
Abstract
BT-11 is an orally active, gut-restricted investigational therapeutic targeting the lanthionine synthetase C-like 2 pathway with lead indications in ulcerative colitis (UC) and Crohn disease (CD), 2 manifestations of inflammatory bowel disease (IBD). In 5 mouse models of IBD, BT-11 is effective at oral doses of 8 mg/kg. BT-11 was also efficacious at nanomolar concentrations in primary human samples from patients with UC and CD. BT-11 was tested under Good Laboratory Practice conditions in 90-day repeat-dose general toxicity studies in rats and dogs, toxicokinetics, respiratory, cardiovascular and central nervous system safety pharmacology, and genotoxicity studies. Oral BT-11 did not cause any clinical signs of toxicity, biochemical or hematological changes, or macroscopic or microscopic changes to organs in 90-day repeat-dose toxicity studies in rats and dogs at doses up to 1,000 mg/kg/d. Oral BT-11 resulted in low systemic exposure in both rats (area under the curve exposure from t = 0 to t = 8 hours [AUC0-8] of 216 h × ng/mL) and dogs (650 h × ng/mL) and rapid clearance with an average half-life of 3 hours. BT-11 did not induce changes in respiratory function, electrocardiogram parameters, or behavior with single oral doses of 1,000 mg/kg/d. There was no evidence of mutagenic or genotoxic potential for BT-11 up to tested limit doses using an Ames test, chromosomal aberration assay in human peripheral blood lymphocytes, or micronucleus assay in rats. Therefore, nonclinical studies show BT-11 to be a safe and well-tolerated oral therapeutic with potential as a potent immunometabolic therapy for UC and CD with no-observed adverse effect level >1,000 mg/kg in in vivo studies.
Introduction
Piperazine-1,4-diylbis((6-(1H-benzo[d]imidazol-2-yl)pyridin-2-yl)methanone) dihydrochloride (BT-11) is a small molecule investigational new drug targeting the lanthionine synthetase C-like 2 (LANCL2) pathway to promote anti-inflammatory effects for the treatment of inflammatory bowel diseases (IBDs). 1,2 BT-11 is an orally active therapeutic that acts locally on cells within the gastrointestinal (GI) tract. As such, it is theorized that this profile will reduce the potential for systemic adverse side effects, such as increased rates of cancer and infection, that present with current standard of care therapeutics for IBD, such as biologics, corticosteroids, and immunosuppressants. Currently, only 10% of patients with IBD have prolonged clinical remission and half of patients will require surgery within 10 years of diagnosis, indicating a clear and severe unmet clinical need. 3,4
Inflammatory bowel disease has a complex etiology involving an interplay between genetics, bacteria, and environmental factors, which results in chronic inflammation within the GI tract. 5 -7 Inflammatory bowel disease encompasses 2 main manifestations, ulcerative colitis (UC) and Crohn disease (CD), that differ in the site of inflammation, penetrance into the tissue, and underlying immunology. The dysregulated immune responses and inflammatory flares experienced in IBD result in severe abdominal pain, diarrhea, and rectal bleeding, which if left untreated can lead to malnutrition, increased risk of colorectal cancer, and the need for intestinal resection of fibrotic lesions. 8 -10
BT-11 is a first-in-class small molecule with a new mechanism of action activating the LANCL2 pathway. 1,2 Lanthionine synthetase C-like 2 is a membrane receptor with a natural ligand, abscisic acid (ABA), that is consumed within the diet and produced endogenously. Beyond BT-11, LANCL2 may serve as a reasonable target for drug development due to lack of observed toxicity with high doses of ABA, a generally recognized as safe compound with 60% bioavailability. 11 Although the pathway has been associated with glycemic control, 12 LANCL2 is also highly expressed within epithelial and immune cells of the GI tract. BT-11 has been shown to modulate late-stage glycolysis in CD4+ T cells to promote stable and potent regulatory phenotypes, through activation of LANCL2, that induce a restoration of homeostasis and decrease in inflammation locally within the GI tract. 2 Meanwhile, the activation of LANCL2 has been shown to be beneficial in various models of respiratory and GI infections including influenza 13 and Clostridium difficile. 14
The efficacy of BT-11 in treating IBD has been validated in 5 mouse models of IBD. 1,2 Due to the multifactorial nature of IBD, a single model of disease is insufficient in demonstrating therapeutic efficacy. Oral dosing of 8 mg/kg/d, from 7 days to 8 weeks in both prophylactic and therapeutic dosing strategies, decreases inflammation, severity of histological lesions, and overall disease activity in dextran sulfate sodium (DSS), interleukin-10 (IL-10)−/−, CD4+ adoptive transfer, MDR1a−/−, and Citrobacter rodentium models of IBD. The collective potential for efficacy demonstrated across these 5 models supports the efficacy of BT-11 in disease diversely initiated by chemical destruction of the epithelial layer (DSS), genetic abnormalities (IL-10−/−, MDR1a−/−), immunological dysregulation (adoptive transfer), and gut microbiota interactions (C rodentium). The LANCL2 pathway is highly conserved across mammalian species. Particularly, between human and mouse, LANCL2 has a 94% similarity upon sequence alignment with no residue differences within the identified binding pocket of BT-11. Indeed, BT-11 has also been tested in peripheral blood mononuclear cells (PBMCs) and colonic lamina propria mononuclear cells (LPMCs) isolated from primary samples from patient with UC and CD donors. 2 Ex vivo BT-11 treatment, on the order of 10 to 100 nM concentrations, decreased tumor necrosis factor α (TNFα), interferon γ, and IL-4 producing cells while increasing IL-10 and FOXP3 expressing cells. Characteristic mechanistic effects in regulatory CD4+ T cell differentiation, stability, and function have been observed in these human PBMCs and LPMCs following treatment with BT-11.
Preliminary safety studies in rats with 30-day dosing suggested that oral treatment with BT-11 has an acceptable safety profile. 15 The purpose of this work was to further characterize the safety profile of BT-11 in a battery of definitive nonclinical toxicity studies evaluating general and specific toxicities to encompass the common modes of safety concerns for pharmaceuticals. The data collected indicate that BT-11 does not induce any forms of general toxicity in rats or dogs at oral doses up 1,000 mg/kg in 90-day repeat-dose toxicity studies. The data also explore specific toxicokinetic (TK), metabolic, genotoxic, respiratory, cardiovascular, and central nervous system responses as part of safety pharmacology studies. Overall, these data support BT-11 as a safe oral investigational therapeutic for IBD with potential for future testing in human clinical trials.
Materials and Methods
General Toxicity
Ten Wistar Han rats and 4 beagle dogs per sex per group were used in 90-day repeat-dose general toxicity studies. The rats were 6 to 7 weeks old and 150 to 320 g at receipt, and the dogs were 5 to 6 months old and 5.2 to 9.1 kg at receipt. The rats underwent a 2-week quarantine period and dogs a 1-week quarantine to ensure suitability for study procedures. Rats and dogs were randomized to groups with normalization of mean body weight per group. During study, rats had ad libitum access to food (certified Harlan Teklad Rodent Diet #8728C) and water. During the study, dogs received a daily ration of food (Harlan Teklad Certified Canine Diet #2025) and had ad libitum access to water. BT-11 was suspended in 0.5% methylcellulose solution at concentrations to provide 0, 250, 500, or 1,000 mg/kg dose in a 10 mL/kg dose volume for rats and a 5 mL/kg dose volume for dogs and delivered by oral gavage daily for 90 days. Doses were selected based on observations from 7-day dose range finding studies in rats and dogs. Rats and dogs were weighed weekly, observed daily for cage-side observations, and observed weekly for detailed clinical signs of dysfunction. Detailed observations in rats were conducted 2 to 4 hours after dosing using a functional observational battery (FOB). 14
Immediately prior to euthanasia at termination of the experiment, blood was collected from the abdominal aorta in rats and jugular vein in dogs for clinical chemistry (albumin, alkaline phosphatase, alanine aminotransferase [ALT], aspartate aminotransferase [AST], total bilirubin [T.BIL], calcium, cholesterol, creatine kinase, creatinine, γ-glutamyltransferase, glucose, lactate dehydrogenase, phosphate, total protein, triglycerides, blood urea nitrogen [BUN], globulin, sodium, potassium, chloride), hematology (white blood cells, red blood cells, hemoglobin, hematocrit, mean corpuscular volume, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, platelets, neutrophils, lymphocytes, monocytes, eosinophils, large unstained cells, basophils, reticulocytes), and coagulation (prothrombin time, activated partial thromboplastin time) parameters. At necropsy, 41 select tissues were macroscopically scored and processed for histopathological examination by blinded, board-certified veterinary pathologists. Examined tissues in both species were adrenals, aorta, brain, epididymides, esophagus, eyes with optic nerve, heart, cecum, colon, duodenum, ileum, jejunum, rectum, kidneys, larynx, liver, lungs with bronchi, mandibular and mesenteric lymph nodes, ovaries, pancreas, pituitary, prostate, salivary glands, sciatic nerve, skeletal muscle, skin with mammary gland, spinal cord, spleen, sternum with marrow, stomach, testes, thymus, thyroid, tongue, trachea, urinary bladder, uterus with cervix, and vagina. The femorotibial joint, Harderian gland, and seminal vesicles were also examined in rats. In dogs, femur with marrow, gallbladder, and lacrimal gland were examined. Adrenals, brain, heart, kidneys, liver, ovaries, testes, and spleen were collected for determination of organ weights in both species.
