
Abstract
The advancement of an investigational new drug in humans is a significant developmental milestone. In first-in-human (FIH)-enabling toxicology studies, the highest dose without a test article–related adverse effect (no-observed-adverse-effect-level [NOAEL]) serves as the basis for deriving a safe FIH starting dose. For anticancer pharmaceuticals, the FIH dose may be calculated using the highest non-severely toxic dose (HNSTD) in nonrodent models or the dose severely toxic to 10% (STD10) in rodents. Given the practice of reporting the NOAEL, but the lack of regulatory requirements to do so for anticancer pharmaceuticals, we conducted an informal survey of 20 companies to answer the question “How is our industry reporting toxic/adverse dose levels in FIH-enabling toxicology studies for anticancer indications?” The data indicated 4 reporting approaches, each providing a path to regulatory acceptance. Within the integrated toxicology study report, 45% of respondents report the HNSTD/STD10, 25% report the NOAEL, 20% report both the HNSTD/STD10 and NOAEL, and 10% do not define either, reserving definitions for regulatory submissions. One reporting approach may be preferred over another for reasons including consistency across indications, repurposing pharmaceuticals, regulatory feedback, or simplicity. The reporting approach should be defined in advance of study initiation, and the pathologist should provide context to support the chosen approach.
Introduction
Adversity in the context of nonclinical toxicology studies has engendered spirited debate within the drug development community for decades. The definition of “adversity,” the criteria by which it is determined, and how adversity should be communicated have been discussed in 2 seminal publications representing the official position of multiple international toxicologic pathology organizations.1,2 According to these papers, in pivotal good-laboratory-practice (GLP) studies, the pathologist’s report or the integrated (main) study report should include an explanation of adverse or nonadverse test article–related effects. Ultimately, these recommended practices for communicating adversity facilitate determination of threshold values such as the no-observed-adverse-effect-level (NOAEL), defined as the highest dose level that does not produce a significant increase in test article–related adverse effects. 3 In nonclinical toxicology, the NOAEL serves as a framework for toxicity assessments and reporting as well as risk assessment strategies.
Adversity in nonclinical toxicology studies is agnostic to therapeutic indication and should not be established differently depending on the indication for the drug. 1 Assignment of the NOAEL is aligned with the International Council for Harmonisation (ICH) Guideline M3(R2) on nonclinical safety studies for the conduct of human clinical trials and with the Food and Drug Administration (FDA) guidance for selection of a safe starting dose.3,4 The NOAEL serves as the basis for deriving a safe starting dose for first-in-human (FIH) clinical trials and informs dose-escalation and dose-discontinuation criteria. The ICH M3(R2) guideline is most often used for small-molecule therapeutics when repeated dosing is necessary to treat acute or chronic diseases that are not immediately life-threatening. Instead of ICH M3(R2), anticancer pharmaceuticals are tested under ICH S9. 5 For pharmaceuticals intended to treat patients with an advanced neoplastic disease and limited therapeutic options, the NOAEL is not considered essential for determining an FIH starting dose. In these cases, the goal is to provide new, effective anticancer drugs to patients expeditiously, at a dose that is expected to have pharmacological effects and is reasonably safe to use. 5 For traditional small-molecule oncology drugs, the starting dose is established as one-tenth of the severely toxic dose to 10% of rodents (STD10) on a body surface area basis, provided that serious irreversible toxicity does not occur in a nonrodent species at this dose level. If serious toxicities occur in nonrodents at the starting dose supported by rodents, or if the nonrodent is known to be the more appropriate animal model, then the starting dose is established as one-sixth of the highest non-severely toxic dose (HNSTD), defined as a dose that does not produce lethal, life-threatening, or irreversible toxicities in nonrodents.6,7 When appropriate, the calculated starting dose may be reduced by additional safety factors. For oncology therapeutics that stimulate the immune system, the starting dose is usually established based on the minimally anticipated biological effect level or pharmacologically active dose (PAD). 8 Accordingly, regulatory guidance allows latitude on the need for determination of the NOAEL for drugs developed for oncology indications.
