adversity and the NOAEL are not required for interpreting nonclinical data sets obtained from exploratory (dose range finder) or early screening studies designed to (1) discriminate between potential drug candidates, (2) establish doses for subsequent regulatory studies, and/or (3) identify potential target organs so as to better design definitive studies. This latter point is important because the outcomes of these early studies may be reported to regulatory agencies as part of product registration packages.
Determining Adverse and “NOAEL”
Administration of a test article may cause one, several, or many changes in the list of anatomic, biochemical, functional, and physiological features that are analyzed in building the data set for a nonclinical study. However, the concept of adversity may be applied properly only to that fraction of changes in which there is an indication that the animal is harmed in some fashion (Figure 1). Defining whether or not a finding is “harmful” (i.e., adverse) or is “not harmful” (i.e., nonadverse) should be done in the context of the individual study; harm should not be extrapolated further than the dose of test article, duration of the study, and so on, employed for a given study. For example, an effect would not generally be viewed as adverse on the assumption that it might prove harmful at higher doses or after a longer duration of exposure if those conditions have not been tested in the study at hand.
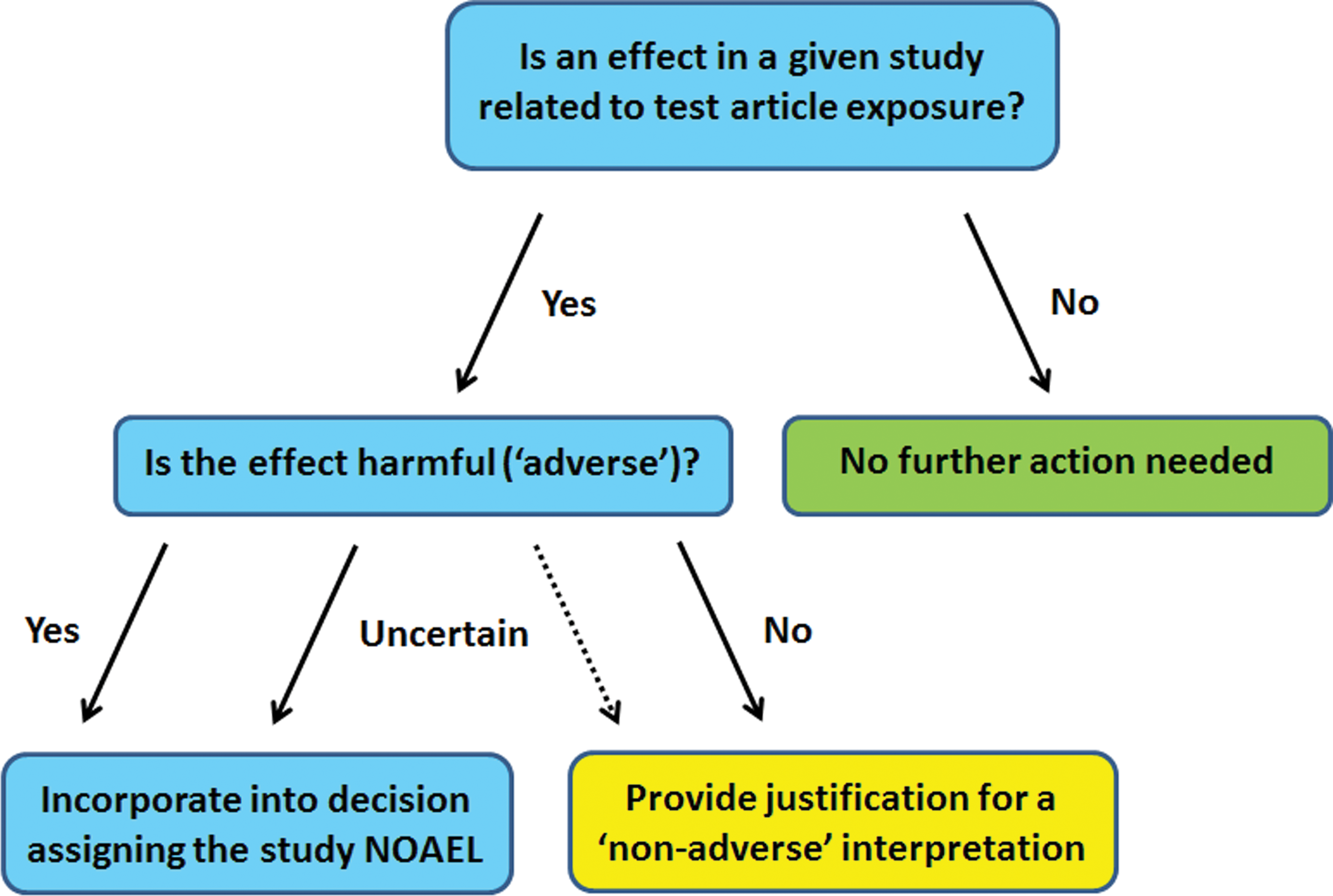
Schematic diagram demonstrating the intellectual steps involved in determining whether or not a test article–related effect will be interpreted as “adverse” (i.e., harmful) or “nonadverse” and the major points that must be considered when communicating such decisions.