Toxicokinetics
Blood samples were collected from the sublingual vein at 0, 0.5, 1, 2, 4, 8, and 24 hours after dosing for rats (3 per sex per group at each time point, forming a separate cohort in parallel to the main general toxicity cohort) and from the jugular vein at 0, 0.5, 1, 2, 4, 8, and 24 hours after dosing for dogs (4 per sex per group) at 250, 500, and 1,000 mg/kg after the first oral dose and again after 89 days of dosing. Blood was collected into K2EDTA tubes and plasma was collected after centrifugation. BT-11 was quantified using a previously developed method 15 that was used and validated for the rat and dog. Plasma samples were mixed in a 2:1 ratio with internal standard (IS; deuterated BT-11, 50.0 ng/mL in acetonitrile) and cold methanol. Samples were shaken, vortexed, and centrifuged. Supernatant was collected, evaporated under nitrogen, and reconstituted in water:acetonitrile:formic acid (90:10:0.1). Samples were analyzed on a Waters Acquity liquid chromatograph with a TSQ Vantage triple quadrupole mass spectrometer (MS) with ESI ionization (Thermo Scientific, Waltham, MA). Samples were injected onto a Water BEH C18 column (2.1 mm × 50 mm; 1.7 µm). Mobile phases were water:formic acid (99:1) and acetonitrile:formic acid (99:1). BT-11 peak was observed with retention time 2.6 minutes, parent ion mass/charge 265.11, and product ion mass/charge 194.08. Concentrations were calculated by linear regression to a calibration curve, linear in the range of 0.5 to 500 ng/mL. Individual plasma concentration–time profiles at scheduled (nominal) sampling times were analyzed by noncompartmental methods using Phoenix WinNonlin version 6.3 (Pharsight Corporation, a Certara Company, St. Louis, Missouri). The TK parameters calculated were tmax, Cmax, AUC0-8 (0-8 hours), area under the curve exposure from t = 0 to t = 24 hours (AUCTAU; where TAU = 24 hours), and tlast. In cases where terminal phase could be determined, t1/2 and Vz/F were also calculated.
Metabolism and Plasma Protein Binding
BT-11 was incubated in duplicate with cryopreserved hepatocytes or liver microsomes (with and without 2 mM nicotinamide-adenine dinucleotide phosphate [NADPH]) at 1 µM at 37°C. At specified time points between 0.25 and 45 minutes (microsomes) or between 0 and 120 minutes (hepatocytes), aliquots were sampled from cultures and mixed with cold methanol to stop reactions. Unmetabolized BT-11 was quantified as described above using liquid chromatography–mass spectrometry (LC-MS)/MS. For plasma protein binding, the test article was added in duplicate to human, rat, or dog plasma (pH 7.4, ±0.1, adjusted as necessary) at a concentration of 5 µM. This mixture was dialyzed in a rapid equilibrium dialysis device (Pierce, Waltham, MA) against phosphate buffer serum (PBS; pH 7.4) and incubated on an orbital shaker for 4 hours at 37°C. At the end of the incubation, aliquots from both plasma and PBS sides were collected, an equal amount of PBS was added to the plasma sample, and an equal volume of plasma was added to the PBS sample. Methanol (3 volumes) containing an analytical IS was added to precipitate the proteins and release the test article. After centrifugation, the supernatant was transferred to a new plate and analyzed by LC-MS/MS to obtain peak area ratios (analyte:IS) for determining the fraction unbound.
Cardiovascular Safety Pharmacology
Four male Beagle dogs at approximately 16 months of age were used in a Latin square design with a 7-day washout between treatments. Dogs were treated with single-dose BT-11 in 0.5% methylcellulose solution by oral gavage with a dose volume of 5 mL/kg to achieve 250, 500, and 1,000 mg/kg oral doses. During telemetry monitoring, the animals were unrestrained and monitored for selected physiological parameters. Body temperature, blood pressure, heart rate (derived from blood pressure and electrocardiogram [ECG]), and the ECG were monitored continuously from at least 2 hours predose until at least 24 hours postdose. On occasion, animals experienced minor temporary intermittent signal disruptions across all groups due to environmental conditions. Data were collected and analyzed in 1-minute intervals and are reported in 1-hour intervals for the first 4 hours and at the 24 hours point. Blood pressure parameters reported include systolic, diastolic, and mean arterial pressures. Electrocardiogram parameters reported include RR interval, PR interval, QRS duration, QT interval, and QTc interval. Temporally correlated blood pressure–derived heart rate and QT interval measurements were utilized for determining QTc interval throughout the study. The QTc interval was calculated using a procedure based on the method described by Spence et al 16 and modified by Miyazaki and Tagawa. 17 Representative ECG tracings were evaluated from each telemetry monitoring period at 1-hour predose (±15 minutes), at 2-hour postdose (±15 minutes), and within the last hour of each recording session (23-24 hours postdose). Qualitative evaluations of these tracings were performed by a board-certified veterinary cardiologist. End points were assessed by descriptive statistics and repeated measures analysis of covariance.
Central Nervous System Safety Pharmacology
Eight male Wistar Han rats were used within each group at 7 weeks of age. Animals were given an 8-day acclimation period after receipt and were randomized to group with normalization of starting body weight. Rats were treated with single-dose BT-11 in 0.5% methylcellulose solution by oral gavage with a dose volume of 10 mL/kg to achieve 250, 500, and 1,000 mg/kg oral doses. Functional observational battery tests were conducted predose and at 24 hours postdose. Functional observational battery included homecage, handling, open-field, and other observations. Parameters were scored as outlined in Moser et al. 18 Forelimb and hind limb grip strength was measured using the procedure described by Meyer et al, 19 and hind limb splay was quantitatively measured as described by Edwards and Parker. 20 Pain perception was assessed by measuring the latency of response to a nociceptive (thermal) stimulus when each animal was placed on a hot plate apparatus set to 52°C (±1°C) as described by Ankier. 21 Continuous end points were tested for significance by group pairwise comparisons (general analysis of variance [ANOVA]) and categorical end points were tested by exact Mantel–Haenszel test with step-down Sidak adjustment.
Respiratory Safety Pharmacology
Eight male Wistar Han rats were used within each group at 7 weeks of age. Animals were given a 7-day acclimation period after receipt and were randomized to group with normalization of starting body weight. Rats were treated with single-dose BT-11 in 0.5% methylcellulose solution by oral gavage with a dose volume of 10 mL/kg to achieve 250, 500, and 1,000 mg/kg oral doses. Each animal was placed in a whole-body plethysmograph chamber at least 2.5 hours prior to dosing for acclimation and collection of predose data. After at least 2.5 hours of time in the chambers, the animals were temporarily removed from the plethysmograph chambers for dosing. Immediately following dosing, the animals were returned to the plethysmograph chambers, and respiratory monitoring was continued for a period of at least 8 hours. Data were collected continuously, logged into 1-minute intervals, and are reported in 1-hour intervals. End points were assessed by descriptive statistics and repeated-measures analysis of covariance.
Reverse Mutation Assay
The tester strains used were the Salmonella typhimurium histidine auxotrophs TA98, TA100, TA1535, and TA1537 as described by Ames et al 22 and Escherichia coli WP2uvrA as described by Green and Muriel. 23 Aroclor 1254-induced rat liver S9 was used as the metabolic activation system at final concentration of 100 µL/mL homogenate with 4 mM β-NADP, 5 mM glucose-6-phosphate, 33 mM potassium chloride, 8 mM magnesium chloride, and 100 mM phosphate buffer. TA98, TA100, TA1535, TA1537, and WP2uvrA were exposed to the vehicle alone, positive controls (2-nitrofluorene for TA98, sodium azide for TA100 and TA1535, 9-aminoacridine for TA1537, methyl methanesulfonate for WP2uvrA, or 2-aminoanthracene with S9 activation for all strains) and 5 dose levels of test article (15-5,000 µg/plate), in triplicate, in the presence and absence of Aroclor-induced rat liver S9 by plate incorporation. Evidence of toxicity was scored relative to the vehicle control plate and recorded along with the revertant count for that plate. Toxicity was evaluated as a decrease in the number of revertant colonies per plate and/or a thinning or disappearance of the bacterial background lawn. For each replicate plating, the mean and standard deviation of the number of revertants per plate were calculated. Determination of a positive result was made by comparison of mean revertants per plate of at least 1 tester strain over a minimum of 2 increasing concentrations of test article equal to or greater than 3.0 times the mean vehicle control in TA1535 and TA1537 and equal to or greater than 2.0 times the mean vehicle control in TA98, TA100, and WP2uvrA.
Chromosomal Aberration Assay
Human peripheral blood lymphocytes (HPBLs) from male and female donors were cultured in complete medium (RPMI-1640 containing 15% heat inactivated fetal bovine serum, 2 mM
Micronucleus Test
Five male Wistar Han rats per group at 6 weeks of age were used. After a 5-day acclimation period, rats were randomized to group with normalization of starting body weight. Rats were treated with 2 doses of BT-11 in 0.5% methylcellulose solution by oral gavage with a dose volume of 10 mL/kg to achieve 500, 1,000, and 2,000 mg/kg doses. A positive control group received cyclophosphamide (25 mg/kg) by oral gavage. At 48 hours postdose, peripheral blood was collected from the retro-orbital sinus, mixed with anticoagulant, and fixed by cold methanol. The cells were resuspended and 20 µL of suspension was added to 80 µL of staining solution containing RNase, FITC-conjugated anti-CD71 antibodies, and PE-conjugated anti-CD61 antibodies. A 1.5 mL of DNA staining solution (propidium iodide) was added; then, the samples were placed on wet ice for at least 5 minutes prior to the flow-cytometric analysis. The frequency of micronucleated reticulocytes in peripheral blood was analyzed after flow cytometry calibration using malaria-infected biostandard and negative control standards provided in the Litron kit. Up to 20,000 reticulocytes per animal, when possible, were analyzed. Proportions of reticulocytes and micronucleated reticulocytes were calculated using standard methods. 25 -27 The group variances for micronucleus frequency for the vehicle and test article groups at the respective sampling time were compared using Levene test (significance level of P ≤ 0.05). If the variation between groups was found not to be significant, a parametric 1-way ANOVA was performed followed by a Dunnett post hoc analysis to compare each dose group to the concurrent vehicle control. If Levene test indicated heterogeneous group variances (P ≤ 0.05), a nonparametric statistical method (Kruskal-Wallis test and/or Mann-Whitney test) was used in evaluation of the data.