The use of nonclinical studies to support clinical development is similar, but not identical, between ICH S9 and ICH M3(R2) indications. Life-threatening conditions with a high unmet medical need can accept greater safety risks than would be appropriate for therapeutics intended for use in patients with non–life-threatening diseases. This approach can extend beyond oncology indications; for example, the FDA issued a guidance in 2019 on the use of the HNSTD/STD10 in severely debilitating or life-threatening hematologic disorders. 9 Accepting greater safety risks in life-threatening indications can be partially mitigated by understanding reversibility and monitorability of adverse findings in nonclinical studies. As such, demonstrating recovery from toxicity may be a more important objective for an anticancer nonclinical study than defining the NOAEL, which may occur below the level of biological activity. Following ICH S9 and 3R guidance, assessment of recovery should be provided before entry into phase 1, but this assessment may be based on scientific judgment rather than demonstrated reversibility. 10
In the authors’ experience, multiple, overlapping approaches inform where (pathology subreport, integrated study report, and/or regulatory documents) and how adversity is defined. This Opinion Piece discusses various approaches for reporting adverse and toxic dose levels in the integrated study report for FIH-enabling nonclinical toxicology studies supporting anticancer pharmaceuticals within the scope of the ICH S9 guideline. To explore the range of approaches used and their associated advantages and disadvantages, an informal cross-industry survey was undertaken. Current practices for reporting of the HNSTD/STD10, NOAEL, both values, or neither are described alongside the reasoning for each. In addition, the pathologist’s role in these reporting practices as a means of communicating adversity is discussed.
Industry Survey
Pathologists from 20 pharmaceutical and biotechnology companies representing the major pharmaceutical markets including the United States, Europe (United Kingdom, France, Germany, and Switzerland), and Asia (Japan and China) were contacted to discuss current practices of reporting the HNSTD/STD10 and NOAEL in toxicology studies for anticancer pharmaceuticals. Company size ranged from approximately 1500 to 130,000 employees with a mean of 57,000 and a median of 51,000 employees.
While conversations began with pathologists, discussions also included toxicologists and oncology portfolio managers. Survey results do not represent official company practices or policies; rather, they describe the general practices currently in use. The survey was conducted as a nonscripted conversation framed by the following questions:
Does your organization typically report the HNSTD/STD10 and/or NOAEL in FIH-enabling study reports for anticancer pharmaceuticals? If so, where are these values reported?
Why is this approach your practice?
What are the reasons for the approach your company is taking?
What, if any, regulatory feedback has your organization received regarding your current practice for reporting adversity?
Answers for these questions were acquired from contact research organizations (CROs) and consultants to investigate additional nuances of adversity reporting decisions, but data from these entities were not included in the survey results because Study Directors at CROs and consultants generally follow instructions provided by the industrial sponsors regarding the inclusion of the HNSTD/STD10 and/or NOAEL.
Results
Overall, the survey identified 4 separate approaches for communicating the HNSTD/STD10 and/or NOAEL of anticancer pharmaceuticals within FIH-enabling toxicology study reports (Figure 1 and Table 1). Nine (45%) respondents report the HNSTD/STD10 within the integrated study report but do not define the NOAEL. Five (25%) report only the NOAEL (when determined). Four (20%) report both the HNSTD/STD10 and NOAEL (when determined), and two (10%) do not report either; rather, these are defined in the regulatory submission (i.e., nonclinical overview) documents. There was no clear association between the company size or geographic location and the preferred reporting approach.