Fundamentally, the decision to designate a test article–related effect as adverse relies on an
It is important to note that each adverse or nonadverse finding does not have to be dealt with separately. Test article–related effects that typically occur in combination should be addressed together as being either adverse or nonadverse (e.g., liver weights with serum liver transaminase enzymes and histological changes; or small effects on serum analytes such as sodium, phosphorus, glucose, and protein decreases associated with reduced food and water intake). Specifically for clinical pathology measurements, test article effects that are adverse in and of themselves (very low platelet or red blood cell counts, etc.) should be addressed within the subreport and the study report. Instances where changes in an analyte are not intrinsically adverse but are markers of an adverse change (e.g., increased serum activity of alanine aminotransferase [ALT] and aspartate aminotransferase [AST] due to hepatocellular necrosis) are properly discussed in the anatomic pathology subreport and/or overall study report in conjunction with the proximate cause (i.e., hepatocellular necrosis) without a requirement for an explicit determination of adversity in the clinical pathology subreport.
In certain instances, minor findings that obviously are part of a single process related to a particular organ may not need to have separate specific statements to justify their lack of adversity, either in the subreport or study report. However, in such cases the text of the subreport and/or study report should address clearly why such clustered treatment-related effects are not considered to be adverse. Similarly, other minor, but possibly unrelated test article effects may be addressed together as being nonadverse due to their small magnitude, transient nature, lack of functional impact, lack of typically associated findings, or some other similar but well-reasoned explanation. Information pivotal to determining adversity may be derived not only from the study data set but also using data from other studies done with the same, related, or unrelated test articles; data from control animals (concurrent or historical), either within or outside of the testing laboratory; published literature; and the collective experience of personnel who participated in the study. Authors of study reports should strive to unambiguously communicate whether or not test article–related effects are interpreted as adverse or nonadverse so that individuals tasked with protecting human health (e.g., authors of nonclinical summary documents, clinicians, and regulators) will be able to appreciate the rationale for such designations when making decisions regarding potential human risk.
The relevance to humans of changes observed in animals represents a major issue because adverse findings in an animal study may not be considered adverse in humans and vice versa. The key choice in this regard is whether or not extrapolations concerning adversity in the test species should also be extended to predict the human response to test article exposure. One perspective on this issue is that anything which induces a harmful effect in the most sensitive of the usual mammalian test species (i.e., dog, nonhuman primate, rabbit, and rodent) should be considered relevant for determining the potential of producing adversity (toxicity) in humans, whether or not data are available to indicate that humans may exhibit a similar response (CLP Regulation 2008).
The opposing perspective holds that toxic effects, well recognized to be restricted to the test species (or at least not having a direct correlate in humans), should not be reported as adverse events when communicating the outcome of nonclinical studies as they have no relevance in estimating the potential risk to humans. Unfortunately, these divergent approaches are not used consistently, thereby creating confusion. Several major difficulties are encountered when trying to decide how to balance these opposing approaches.
a. When toxicity in a test animal is interpreted as being specific to that species and lacking relevance to humans, the test article effect may still be an adverse response for the species being tested.
The importance of this issue is founded on 2 related questions that seek to prescribe the limits surrounding interpretations of species-specific findings. May a severe toxic effect in the test species (even those that cause debilitation or death) ever be considered as nonadverse just because the literature suggests that it is a species-specific toxicity, which thus will be irrelevant to human biology? By extension, if a decision is made to consider a harmful finding as specific to the test species and not relevant to humans, what criteria would provide sufficient weight of evidence to support this interpretation? The perspective one takes regarding these 2 questions will influence decisions regarding adverse and nonadverse when interpreting nonclinical data sets.
A pharmaceutical product development example related to the first question is the occurrence of test article changes within a tissue or tissue substructure that is present in the animal species but is either absent or morphologically different in the human. The azalide antibiotic azithromycin produces phospholipidosis in multiple species, but a unique feature of the test article is striking accumulation within the tapetum lucidum of dogs, which is accompanied by histological and ultrastructural changes in tapetal cells (Fortner et al. 1993). Studies conducted with mutant dogs lacking a developed tapetum lucidum revealed mild morphologic changes in residual, incompletely developed tapetal cells; however, the presence of an incompletely developed tapetum lucidum resulted in the absence of severe test article accumulation because the volume of the putative reservoir was itself not present (Fortner et al. 1993). Unfortunately, predicaments in the use of the adversity term are commonly “resolved” inappropriately by a compromise approach in which mild findings that may be declared as animal-specific are discounted as being nonadverse (purely on the basis of presumed lack of human relevance) while more serious findings are judged to be adverse because of functional consequence to the animal (e.g., impairment of vision being a deleterious outcome). This compromise approach to assigning adversity should be avoided because it introduces subjectivity that is inconsistent with an evidence-based application of animal data for human risk assessment. Instead, the adversity of a test article–related effect should be defined in the study report based on its impact in the test species alone. This scenario reinforces the need for more uniform communication in identifying adversity. Conversely, findings that are within acceptable limits for a nonclinical species are sometimes at risk of being deemed adverse because their occurrence in human patients would be unacceptable. For example, mild test article–related emesis in dogs is not uncommon as they are especially responsive to emetic stimuli; indeed, in this species, such an effect may not cause debilitation or even alter food consumption (Bowen 1996; Elwood et al. 2010; Bassett et al. 2014). Therefore, emesis should not be considered adverse in the context of a dog toxicity study unless body weight (or another parameter, such as hydration status) is significantly affected. Although relevance to human does not contribute to the adversity designation, study report authors may find it useful to cite relevant literature or other information that provides context for test species–specific findings.