Test Article
The test article in these studies was BT-11, an identifier for a dihydrochloride salt of piperazine-1,4-diylbis((6-(1H-benzo[d]imidazol-2-yl)pyridin-2-yl)methanone). BT-11 was produced for these studies in a similar manner as previously described. 1 Produced material was purified and determined to be ≥98.9% purity by high performance liquid chromatography (HPLC). Final material was absent any individual organic impurities >0.1% with known toxicity concerns, heavy metals above detection limit, or microbial contamination and was within acceptable limits for residual solvents. All solution concentrations were validated to be accurate to nominal value by HPLC. The test article was compliant with ICH Q3A and Q3C for drug product and residual solvents.
Statistics
Treatment emergent trends were analyzed by varied statistical and comparative methods described within individual sections above chosen based on the accepted standard of analysis for each respective assay. Statistical differences were, in general, indicated by P ≤ 0.05.
Results
General Toxicity
The 90-day repeat-dose general toxicity study in Wistar Han rats indicates that BT-11 was well tolerated when administered via oral gavage to male and female rats in doses of 0, 250, 500, or a 1,000 mg/kg/d for 90 consecutive days. No treatment-related deaths or test article–associated clinical signs of toxicity were noted. Recorded observations were limited to red pigmented ears, small scab on head, and swollen ears in single rats with no dose- or treatment-dependent trend. These findings were not considered to be treatment-related as they were not observed in a dose-related manner, occurred only in intermediate groups, and/or the changes were not observed in both sexes. Ophthalmological examinations were conducted and no differences between control and treated animals were observed. Body weights and body weight gains were unaffected by treatment (Figure 1). No changes in food consumption were observed. Evaluation of clinical pathology profiles, with representative parameters presented in Table 1, did not reveal any test article–related changes or dose associated trends. In addition, no test item–related gross necropsy findings or microscopic changes were observed at necropsy suggesting a lack of target organ toxicity with oral dosing. Further, no organ weight changes were observed. The no-observed adverse effect level (NOAEL) for BT-11 was considered to be 1,000 mg/kg/d in rats.
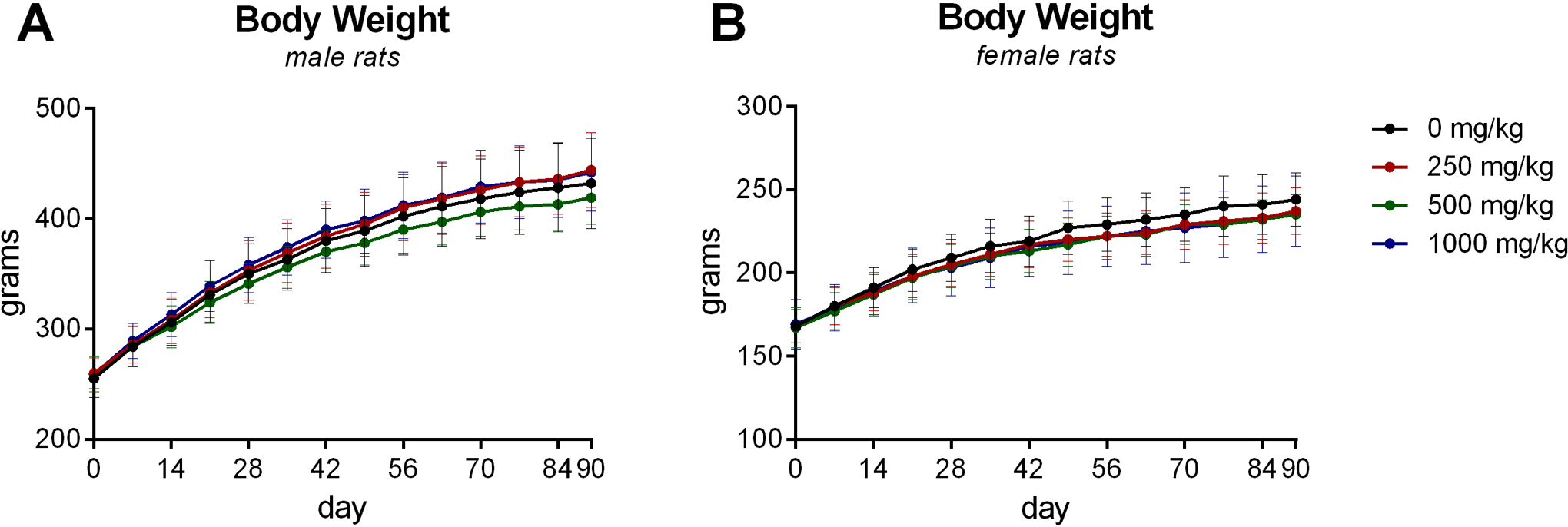
Body weight of male and female Wistar Han rats treated daily with BT-11 (0, 250, 500, 1,000 mg/kg/d) for 90 days. Data presented as mean value with error bars representative of standard deviation. No statistical significance was noted among test and control groups (n = 10/sex/group).
Representative Rat Clinical Pathology and Organ Weights.a
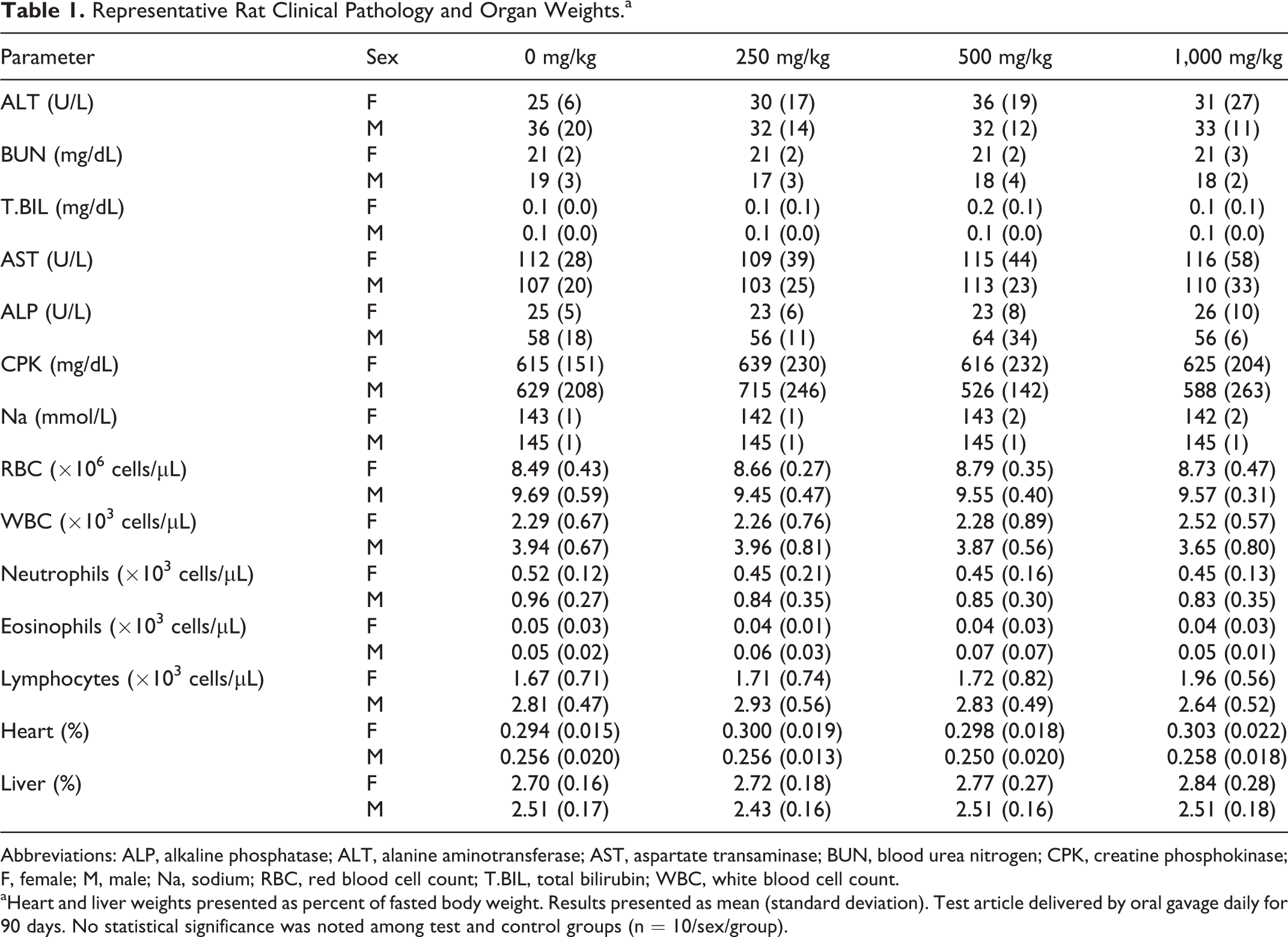
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate transaminase; BUN, blood urea nitrogen; CPK, creatine phosphokinase; F, female; M, male; Na, sodium; RBC, red blood cell count; T.BIL, total bilirubin; WBC, white blood cell count.
aHeart and liver weights presented as percent of fasted body weight. Results presented as mean (standard deviation). Test article delivered by oral gavage daily for 90 days. No statistical significance was noted among test and control groups (n = 10/sex/group).
The results of the 90-day repeat-dose general toxicity study in Beagle dogs provided similar results that oral BT-11 was well tolerated when administered via gavage to male and female Beagle dogs in doses of 0, 250, 500, or a 1,000 mg/kg/d for 90 consecutive days. No treatment-related deaths or treatment associated clinical signs of toxicity were noted. Observations were limited to a few dogs noted with isolated episodes of emesis and salivation with no dose- or treatment-dependent trend and one female noted with a sore above the right eye that resolved by day 35. Ophthalmological examinations were conducted and no differences between control and treated animals were observed. Body weights and weight gains were unaffected by treatment (Figure 2). No changes in food consumption were observed. Evaluation of clinical pathology profiles (hematology, coagulation and clinical chemistry), representative parameters in Table 2, did not reveal any test article–related changes. No significant trends in liver enzymes (ALT, AST), kidney function (BUN), ion levels (potassium, sodium, chloride, calcium), or signs of liver damage (T.BIL) was noted during the study. There was also no evidence of systemic immune effects as indicated by no changes in white blood cell counts and specific white blood cell subsets (ie, neutrophils, eosinophils, lymphocytes) after 90 days of dosing which was in agreement with the lack of observed changes microscopically and by weight to lymphoid organs. Cardiovascular evaluations were normal in all test animals at termination. No test article–related gross necropsy findings or microscopic changes were observed at necropsy. Organ weight differences were observed in 500 mg/kg dosed females as increased mean absolute heart weights and increased mean liver weights relative to brain; however, differences were nonsignificant relative to body weight and were diminished at the higher 1,000 mg/kg dose level (Table 2). These changes were not observed with males dosed with 500 mg/kg or any dog dosed with 1,000 mg/kg respective to controls. No significant change in liver- or heart-associated enzymes or microscopic changes in respective organs were associated with the change in organ weight. Based on the results of this study, BT-11 administration results in no test article–related clinical signs, body weight changes, clinical pathology changes, or gross or microscopic tissue changes when given via oral gavage at dose levels up to 1,000 mg/kg/d to male and female dogs. This would make 1,000 mg/kg/d the NOAEL for BT-11 in beagle dogs.