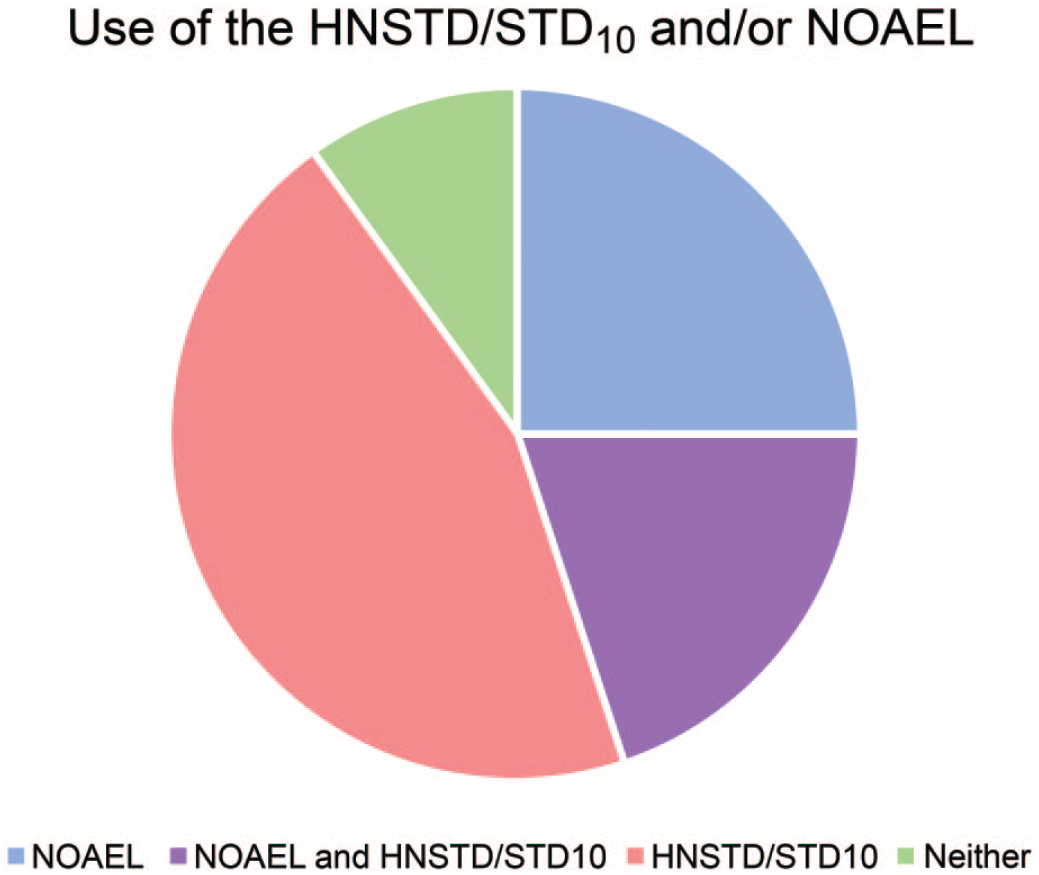
Pie chart showing the percentage distribution of survey respondents by reporting style: 45% report only the HNSTD/STD10; 25% report only the NOAEL; 20% report both the HNSTD/STD10 and NOAEL; and 10% do not report the HNSTD/STD10 or NOAEL.
Approaches for reporting toxic and adverse dose levels in nonclinical toxicology studies supporting development of anticancer pharmaceuticals by geographic region.
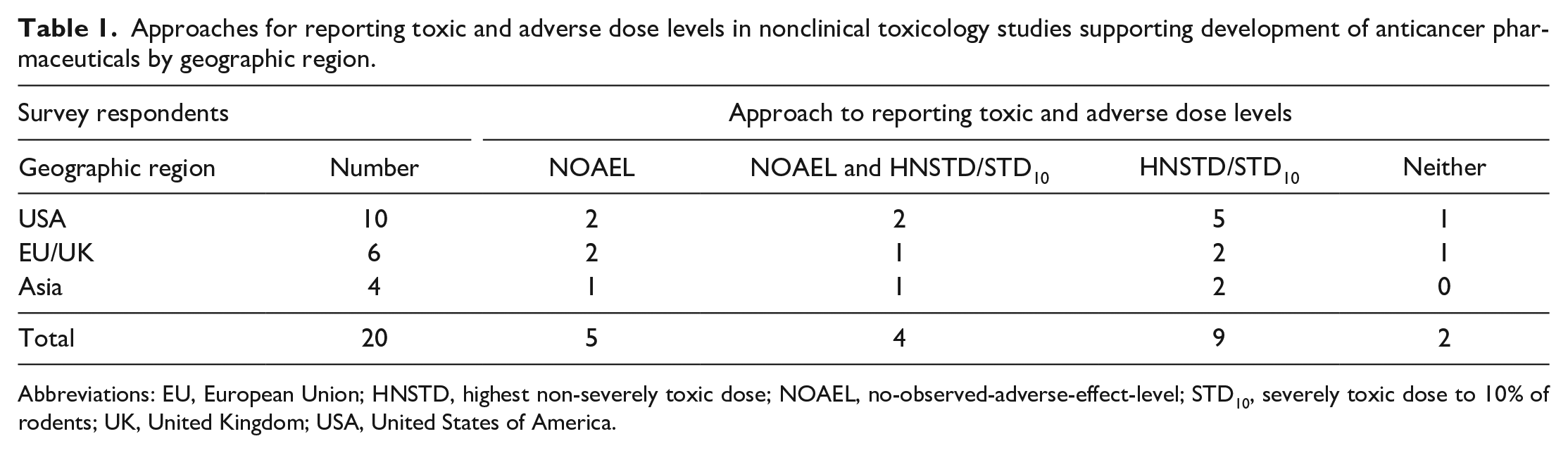
Abbreviations: EU, European Union; HNSTD, highest non-severely toxic dose; NOAEL, no-observed-adverse-effect-level; STD10, severely toxic dose to 10% of rodents; UK, United Kingdom; USA, United States of America.