In development of products where deliberate exposure of humans is often not feasible to evaluate “efficacy” and/or safety, such as environmental chemicals, foods, and so on, it may be valid to exempt a finding from calculations used for setting allowable human exposure thresholds. This decision should be accompanied by a sound rationale. For example, chemical modulation of spontaneous α 2u globulin accumulation, leading to renal tubular cytotoxicity, may be adverse for rats in a study (Hard et al. 1993) and contribute to the study NOAEL. However, this male rat-specific process is not present in humans, which may provide
b. Neither the therapeutic indication nor the patient population should influence adversity decisions in the test species.
A test article–related finding in animals should not be considered to have a different degree of adversity in nonclinical reports with regard to separate subpopulations of human patients simply because the severity of the change is deemed to be less objectionable for individuals with particularly severe disease. Accepted drug development practice often leads to test article evaluation as a therapy for several distinct diseases and indications. However, toxicity that would be permitted as allowable for one indication (e.g., cancer) could be considered an inappropriate risk to another indication (e.g., diabetes). Since the report for a nonclinical study may be used to support both of these therapeutic indications, any text in a nonclinical study report that states a decision about adversity and the NOAEL in terms of clinical indication rather than the objective animal data creates a dilemma. The desire to avoid such challenges may lead to substantial pressure to declare such lesser findings as nonadverse from the beginning. In general, it is difficult to craft a single study report in which such a finding may be classified as both adverse and nonadverse depending on context; such nuanced discussions instead belong in the nonclinical overview document that integrates the data from multiple studies in multiple test species to help assess risk in humans receiving the test article for particular therapeutic indication (Figure 2).
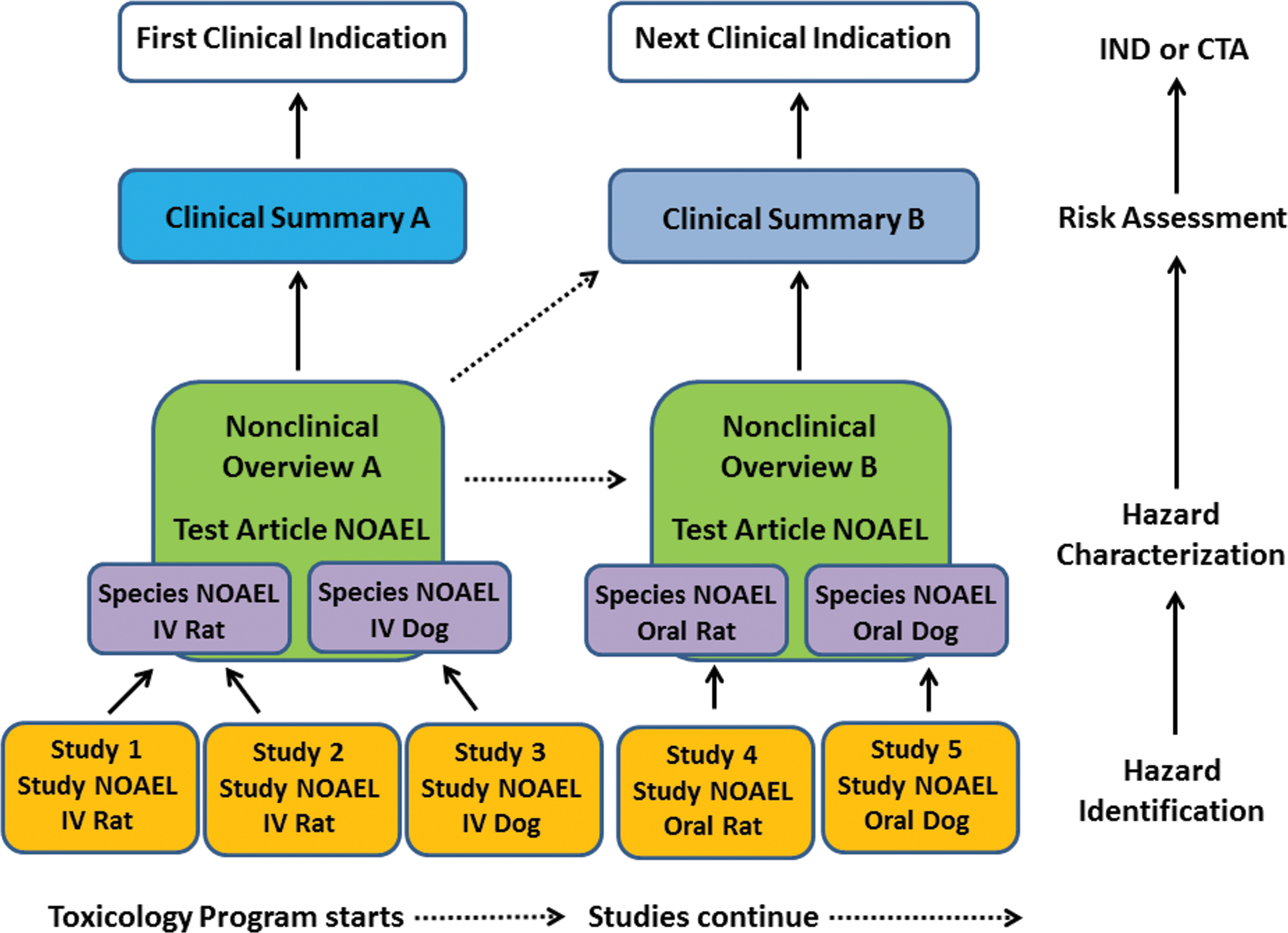
Schematic diagram delineating the hierarchy (built here from the bottom up) by which “adverse” test article–related effects are communicated when assembling a regulatory submission for an agent (shown here for a pharmaceutical candidate). For hazard identification, a series of nonclinical studies (yellow boxes)—typically combining data from multiple subreports: absorption/distribution/metabolism/excretion, pathology, pharmacology, pharmacokinetics, and so on—will be undertaken for one or more clinical indications (white boxes), each of which will yield a study-specific “no observed adverse effect level” (NOAEL) that is applicable only to the species, formulation, and regimen under the conditions of the study in question. For each indication, the NOAELs from different studies for a given animal species are compared to define the species-specific NOAEL (purple boxes), which then are considered together with other relevant data (e.g., historical control values, literature reports) in preparing an integrated nonclinical overview document (green boxes) that assigns a test article–specific NOAEL and affords a detailed characterization of the in vivo hazard as indicated by responses in test animals. In pharmaceutical development, the nonclinical overview will be used in combination with a clinical summary (blue boxes) relating the outcome of human clinical exposures in assessing human risk; for products in which human trials are not performed routinely (e.g., environmental chemicals and food additives), the clinical summary may be replaced by human epidemiologic data but often is absent, such that the nonclinical overview