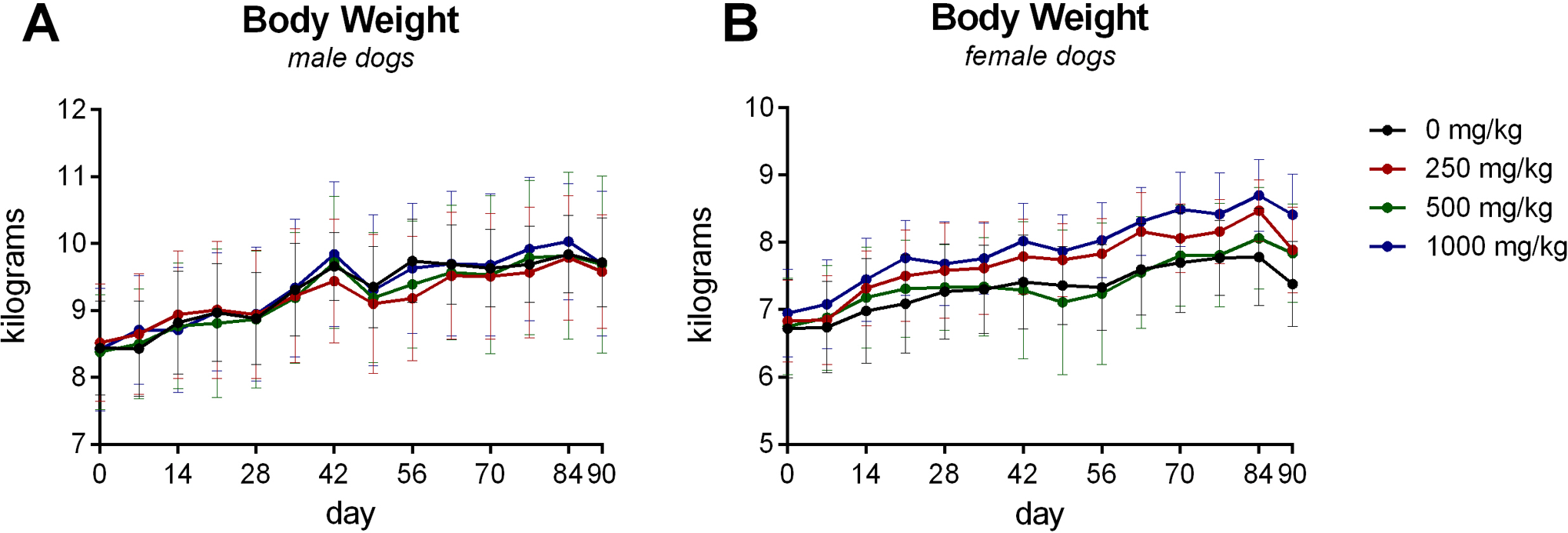
Body weight of male and female Beagle dogs treated daily with BT-11 (0, 250, 500, 1,000 mg/kg/d) for 90 days. Data presented as mean value with error bars representative of standard deviation. No statistical significance was noted among test and control groups (n = 4/sex/group).
Representative Dog Clinical Pathology and Organ Weights.a
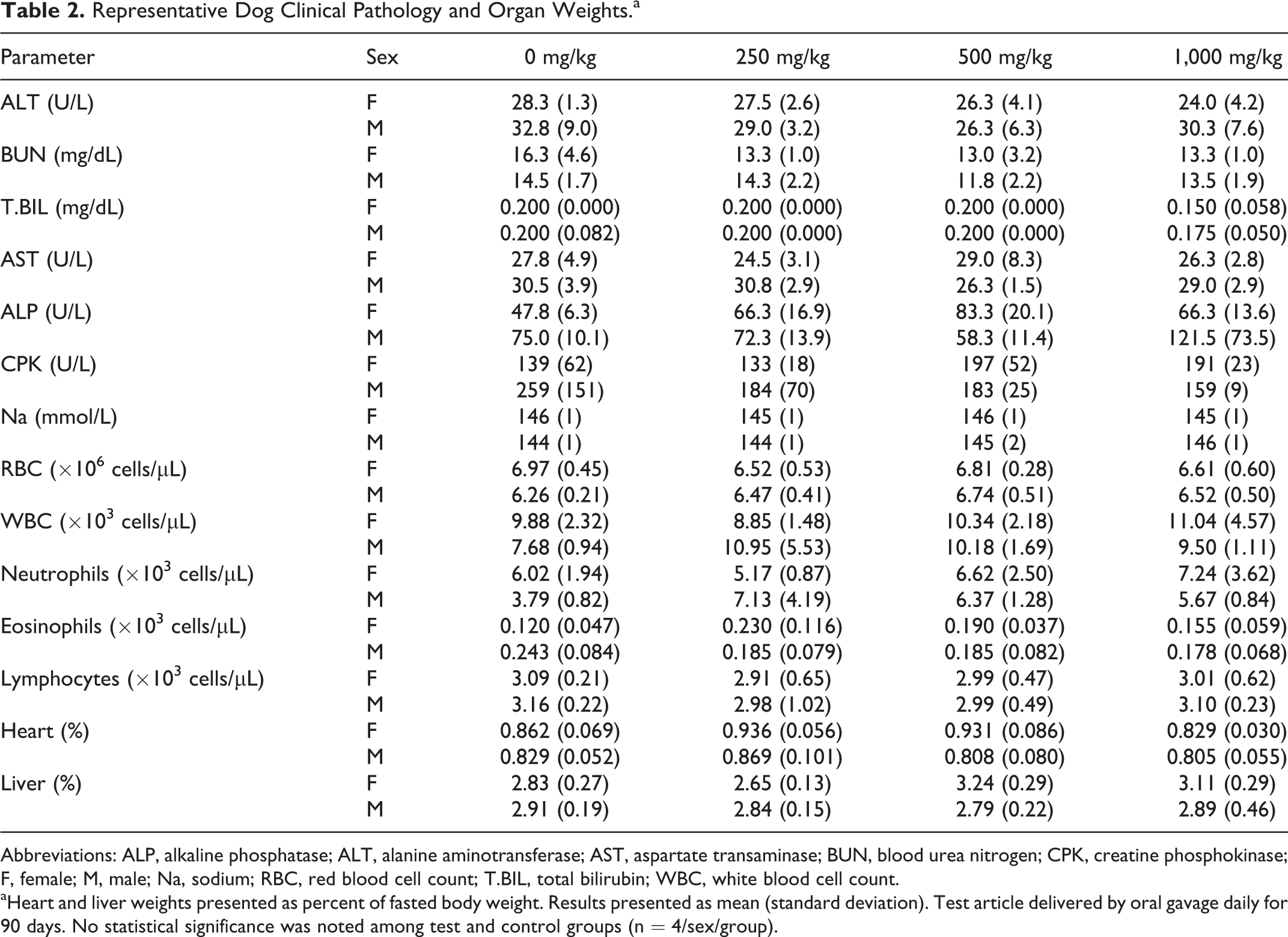
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate transaminase; BUN, blood urea nitrogen; CPK, creatine phosphokinase; F, female; M, male; Na, sodium; RBC, red blood cell count; T.BIL, total bilirubin; WBC, white blood cell count.
aHeart and liver weights presented as percent of fasted body weight. Results presented as mean (standard deviation). Test article delivered by oral gavage daily for 90 days. No statistical significance was noted among test and control groups (n = 4/sex/group).
Toxicokinetic and Metabolism
The TK of BT-11 was assessed in rats (Table 3) and dogs (Table 4) after administration after the first oral dose and again after 89 days of dosing with 250, 500, or 1,000 mg/kg/d. In both rats and dogs, TK were similar between males and females at each dose level. In rats, the profile was similar across all doses examined (250, 500, and 1,000 mg/kg/d) with a maximum concentration occurring between 0.5 and 1 hours postdose. Clearance from the plasma by 8 hours postdose suggested a half-life between 1.91 and 2.90 hours in all but 1 group. Females dosed with 1,000 mg/kg had a half-life of 4.77 hours. The rapid clearances indicate a low potential for dose accumulation. Oral BT-11 had low systemic exposure, when comparing total dosage and plasma concentrations. This was indicated in rats as they had AUCTAU ranging between 93.0 and 281 h × ng/mL and mean Cmax of 19.3 to 123 ng/mL across groups. Further, BT-11 concentrations did not increase in a dose-proportional manner, with 1,000 to 250 mg/kg ratios in AUCINF of 2.16 in females and 0.87 in males. When tested in dogs, BT-11 was detectable for a longer period of time compared to rats, with a tlast ranging from 12 to 24 hours in dogs. As such, the AUCTAU was slightly higher in dogs ranging from 379 to 1101 h × ng/mL. Mean group Cmax ranged from 61.4 to 210 ng/mL and occurred between 2 and 4 hours postdosing. When comparing dose groups in female dogs, no dose proportionality using AUCTAU was observed as the 1,000 to 250 mg/kg ratio was 0.9. However, comparing dose groups in males resulted in a near dose-proportional relationship both between 250 and 1,000 mg/kg and 500 and 1,000 mg/kg dosed groups. Toxicokinetic assessment was conducted both at the beginning and at the end of study for rats (Table 3) and dogs (Table 4). No accumulation in plasma, based on the accumulation ratio in maximum concentrations and AUC, was observed over the 90-day dosing interval in either rats or dogs.