Respondents That Report Only the HNSTD/STD10 in the Integrated Toxicology Study Report
Across companies surveyed, the HNSTD/STD10 was the most commonly reported dose level for anticancer test articles, which is consistent with ICH S9 guidance. Survey respondents following this approach reported 3 main reasons for doing so:
Simplicity. This approach was considered the most straightforward reporting style. Some respondents felt that using only the HNSTD/STD10 facilitated clear communication of toxicology data within research teams.
Regulatory feedback. Submissions using this approach have been accepted by global regulatory agencies. Two respondents believed that the key to regulatory acceptance of an HNSTD/STD10-only approach is determining that test article–related findings are reversible. For most anticancer pharmaceuticals, reversibility and the ability to monitor the extent of toxic findings (and their regression) facilitate entry into the clinic. As long as recovery from toxicities was demonstrated in nonclinical studies, understanding the NOAEL was not considered essential for setting an FIH starting dose.
Inconsistent availability of the NOAEL. Toxicology studies performed in support of an anticancer pharmaceutical are not always able to identify the NOAEL. Blinded data on 92 small-molecule anticancer pharmaceuticals from 11 companies were evaluated by the International Consortium for Innovation and Quality, DruSafe Leadership Group. 11 In this review, it was possible to determine the NOAEL for 72% of compounds, indicating that a NOAEL was not achieved in 28% of the studies surveyed. Specific examples in which the NOAEL was requested during regulatory review are detailed below.
Respondents That Report Both the HNSTD/STD10 and the NOAEL in the Integrated Toxicology Study Report
Justification for this practice included the following reasons:
Repurposed molecules. The most frequently cited reason was the potential need to adapt studies initially performed for oncology indications to support non-oncology and/or adjuvant indications in the future. Interestingly, most respondents with this concern have successfully submitted an oncology pharmaceutical for a non-oncology indication with study reports that did not include the NOAEL. Respondents stated that secondary submissions following the ICH M3(R2) guidance have not required report amendments to include the NOAEL because (1) the NOAEL was discussed within the regulatory submission documents; (2) human data were available at the time of submission for the non-oncology indication and held more weight than the nonclinical data alone; and/or (3) new regulatory submissions had a lower starting dose than those already used in the clinic. Despite these facts, respondents that report both the HNSTD/STD10 and NOAEL in the integrated toxicology report felt that pharmaceutical-specific details and reviewer requests may influence the need for the NOAEL when repurposing an anticancer pharmaceutical for a non-oncology indication. Therefore, including the NOAEL in addition to the HNSTD/STD10 is their current practice.
Regulatory feedback. Multiple respondents described individual instances in which they have been asked for the NOAEL by reviewers. In most cases, the request was adequately answered with a statement that the NOAEL was not achieved or was not the goal of the study. Despite the acceptability of such answers, some companies prefer to declare the NOAEL in study reports to preempt these time-consuming regulatory interactions.
Evolving regulatory focus. As cancer survival rates improve, the quality of life after treatment has become more important to oncology patients and regulatory agencies. For example, infertility and cardiotoxicity may no longer be acceptable side effects in patients with improved survival. One respondent receiving a reviewer’s request for the NOAEL interpreted the request to be prompted by an increased focus on sublethal, persistent side effects of an oncology drug that could affect the quality of life over time.
Controlling the narrative. Multiple respondents report the NOAEL to ensure that interpretation of adversity decisions is clear. Understanding that regulatory reviewers will make their own decisions based on the data presented, there was generalized agreement that reviewers would benefit from the interpretations of adversity made by the Study Director and contributing scientists.
The request of the clinician. Multiple respondents report the NOAEL at the request of the clinician responsible for clinical protocol development. (NOTE: Clinicians were not surveyed for this opinion piece.)