will assume magnified importance in the risk assessment process. Two essential points must be recognized by individuals using nonclinical study reports and nonclinical overviews. First, these documents, which are based on empirical measurements, will not change once they have been completed, so nonclinical documents prepared early during a development program may be used to inform (dotted lines) the integration of nonclinical overviews and clinical summaries that are assembled later in the program. Second, interpretations regarding a particular clinical use, such as prediction of an acceptable safety margin and dose range, are best incorporated in the clinical summary rather than in the nonclinical documentation because such human-specific issues may change dramatically depending on the human population at risk. Adoption of this latter point will ensure that previously finished nonclinical reports and overviews do not need to be revisited and revised if different clinical summaries (indicated by the differing shades of blue) must be assembled to support distinct clinical indications are devised in the future. (IND = Investigational New Drug Application for US FDA; CTA = Clinical Trials Authorization for European Medicines Agency).
c. Test article–related exacerbations of background lesions in animals can be considered adverse.
The incidence and/or severity of common background (“incidental”) findings in animals may be changed by test article administration. In most cases, such alterations present as increases, typically of minimal to mild degree. When they occur, these shifts often are postulated to result secondarily from general debilitation rather than as a primary outcome of direct target organ toxicity. However, the many cases where similar levels of debilitation in other studies did not result in increases in the same background change are rarely brought forward. Furthermore, in some cases, the increased incidence of the finding of interest may represent evidence of an otherwise occult toxicity. For example, rats may be more sensitive to the effects of a myocardial toxicant than other species due to their propensity to develop cardiomyopathy, a rat-specific background lesion; in such cases, an increased incidence of cardiomyopathy may indicate a direct test article–related effect rather than a secondary consequence of other toxicity. Because there is rarely any definitive evidence to demonstrate the lack of a direct effect by a test article, the best approach usually is to discuss how the test animals were affected and whether or not they were “harmed” by exacerbation of the background change (i.e., an adverse effect or not). Further evidence of the presence or absence of similar effects in other studies with other species may support or refute this interpretation for a single study report once the nonclinical overview documents have been assembled.
Conclusion for Recommendation 3
Considering the scenarios explored above, melding empirical data from a toxicity study with subjective estimates of potential human biological responses (including whether or not exacerbations of otherwise species-specific background changes may represent a risk to humans) is not recommended when considering how to communicate adversity interpretations in individual nonclinical reports. Instead, nonclinical study reports and subreports should limit decisions regarding adversity to the test species used during the study. When there is a clear understanding in the literature of differential responses across species with a specific class of test articles, it may be appropriate to introduce that information within a subreport and/or study report, to support subsequent preparation of the nonclinical overview document. Interpretations regarding potential human risk should be communicated in the nonclinical overview (Figure 2).