Rat Toxicokinetics.a
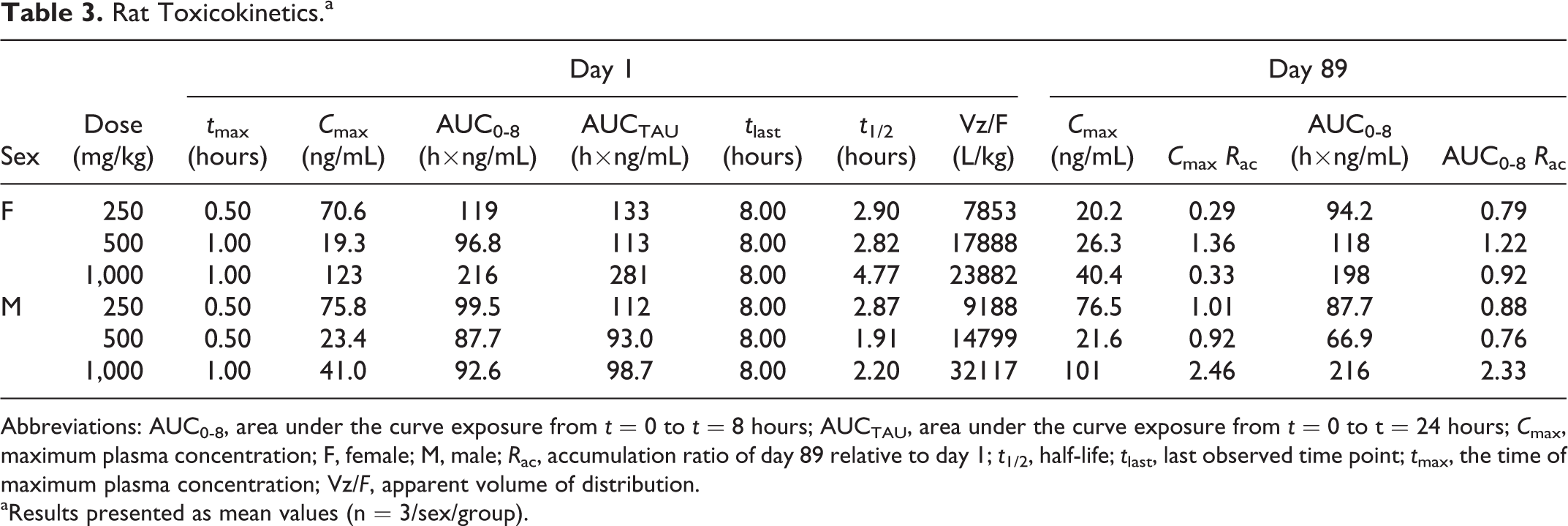
Abbreviations: AUC0-8, area under the curve exposure from t = 0 to t = 8 hours; AUCTAU, area under the curve exposure from t = 0 to t = 24 hours; Cmax, maximum plasma concentration; F, female; M, male; Rac, accumulation ratio of day 89 relative to day 1; t1/2, half-life; tlast, last observed time point; tmax, the time of maximum plasma concentration; Vz/F, apparent volume of distribution.
aResults presented as mean values (n = 3/sex/group).
Dog Toxicokinetics.a
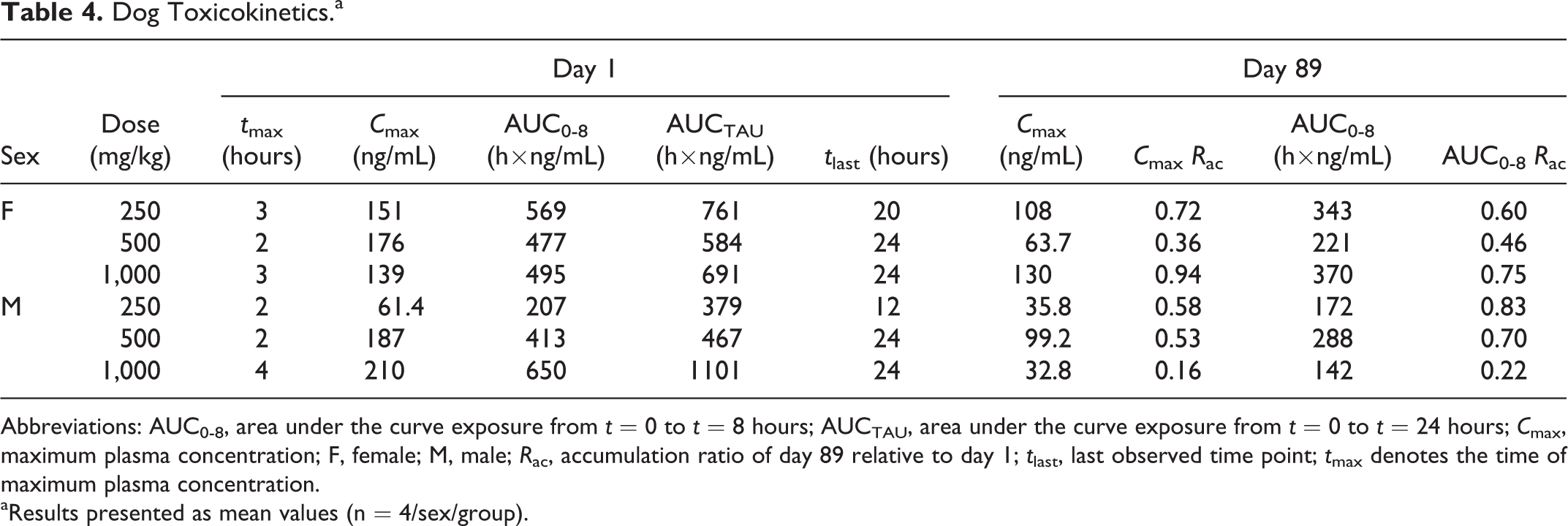
Abbreviations: AUC0-8, area under the curve exposure from t = 0 to t = 8 hours; AUCTAU, area under the curve exposure from t = 0 to t = 24 hours; Cmax, maximum plasma concentration; F, female; M, male; Rac, accumulation ratio of day 89 relative to day 1; tlast, last observed time point; tmax denotes the time of maximum plasma concentration.
aResults presented as mean values (n = 4/sex/group).
For metabolism experiments, BT-11 (1 µM) was incubated in duplicate with pooled liver microsomes and pooled hepatocytes from humans, rats, and dogs (Table 5). For microsomes, analysis by LC-MS/MS indicated that no metabolism of BT-11 occurred in the absence of NADPH. In the presence of NADPH, BT-11 was observed to have a mean half-life of 54.9 minutes (human), 116.0 minutes (rat), and 73.6 minutes (dog) in microsomes with mean clearance rates of 42.1 (human), 20.0 (rat), and 31.4 (dog) µl/min/mg. For hepatocytes, LC-MS/MS analyses indicated that BT-11 had a mean half-life of 285.2 minutes (human), 320 minutes (rat), and 247 minutes (dog) in hepatocytes with mean clearance rates of 4.9 (human), 4.33 (rat), and 5.61 (dog) µL/min/million cells. Additionally, plasma protein binding of BT-11 was tested in duplicate in human, rat, and dog plasma at a 5-µM concentration. Propanolol and warfarin were used as representative controls. BT-11 was observed to be >97% plasma bound in all 3 tested species (rat, dog, human) with >99% protein binding in humans (Table 6).
Microsomal and Hepatocyte Stability.a
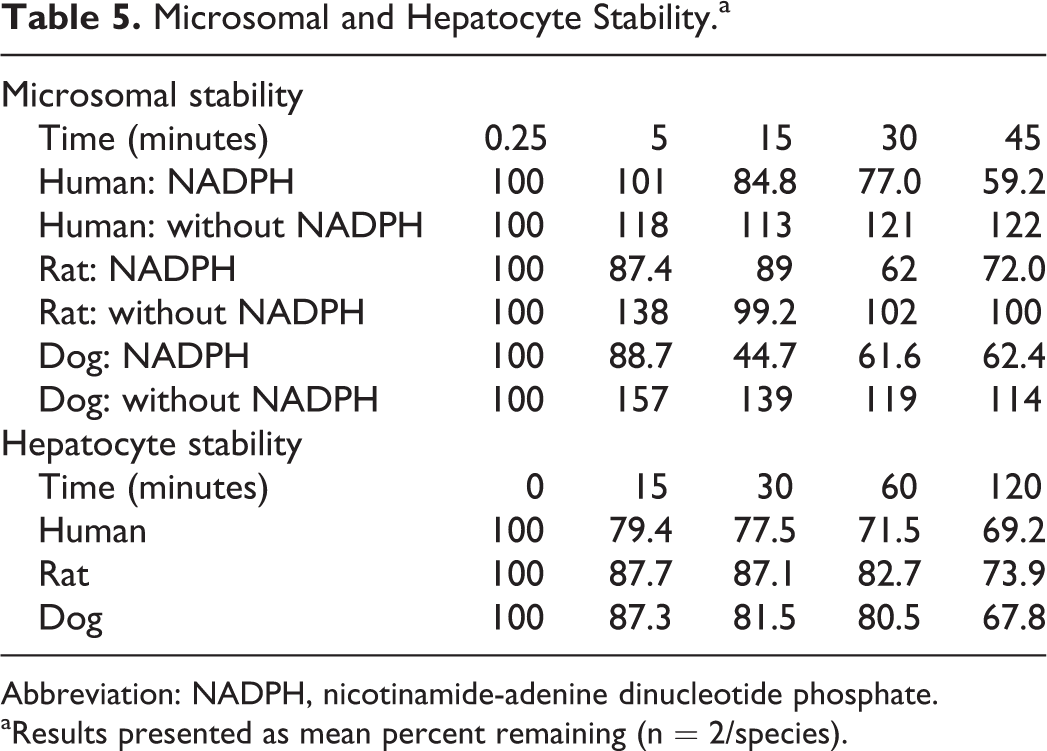
Abbreviation: NADPH, nicotinamide-adenine dinucleotide phosphate.
aResults presented as mean percent remaining (n = 2/species).