Dose response. One respondent indicated that reporting of both the HNSTD/STD10 and NOAEL helps define the steepness of the dose-response curve. The relationship between the lethal dose (if available) and these two values may inform the dose-increment and dose-escalation methods used in the phase 1 study. For example, when the HNSTD/STD10 and NOAEL are very low and occur close together, a traditional rule-based clinical trial design (e.g., “3+3” design) with more conservative increments may be favored over an accelerated titration design or a model-based continuous reassessment method. 12
Healthy volunteers. Some respondents have found it expedient and cost-effective to use healthy volunteers for clinical pharmacology studies run concurrent with the phase 1 study. To enable dosing in healthy volunteers, a robust nonclinical package must be generated, consistent with ICH M3(R2) guidelines. 13 The NOAEL and PAD doses (along with pharmacokinetic modeling) are used to calculate the acceptable starting dose in healthy volunteers. The NOAEL would need to be deduced from the nonclinical toxicology data for this purpose.
Occupational health. Survey respondents discussed the need to protect patients from residual drug carryover in multiproduct manufacturing and quantify potential worker exposure during the handling and manufacturing of drugs. For these purposes, toxicologists calculate acceptable daily exposure (ADE) limits and occupational exposure limits (OELs) from robust nonclinical and clinical data sets. While NOAELs from investigational new drug (IND)-enabling studies could be used in these calculations, early nonclinical data sets result in significant uncertainties; thus, in most cases, default values are assigned to both the ADEs and OELs during nonclinical development.
Data mining. Inclusion of the NOAEL as a standard practice across studies/indications allows for data mining across compounds.
Completeness. Some respondents felt that if the NOAEL can be declared, it should be reported for completeness of the integrated study report.
Respondents That Report the NOAEL Only
In addition to the reasons discussed above for including the NOAEL, respondents that excluded the HNSTD/STD10 from the integrated study reports did so for consistency across drugs and indications. In these cases, the HNSTD/STD10 was discussed in the regulatory documents.
Respondents using this reporting style design nonclinical studies to identify the maximum tolerated dose and assess toxicity over a range of doses, with the understanding that the NOAEL may or may not be achieved. When possible, a dose level anticipated to be an NOAEL is included. For compounds where achieving an NOAEL is not anticipated (e.g., a cytotoxic compound), dose levels are set to help inform the HNSTD/STD10.
Respondents That Do Not Report the HNSTD/STD10 or the NOAEL
For respondents reporting neither the HNSTD/STD10 or NOAEL in the integrated study reports, interpretation of severely toxic (and/or adverse) dose levels is made using a weight-of-evidence approach across studies and species within the regulatory documents. While this approach is in the minority, survey respondents using this approach shared that it has historically been acceptable by global regulatory bodies.
General Results
More than a quarter of the survey respondents discussed a recent history of change in their reporting practices for the HNSTD/STD10 and NOAEL, most commonly associated with a change in leadership. If leadership change was due to a merger/acquisition, the acquired company most commonly (but not always) adopted the practices of the acquiring company over time. Within stable companies, leadership attrition, new management, or reorganization frequently resulted in an evaluation and subsequent modification of reporting practices. Given this ebb and flow of reporting approaches, the results of this survey represent a “snapshot” in time.
In addition to the 20 companies surveyed, CROs and pathology consultants were included in discussions of adversity-reporting decisions. For these CROs and consultants, the reporting approach varied based on the preference of the client. In general, adversity is defined within the pathology report to enable the use of the NOAEL in the integrated study report. Unless otherwise directed by the client, repeat-dose GLP-compliant nonclinical studies usually report the NOAEL within the integrated study report. When a client shares that the test article is for an ICH S9 anticancer indication, reports usually include the HNSTD/STD10, and the integrated report may also include the NOAEL, if it is identifiable, unless otherwise requested. As recommended by a CRO respondent, the client should communicate the need to include the HNSTD/STD10 and/or NOAEL in the report text to the Study Director as early as possible. Furthermore, the study pathologist should be informed prior to the initiation of their evaluations.