Adversity decisions should normally be based upon the actual measured or observed effect itself. Speculation about the biologic process(es) involved in formation of the observed effect should generally not be used to either rule in or rule out a call of adversity. Conversely, there will be few cases in which well-defined biological processes are known to either cause or contribute to an observed effect, and these should be included with a literature citation when invoked as a factor for consideration when making a call on adversity. An important distinction must be made regarding “markers of toxicity” that are not in and of themselves adverse, such as with many clinical pathology analytes. For example, an increased serum creatinine level coincident with renal tubular damage contributes to an overall call of adversity because the relationship between the 2 is well known. In contrast, a creatinine signal occurring in the absence of other correlating changes may be nonadverse. Other examples of this principle follow.
a. Effects believed to be suprapharmacological should be considered either adverse or nonadverse.
Interpretations of a change as adverse or nonadverse have been made based on judgments regarding whether or not the effect originates as an expected pharmacologic (exaggerated or unwanted, but still anticipated) effect versus a nonexpected (off-target toxicologic or unanticipated pharmacologic) event. Two different approaches to this issue have been presented in the literature.
One view is that [a]ny effect seen in a nonclinical toxicology study, whether it is broadly defined as pharmacology (on-target) or toxicology (off-target), may be considered undesirable and therefore adverse. (Dorato and Engelhardt 2005) [i]n the development of pharmaceuticals, some changes could be expected as a result of the pharmacology of the drug, and these pharmacologic changes would not be considered adverse, within certain limits, as the compound is designed to produce these changes. (Holsapple and Wallace 2008)
Therefore, the more conservative way to protect humans who are being treated with novel test articles is to independently judge and fully disclose the full spectrum of substantial biological changes identified during the nonclinical studies. If exaggerated pharmacology is adverse in a test species, this implication regarding adversity and its contribution to defining the study NOAEL should be included in the study report. The explanation will need to include the rationale regarding why the adverse effect is known (or thought) to be a suprapharmacologic one. Findings caused by presumptive exaggerated pharmacology should be treated as adverse when they cause harm, just as for any other test article–related effect in a study. Conversely, test article–related changes resulting in expected pharmacologic effects are not adverse when they do not cause harm to the test species.
b. All relevant effects should be considered in the interpretation of adversity, regardless of whether they are perceived to be primary, secondary, or tertiary.
A common distinction in the determination of adversity for a change due to a test article is often made by differentiating among “primary, secondary, and tertiary effects” or “direct versus indirect toxicity,” especially when toxic effects involve the endocrine and immune systems (Kimber and Dearman 2002). However, it is important to distinguish the concept of primary, secondary, and tertiary toxicity, which define damage to one organ system or cell type caused by toxicity in another cell or system, from “markers” of toxicity, which are expected alterations of analyte levels that occur as a consequence of toxic damage. For example, primary renal damage may result in secondary metastatic calcification and potentially gastric dysfunction due to soft tissue mineralization (a tertiary effect). Such constructs of biologic processes may be very simple and thereby appropriate to make. On the other hand, higher serum levels of urea nitrogen and creatinine in such animals are markers of the toxicity that are well recognized and, consequently, appropriate to use within reports.
Discriminating primary changes that are directly caused by xenobiotic exposure from secondary and tertiary changes that arise as indirect consequences in response to a primary treatment-induced effect may appear obvious within a study data set, or they may be inferred from the literature. However, such determinations may be purely speculative and could lead to confusion when decisions regarding adversity are being made. It is often important to articulate in the final report the understood biologic processes and possible origins of the effects observed in the study. However, in the absence of data or a very compelling argument such as literature citations, it is not recommended to dismiss an otherwise adverse effect on the basis of speculation. For example, liver necrosis may be a secondary consequence of cardiac damage, but it also may signal that the test article is a primary hepatotoxicant. The distinction is important in that it assists in the risk assessment, but in itself, the distinction does not change the adversity designation of either the cardiac or the hepatic effects.
c. Responses interpreted as being “adaptive” should be considered either adverse or nonadverse
Another example in which speculation is used in assigning adversity to nonclinical data sets is the question of whether or not effects related to test articles represent an adaptive response (i.e., one presumed useful in allowing an organism to successfully adjust to xenobiotic exposure). In the context of toxicology, an adaptive response may be defined purely in terms of maintaining normal function as … the process whereby a cell or organism responds to a xenobiotic so that the cell or organism will survive in the new environment that contains the xenobiotic without impairment of function. (Keller et al. 2012) [a]daptive changes are those that the organism makes to allow continued normal function in the face of a stimulus. A nonfunctional change is a detectable change in a measured parameter which is of insufficient magnitude to result in a change in function. (Holsapple and Wallace 2008)
The intracellular accumulation of lipid, especially in hepatocytes, can also occur as adaptive change under conditions unrelated to enzyme induction. Examples include chronic overnutrition (resulting from inability to fully export recently processed lipids) and long-standing undernutrition (due to extensive fat mobilization with transport to the liver for processing). The recommendation with hepatic lipid accumulation, as with other potential adaptive responses (such as cell death as discussed above), is that determination of adversity is not predicated on presumptive biologic process(es) involved but rather is based on the nature and severity of the change itself. Lipid accumulation may not be judged to be adverse if it is of modest degree and does not progress over time toward marked fatty degeneration, pronounced metabolic disruption, or, eventually, cell death.