Plasma Protein Binding.a
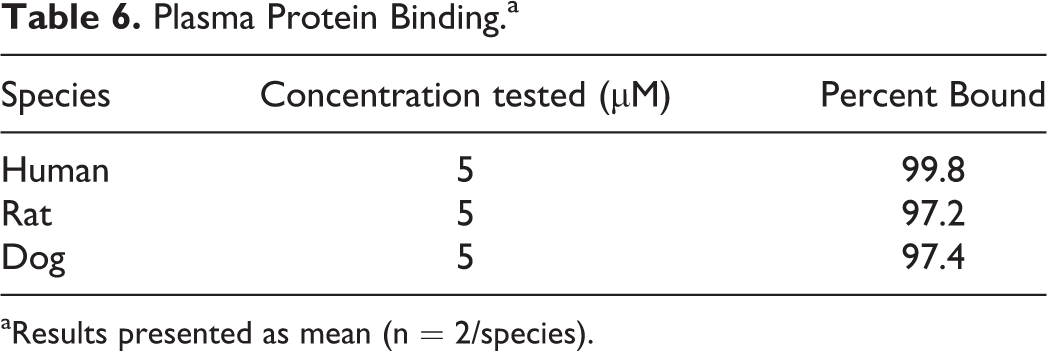
aResults presented as mean (n = 2/species).
Safety Pharmacology
In the cardiovascular study, telemetered dogs were given a single oral dose of BT-11 at 250, 500, or 1,000 mg/kg. A negative control receiving vehicle was included. BT-11, administered orally at single doses of 250, 500, or 1,000 mg/kg to dogs, did not produce mortality, clinical signs, or effects on body temperature, blood pressure, heart rate, or the evaluated ECG parameters (Table 7). There were no BT-11-related changes noted in systolic, diastolic, or mean arterial blood pressures during the study. Occasional fluctuations in all blood pressure parameters were noted during the monitoring periods. Increases in blood pressure were noted for all treatments, including vehicle control, at the time of dosing and then began to return to predose levels by 30 minutes postdose. These increases were considered related to environmental stimuli related to the general restraint and handling of the animals. There were no BT-11-related changes noted in heart rate during the study beyond occasional fluctuations in all animals observed at times of environmental stimuli, such as dosing. No treatment-related significant trends were observed in ECG parameters. Slight elevations in PR interval, across treated groups, were observed at 3 and 4 hours postdosing that resolved by 24 hours postdosing with treatment. However, these changes were largely influenced by single animals that displayed higher predose PR intervals on day of dosing with test article relative to days dosed with vehicle. Importantly, BT-11 did not influence corrected QT interval at 1,000 mg/kg, with overall mean 238.5 milliseconds relative to 235.9 milliseconds. Accordingly, oral administration of BT-11 at doses up to and including 1,000 mg/kg was not associated with any effects on cardiovascular function in telemetered dogs.
Cardiovascular Safety Pharmacology in Dogs.a
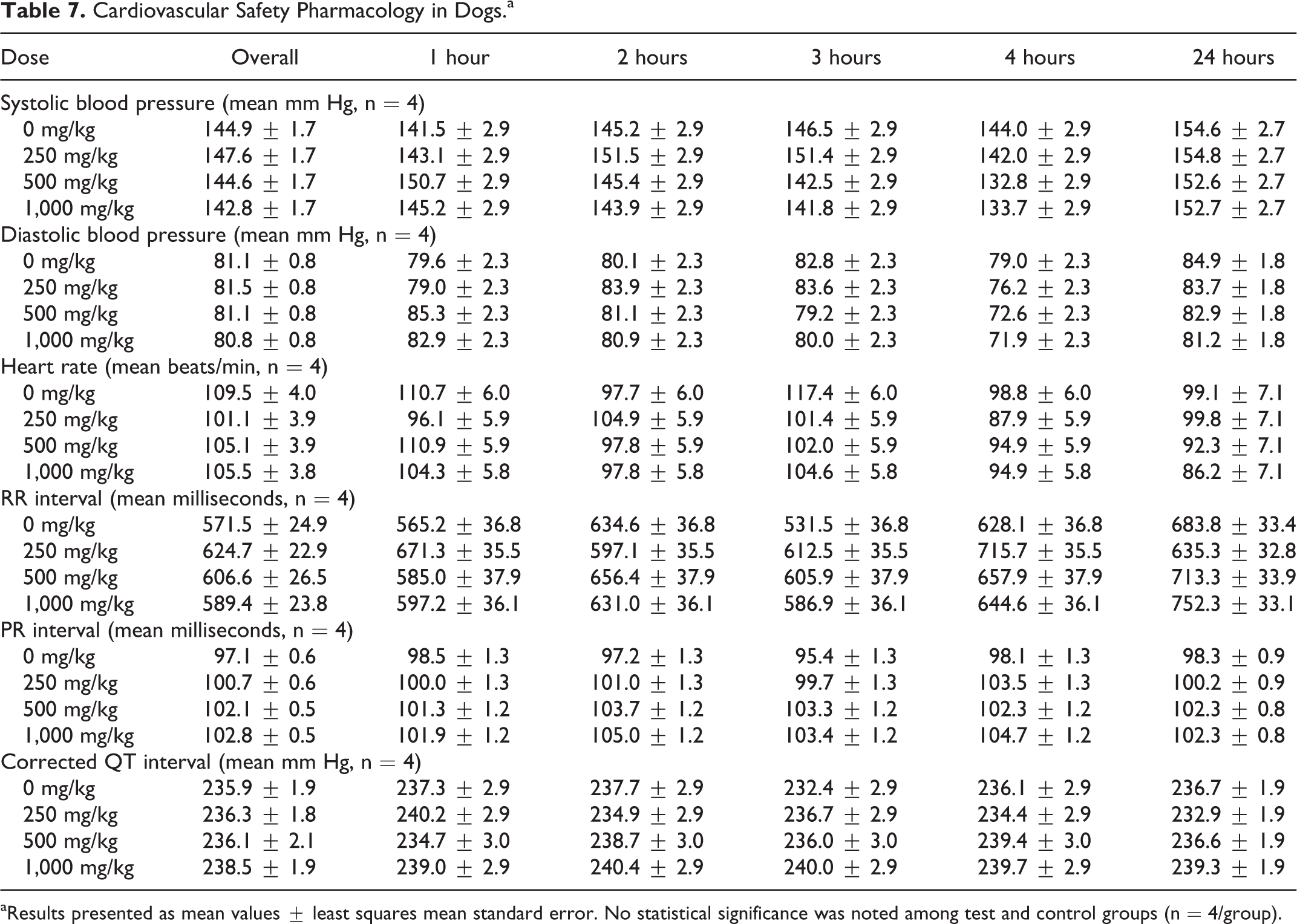
aResults presented as mean values ± least squares mean standard error. No statistical significance was noted among test and control groups (n = 4/group).
In the central nervous system study, rats were given a single oral dose of BT-11 at 0, 250, 500, or 1,000 mg/kg, then monitored by FOB at 2 and 24 hours postdose. Data from 24 hours postdose are provided in Table 8. No deviations from normal behavior were observed in any group within posture, vocalization, exophthalmos, lacrimation, palpebral closure, piloerection, pupil response, salivation, bizarre behavior, clonic behavior, gait, mobility, righting, tonic movements, and respiration parameters. Thus, these parameters were scored as 0 across all tested doses. BT-11, administered orally to male rats at single doses of 250, 500, or 1,000 mg/kg and did not produce mortality, clinical signs, or effects on the evaluated neurobehavioral parameters. No significant trends in neurobehavioral parameters were noted. Further, no trend in activity, homecage, or handling observations and autonomic, neuromuscular, or physiological measurements were noted. A single sensorimotor statistical difference was noted in the lack of a tail pinch response in the 1,000 mg/kg dosed group. However, this may be a normal response in rats as the lack of change in touch and thermal responses suggest this is not associated with treatment. Relative to vehicle, the 1,000 mg/kg dosed group was observed to have slight numerical increases in ease of removal, approach, and touch response scores. However, these scores did not statistically differ from vehicle-treated rats or baseline and did not vary outside the normal range. No rat was observed to be overtly aggressive in any group. Accordingly, oral administration of BT-11 at doses up to and including 1,000 mg/kg was not associated with effects on neurobehavioral function in rats.