After the determination of adversity and the HNSTD/STD10 or NOAEL, the pathologist should consider the translation of study data from test animals to patients. While a pathologist may know how a particular lesion is adverse to study animals, they should also be prepared to comment on the relevance (if any) to humans. Using a weight-of-evidence approach, the pathologist may be asked to communicate their understanding of translatability for inclusion within a risk-benefit analysis in regulatory documents. For example, if the HNSTD in a monkey study evaluating a human-derived protein therapeutic is set based on perivascular immune complex deposition (due to formation of antidrug antibodies), the lesion is not anticipated to be predictive of a clinical outcome. This assessment is a valuable final step in the interpretation of pathology data in which toxicologic pathologists should be intimately involved.
Discussion
The survey results demonstrated the flexible and fluid nature of approaches for communicating the HNSTD/STD10 and/or NOAEL for anticancer pharmaceuticals within FIH-enabling toxicology study reports. Each of the four approaches described here is appropriate and acceptable to regulatory bodies. Many participants in this survey advocated strongly for their current reporting practices; however, the cumulative responses show that there is no right or wrong reporting approach. For some respondents, the reporting approach may be tailored based on pharmacologic activity (i.e., immune-oncology) or mechanism of action.
Over time, practices for reporting the HNSTD/STD10, NOAEL, or both are revisited and revised within individual companies and across the industry. Evaluation and alteration of current practices most commonly occur after changes in leadership, regulatory feedback, or a shift in company focus (i.e., bringing on additional modalities). The standard reporting approach for a company may be influenced by the regulatory environment and new/emerging approaches within the field. Through the International Consortium for Innovation and Quality, DruSafe Leadership Group, eleven companies have participated in a data-mining effort to explore ways of reducing first-in-patient dose-escalation duration and the number of cancer patients exposed to subtherapeutic doses by proposing alternative approaches to determining a safe starting dose. This industry analysis has demonstrated the potential utility of the NOAEL, without additional safety factors, in calculating safe starting doses that reduce the number of cohorts required to reach a maximum tolerated dose.11,14 If efforts to redefine the determination of acceptable starting doses are promising, then defining a NOAEL in reports for some anticancer pharmaceuticals may become more common.
Regardless of the reporting approach, defining adversity within the pathology report facilitates determination of the NOAEL prospectively and retrospectively. Many survey respondents discussed the importance of clearly understanding the pathologist’s interpretation of adversity and stated that the primary and peer review pathologists are integral members of the study team that determine the HNSTD/STD10 and/or NOAEL, a sentiment supported by the literature. 1 Based on the variety of reporting approaches, it is the authors’ opinion that sponsors should share the reporting approach (NOAEL or toxic dose) with the Study Director and primary pathologist in advance, and the primary pathologist’s report should provide the basis for the dose levels reported in the integrated study report. There are challenges associated with post hoc deliberation/determination of the HNSTD/STD10 and/or NOAEL, including inefficient use of pathologist’s time, potential delays in communicating results, and additional cost. From the perspective of the CRO pathologist, significant changes in reporting approach (i.e., removing or including an HNSTD/STD10 or NOAEL) after draft report generation are frustrating and costly investments in time. It is important to remember that future access to the study pathologist may not be possible; therefore, early and clear communication within the study team is critical to ensure that appropriate methods of defining adversity are included in the final study report.
In conclusion, there are several approaches to reporting adverse or toxic dose levels in FIH-enabling nonclinical toxicology study reports for anticancer pharmaceuticals, including the HNSTD/STD10, NOAEL, both values, or neither. The variable practices highlight the flexibility in defining how and where nonclinical safety scientists define adversity. All approaches described herein are acceptable under ICH S9 guidance, but there may be specific reasons for selecting one approach over another in developing anticancer pharmaceuticals. The need to include the HNSTD/STD10 and/or NOAEL in the report text should be communicated to the Study Director and study pathologist as early as possible, and the primary pathologist should provide the context to support the chosen approach.
Footnotes
Acknowledgements
The authors thank Bart Jessen and Timothy Hart for their helpful discussions and Brad Bolon, Tom Monticello, Whitney Helms, and John Vahle for their expert review of the final manuscript.
This is an opinion article submitted to the Toxicologic Pathology Forum. It represents the views of the authors. It does not constitute an official position of the Society of Toxicologic Pathology, British Society of Toxicological Pathology, or European Society of Toxicologic Pathology, and the views expressed might not reflect the best practices recommended by these societies. This article should not be construed to represent the policies, positions, or opinions of their respective organizations, employers, or regulatory agencies.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.