Conclusion for Recommendation 4
To avoid confusion in attempting to classify test article–associated effects, salient changes related to test article exposure should be addressed within reports regardless of their designation as adverse or nonadverse (Figure 1). Decisions regarding the extent to which test article–related changes are to be deemed adverse should be independent of unsubstantiated speculation about pharmacologic versus toxicologic changes, primary or subordinate (secondary or tertiary) effects, or purposeful adaptations. Study reports should clearly articulate logical and scientifically cogent reasons for discriminating among findings as primary toxicities, other corollary consequences (indirect effects or adaptive responses), or exacerbations of species-specific conditions in order to appropriately inform subsequent positioning of these effects within nonclinical overview documents.
Communicating Adverse and NOAEL
Once agreement has been reached regarding which findings in a nonclinical data set are adverse and which are not adverse, these interpretations may be written in many ways. Regardless of the chosen format, clear communication of both criteria and the reasons for making adversity interpretations is essential to avoid confusion among various constituencies who use nonclinical data to assess human risk.
The scientific judgment needed to interpret test article–related findings and to assign a NOAEL for a nonclinical study requires communication among key members of the study team responsible for data interpretation. In such deliberations, due weight must be given to the opinions of the experts who wrote each subreport rather than deferring the decision solely to the study director. In this fashion, the determination that a pathology finding should be considered the basis for setting the NOAEL would be informed by the study pathologist who made the original observations and interpretations.
Recommendation 5 includes multiple components that are addressed separately to facilitate the utmost clarity and consistency for individuals and institutions seeking to communicate decisions and justifications regarding adversity or lack thereof.
a. Test article–related changes should be documented in subreports, regardless of whether they are considered to be adverse or nonadverse.
All subreports should clearly define whether or not apparent effects of treatment are related to test article. Explanations will frequently be study-specific. For example, a spurious finding in a single animal is 3 times more likely to occur in a treated animal than in a control animal, assuming the study design has equal numbers of animals per group and possesses the standard design of 1 control group and 3 treated groups. Typical arguments made in explaining away spurious effects include lack of dose exposure/response, unusually low or high values in one or several animals of a concurrent control group, random variations within the range observed in historical data, literature references, and so on. Effects dismissed in a subreport as spurious do not need to be considered further in the overall study report.
Once a test article effect is declared to exist, authors should consider relationships with other findings in the study when determining adversity. If a determination of adversity is made, it should be communicated within the study report and/or subreport. Where the same finding is deemed as either adverse or nonadverse at different doses within the same study, evidence (such as specific criteria for setting lesion severity grades) should always be provided to support the discrimination, focusing on the reasons why effects at higher doses are to be considered adverse.
b. Test article–related adverse findings considered to be part of a constellation of related effects should be discussed together.
Although it should be clear to the reader of the final study report which test article effects are adverse or associated with adverse effects, and which are not, it is important to note that each individual effect does NOT have to be separately dealt with and justified either within subreports or study reports. For example, a modest increase in serum AST activity on its own would likely not be adverse per se, and it may not be useful to make a statement in the clinical pathology subreport to that effect in the text if it occurs as an isolated finding. However, when coupled with morphologic evidence of skeletal muscle necrosis, the AST alteration becomes part of a collection of related findings indicating adversity and is thereby communicated in the overall study report. In another example, clinical chemistry alterations that serve as biomarkers of hepatic damage (elevations in serum ALT activity or bilirubin concentration, etc.) should be discussed in relation to liver weight measurements and morphologic observations when evaluating whether or not the biochemical and/or structural changes observed in animals should be considered together in the determination of adversity. Such relationships between end points that are well understood to be causally related should be handled together in respect to adversity decisions.
If toxicity is considered to be unequivocally the result of a single process, the adversity decision can be applied to the single effect. For example, it is expected that liver necrosis also will lead to changes in ALT and AST activities, liver weights, and so on, so it makes no sense to deal with each of these items separately with respect to decisions about adversity. Accordingly, the consideration of adversity may be focused on necrosis as the primary event of interest. This concept should be communicated clearly in reports.