Functional Observational Battery in Rats.a
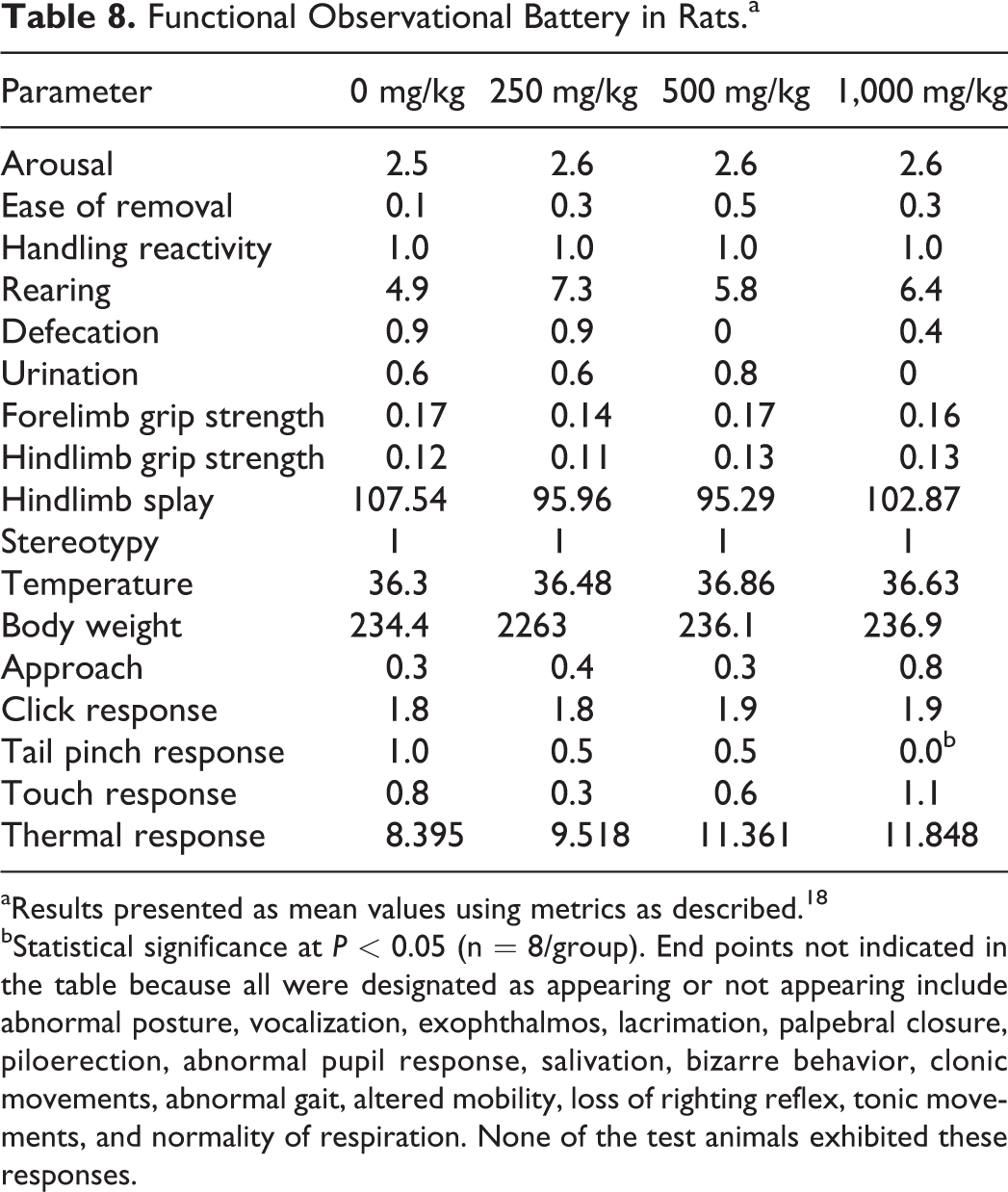
aResults presented as mean values using metrics as described. 18
bStatistical significance at P < 0.05 (n = 8/group). End points not indicated in the table because all were designated as appearing or not appearing include abnormal posture, vocalization, exophthalmos, lacrimation, palpebral closure, piloerection, abnormal pupil response, salivation, bizarre behavior, clonic movements, abnormal gait, altered mobility, loss of righting reflex, tonic movements, and normality of respiration. None of the test animals exhibited these responses.
In the respiratory study, rats were administered a single oral dose of BT-11 at 250, 500, or 1,000 mg/kg, then monitored in a whole-body plethysmograph chamber for 8 hours. BT-11, administered orally to male rats at single doses of 250, 500, or 1,000 mg/kg, did not produce mortality, clinical signs, or effects on respiratory function (Table 9). No trends were observed at individual intervals between 1 and 8 hours postdose in respiratory rate, tidal volume, or minute volume. Similarly, overall averages had no treatment-related trends with minimal differences between groups in respiratory rate (115 breaths/min [1,000 mg/kg] to 115 [0 mg/kg]), tidal volume (1.134 mL [1,000 mg/kg] to 1.109 [0 mg/kg]), and minute volume (123.033 mL/min [1,000 mg/kg] to 121.019 [0 mg/kg]). Accordingly, oral administration of BT-11 at doses up to and including 1,000 mg/kg was not associated with effects on respiratory function in rats.
Respiratory Safety Pharmacology in Rats.a
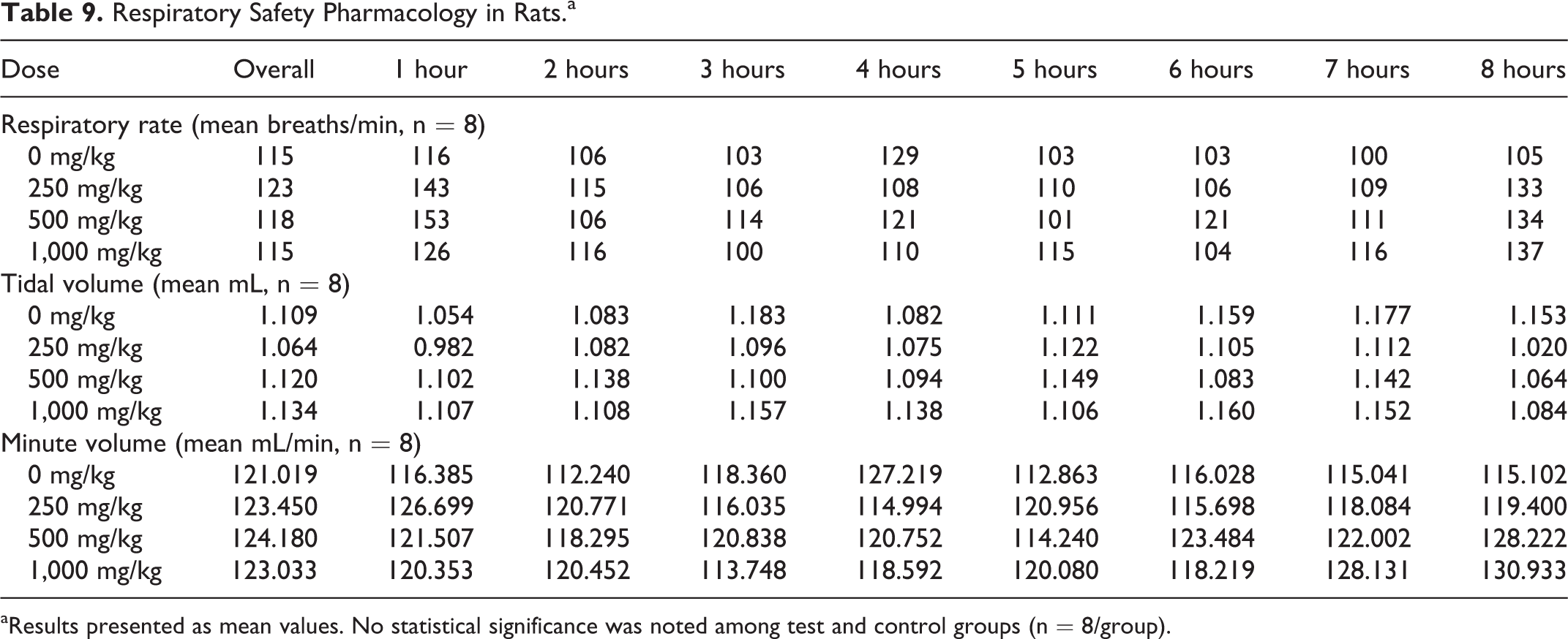
aResults presented as mean values. No statistical significance was noted among test and control groups (n = 8/group).
Genotoxicity
The genotoxicity of BT-11 was tested in 3 assays: the bacterial reverse mutation assay (Ames test), the chromosomal aberration assay, and the micronucleus test. The bacterial reverse mutation assay, in TA98, TA100, TA1535, TA1537, and WP2uvrA, was conducted at dose levels of 15, 50, 150, 500, 1,500, and 5,000 µg/plate. The results of the bacterial reverse mutation assay indicate that BT-11 did not cause a positive mutagenic response with any of the tester strains in either the presence or absence of Aroclor-induced rat liver S9 (Table 10). Additionally, the data showed no numerical increase in colonies on plates with BT-11 relative to vehicle control.
Ames Test.a
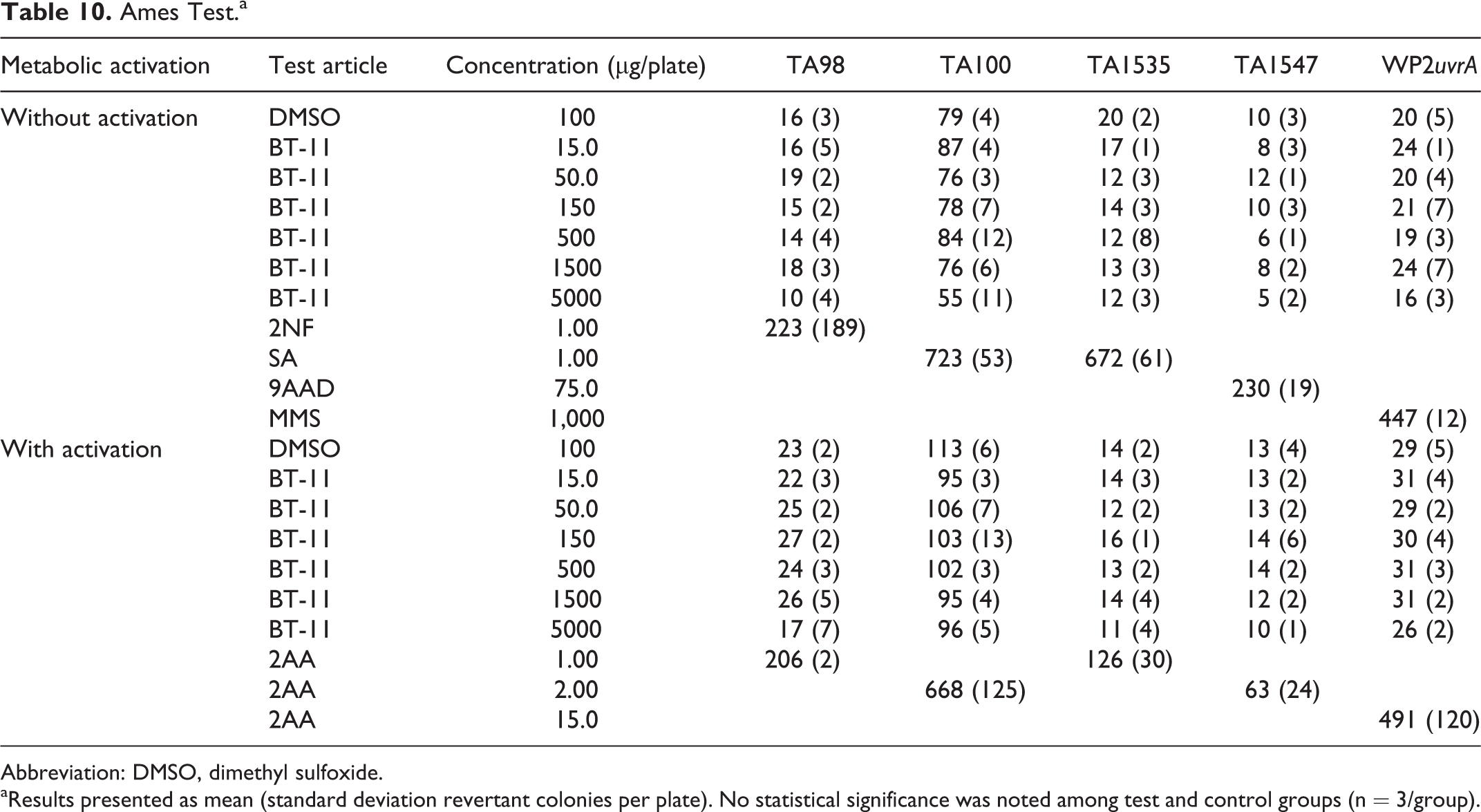
Abbreviation: DMSO, dimethyl sulfoxide.
aResults presented as mean (standard deviation revertant colonies per plate). No statistical significance was noted among test and control groups (n = 3/group).