An adverse effect should be communicated in the appropriate subreport and again in the overall study report. A test article effect in a subreport that is considered to be a marker of a potential adverse effect may be described as adverse in a subreport if other evidence is available showing that the marker occurs concurrently with the adverse effect. For example, the clinical pathology subreport might state, “Increased serum ALT activity in high-dose rats (4.2× the control values) corresponds to periportal hepatocellular necrosis”; whereas the related anatomic pathology subreport might state, “Periportal hepatocellular necrosis in high-dose rats (8/10) corresponded to increased serum ALT activity.” If the full information is not available when a subreport is completed (e.g., when a clinical pathology subreport is finalized in advance of the anatomic pathology subreport), the author of a subreport may communicate that an effect is test article–related without making a decision regarding its adversity, with the understanding that the adversity decision will be communicated in the overall study report.
c. The NOAEL is to be assigned for the study as a whole rather than to subreports.
The finding or group of findings that will be used to establish the NOAEL for a whole study typically should not be predicted in advance by viewing adverse findings in isolation as defined in individual subreports but rather should await the opportunity to view the entire study data set, which is assembled from the complete collection of subreports. A single test article–related change in a critical organ may be the key finding used to set the NOAEL (e.g., neuronal necrosis in the brain). In other instances, a related spectrum of changes might be used collectively to establish the NOAEL even though each finding might be viewed as inconsequential if each occurred in isolation. A test article effect described in a subreport as adverse may have less relevance for establishing an adverse response in animals in light of the complete data set from all subreports assembled and integrated in the overall study report.
d. Vague statements such as “not biologically relevant” or “not toxicologically important” should only be used in the study report when defined and supported by a sound rationale.
The phrases not biologically relevant, “not toxicologically significant,” and similar wording should not be used as a substitute for the more direct phrase nonadverse. However such ambiguous concepts as relevance and significance, as well as many others resembling them, may be used if they are explained using a sound rationale, and used consistently throughout the text. A means of bolstering interpretations of this nature is to provide, when appropriate and available, authoritative references (i.e., a confirmatory data set, ideally communicated as a literature citation) to support statements that are otherwise subjective opinions.
Discussions that transcend scientific disciplines are needed to foster the complete understanding of the spectrum and importance of test article–related effects needed to prepare high-quality study reports. Discussions should be undertaken by the entire team of contributors who prepared subreports. Such conversations are necessary since a single test article effect may manifest in different fashions to contributing scientists preparing individual subreports in relative isolation. An understanding of the effect and its implications needs to be considered and understood as a whole by all study personnel to insure clarity and consistency within and between subreports and the main study report. As an example, a hepatotoxic effect may be most evident histologically but likely also will be reflected in liver weight changes at necropsy, elevations in serum activities of hepatic enzymes, and perhaps clinical signs such as icterus and/or neurological dysfunction. Concordance of findings across multiple subreports, especially if such findings would be deemed of modest import when considered individually, will lend greater weight to adversity decisions communicated in the overall study report.
The terminology “overview document” as applied to animal-derived toxicity data is to be found in nonclinical sections of regulatory submission documents. Examples include investigator’s brochures, investigational new drug applications (IND), clinical trials application, new drug application, biologic licensing applications, and similar products. Integration of data within the nonclinical overview document is necessary (Figure 2) because a NOAEL identified for an individual study may be discounted as irrelevant in the overview document by using data from another nonclinical study (e.g., where an adverse effect in one species is not observed in a species where relevance to human responses for this effect is well documented, or in the case that longer multidose studies demonstrate adverse findings at lower doses than shorter studies). Alternatively, a finding determined to be nonadverse within an individual study may assume greater importance in defining an adverse test article–related effect in an overview document (e.g., where a similar test article effect on a particular organ system is identified in multiple species). As an example, treatment-related exacerbation of chronic progressive nephropathy resulting in sick rats will have been recorded as an adverse effect in a study report; however, the absence of such effects in nonhuman primates at much higher exposure levels might provide a scientific justification for discounting (in the overview document) its relevance as a predictor of risk to humans. If data from other nonclinical studies show that rats, nonhuman primates, and humans all express the receptor for the test article equally throughout their renal parenchyma, then the rationale for this interpretation is strengthened. In this case, a scientifically sound argument may be made that any NOAEL based on renal lesions described in the rat study may be discounted in the overview document used to set the starting dose for human trials.
The test article NOAEL communicated in the nonclinical overview document serves as only one element in evaluating the potential for human risk. The complete analysis of human risk also considers likely exposure, any available clinical information as well as other available information (e.g., published class effects) that might be accessible to individuals performing the assessment. The amount of nonclinical and clinical data, as well as other information, will increase as the development program proceeds.
Final study reports and nonclinical overview documents should facilitate a clear appreciation for the judgments made in setting a NOAEL to define limits for human exposure. Nuances within the text often provide critical insight necessary for the rational use of a NOAEL in human risk assessment. Placement of NOAEL values inside data tables without necessary context (such as cross-referencing to important text passages or inclusion of explanatory footnotes) encourages the quick but unthinking use of such values to set human doses. This does not provide the rationale utilized for establishing the NOAEL.
Using Adverse and NOAEL in Assessing Potential Human Risk
The identification of adverse findings and the designation of a NOAEL in nonclinical studies provide useful information for the assessment of human risk, which is the ultimate use of the nonclinical study data. To obtain the maximum use or value from the nonclinical data, it is essential that this information be thoroughly and appropriately considered through the application of all available scientific expertise.
Decisions about human risk determined from nonclinical study data should incorporate the assessments of experienced scientists, ideally those who generated the relevant subreports and final study reports but at minimum subject matter experts who evaluated the data and reports. These scientists, typically including toxicologists, pathologists, safety pharmacologists, and sometimes other nonclinical scientific experts should interact closely with the clinical research team charged with designing human trials for test articles before finalizing overview documents that will be used to support an IND dossier and human clinical trials; such interactions may take place in formal or informal settings, but they should take place on a regular basis. In particular, toxicologists and pathologists are likely to be most aware of interspecies differences that may render certain effects in animals irrelevant to human risk assessment. The intent of this recommendation is that scientists with relevant training and experience in the evaluation and interpretation of nonclinical data are well positioned to address the implications of any findings observed in animals that may create concerns about human risk.
An adverse test article–related effect in any single subreport or a NOAEL derived from any one nonclinical study should not be considered in isolation as definitive evidence of the effects likely to develop in humans. The risk/benefit profile may change as the test article is used in multiple species or in studies of longer duration. Furthermore, NOAELs in study reports may differ as diverse end points are measured in other types of studies (i.e., reproductive and developmental toxicity or carcinogenicity studies, which usually take place later in drug development). Additionally, the test article may be used in new clinical indications targeting different patient populations, possibly with other routes of administration resulting in dissimilar tissue levels, and therefore leading to a different risk/benefit profile.
Assessments of probable human risk should be based on the entire portfolio of available nonclinical and clinical data from all existing studies as well as any literature for structurally related or similar acting agents. For example, experiments designed to understand the biologic process(es) related to a finding in nonclinical studies may profoundly influence the risk profile of a test article. Establishing that a receptor on a cell type that serves as a target for toxicity in a nonclinical species is not present in humans may mitigate the perceived risk to human subjects even if a test article produces severely adverse effects in animals. Similarly, knowing whether or not toxicities are due primarily to the parent drug or an animal-specific metabolite may provide an opportunity to mitigate the potential risk of humans developing the same toxicities observed in nonclinical species. Thus, risk assessment is an iterative process through which the risk/benefit profile may change as additional nonclinical and clinical data accumulate over time.
Conclusions and Forward Vision
These 10 recommendations have been designed to produce a more consistent approach for determining and communicating adverse findings in the context of nonclinical toxicity studies for use in risk assessment. Efforts to protect the health of humans exposed to new chemical or pharmaceutical entities depend heavily on predictions made using nonclinical data derived from animal testing. Common species of test animals and humans share orthologous genes and molecular pathways largely due to shared evolutionary history. However, toxicity profiles in laboratory animals do not always translate closely to clinical outcomes in humans (Fletcher 1978; Freireich et al. 1966; Greaves, Williams, and Eve 2004; Olson et al. 2000; Owens 1962; Schein et al. 1970). Such discrepancies necessitate that all test article–related findings in animals be described carefully and completely. The communication of the rationale for classifying such effects as adverse or nonadverse is critical since interpretations must be understandable to all potential reviewers. This may include individuals with little or no familiarity with laboratory animal biology, species sensitivity, comparative pathology and its terminology.
When a NOAEL derived from a nonclinical safety study report is used in isolation as a principal component for the assessment of potential human risk, it is of limited value because NOAEL lacks a universally accepted definition, is variably applied, and often is poorly communicated. In the future, the concept of adversity may be expanded and better defined by using additional types of data: transcriptomic and proteomic profiles, epigenetic changes, in vitro bioactivity, and so on (Keller et al. 2012). For now, though, the NOAEL and the concept of adversity, defined using in-life observations as well as anatomic and clinical pathology end points, will remain the principal means for communicating the scope of potential toxic effects in humans exposed to new test articles. As researchers, institutions, and regulatory agencies that produce and evaluate NOAEL values over time adopt these recommended approaches to describing, defining, and communicating adversity, there will be less dependence on the NOAEL as a stand-alone term to define risk, favoring a more comprehensive and complete communication of the estimated risk to humans exposed to xenobiotics.
Footnotes
Acknowledgments
Author Contributions
All authors (RK, BB, JB, SF, PG, VM, and JP) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Authors’ Note
The views expressed in this article are those of the authors and do not necessarily represent the policies, positions, or opinions of their respective employers and agencies.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This document was sponsored and is supported by the Society of Toxicologic Pathology and is endorsed by other professional organizations involved with toxicologic pathology, including The American College of Veterinary Pathologists, The American Society for Veterinary Clinical Pathology, Société Française de Pathologie Toxicologique” (French Society of Toxicologic Pathology), The British Society of Toxicologic Pathology, The European Society of Toxicologic Pathology, The Japanese Society of Toxicologic Pathology, and The American College of Toxicology).