For the chromosomal aberration assay, no significant differences in mitotic index or chromosomal aberrations were noted with BT-11 up to the tested limit concentration of 125 µg/mL (Table 11). BT-11 did not produce any significant concentration-dependent trend in cytotoxicity with either 4-hour treatment and 16-hour recovery or 20 hours of continuous treatment. All BT-11-treated groups had equal or lesser mean percentages of numerical and structural aberrations compared to negative vehicle control (dimethyl sulfoxide). Validity of the assay was confirmed with positive controls, mitomycin C (without metabolic activation), and cyclophosphamide (with metabolic activation).
Chromosomal Aberration.a
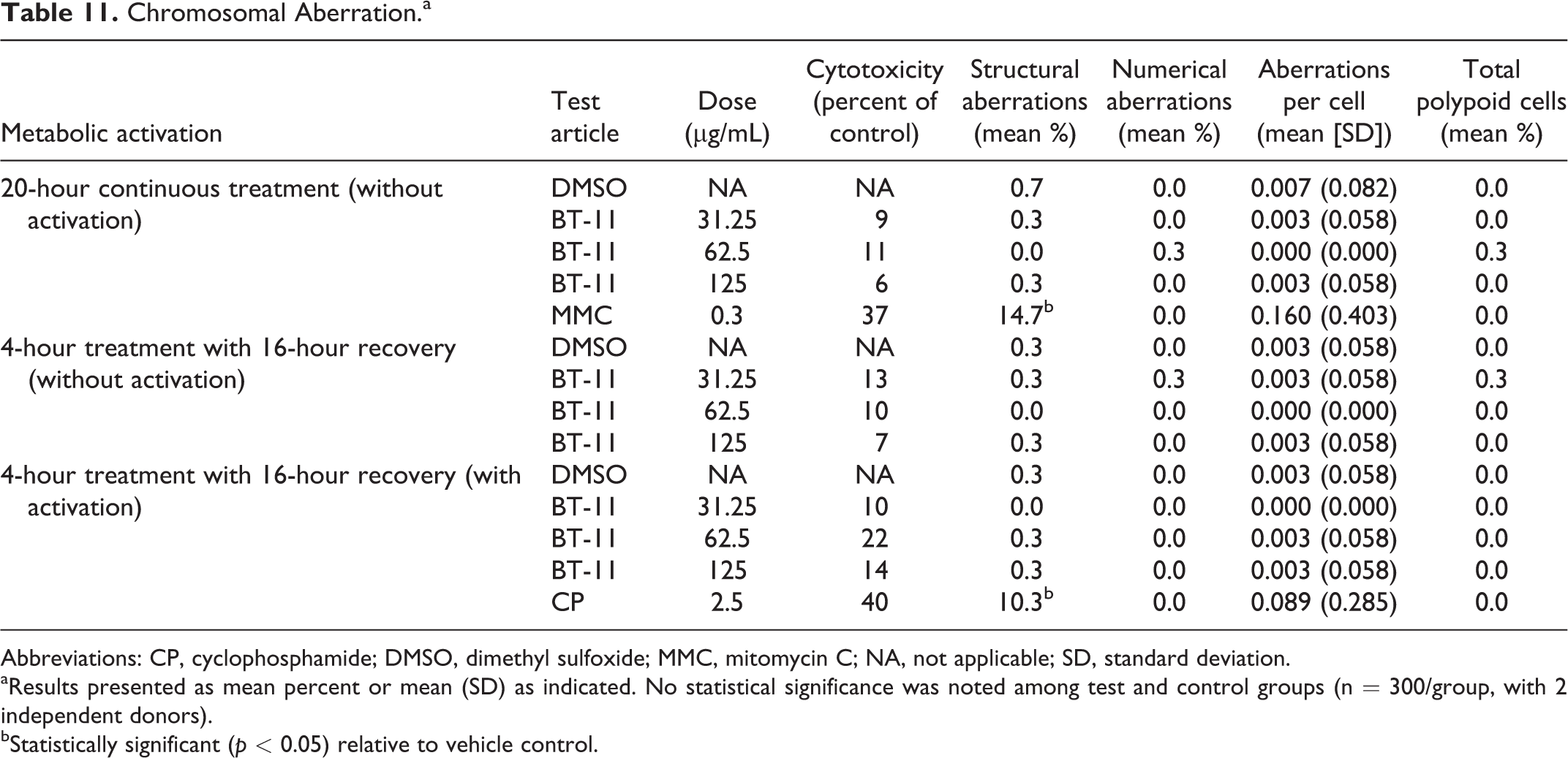
Abbreviations: CP, cyclophosphamide; DMSO, dimethyl sulfoxide; MMC, mitomycin C; NA, not applicable; SD, standard deviation.
aResults presented as mean percent or mean (SD) as indicated. No statistical significance was noted among test and control groups (n = 300/group, with 2 independent donors).
bStatistically significant (p < 0.05) relative to vehicle control.
For the micronucleus assay, 6-week-old male Wistar Han rats (n = 5/group) were dosed with vehicle, BT-11 at 3 dose levels (500, 1,000, 2,000 mg/kg), or positive control (cyclophosphamide). All dose formulations were administered once per day on 2 consecutive days. Under the conditions of the micronucleus assay described, BT-11 was negative for the induction of micronucleated reticulocytes up to the oral dose of 2,000 mg/kg (Table 12). No changes in percent reticulocytes were observed at any of the 3 BT-11 doses tested, indicating that oral BT-11 up to 2,000 mg/kg did not induce cytotoxicity. Further, no statistically significant increase in micronucleated reticulocytes was observed in BT-11-treated groups. The cyclophosphamide positive control did induce a significant increase in micronucleated reticulocytes.
Micronucleus Test in Rats.a
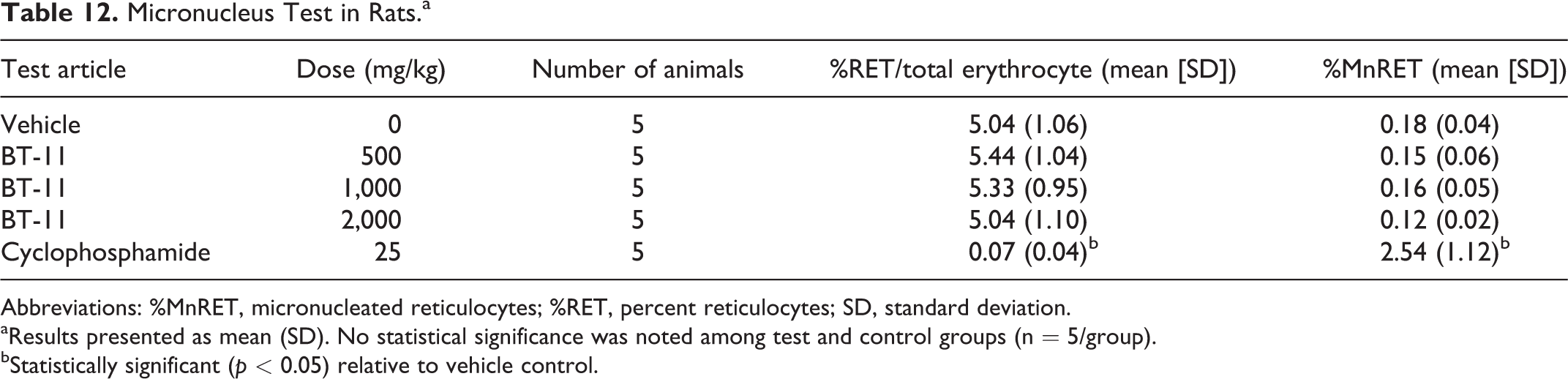
Abbreviations: %MnRET, micronucleated reticulocytes; %RET, percent reticulocytes; SD, standard deviation.
aResults presented as mean (SD). No statistical significance was noted among test and control groups (n = 5/group).
bStatistically significant (p < 0.05) relative to vehicle control.
Discussion
In the nonclinical Good Laboratory Practice (GLP) studies presented, BT-11-dosed animals showed no apparent signs of toxicity at any of the doses tested (250, 500, or 1,000 mg/kg/d). The results of 90-day repeat-dose GLP toxicity studies in rats and dogs, combined with targeted cardiovascular, respiratory, and central nervous system safety pharmacology studies, indicate that oral dosing of BT-11 was well tolerated in these studies, allowing for further clinical development. Additional testing of genotoxicity in 2 in vitro (bacterial and mammalian) and 1 in vivo assay indicates that BT-11 has no mutagenic or genotoxic potential under the conditions of the tested assays.
The overall risk of systemic side effects with BT-11 is suggested to be minimal due to the low systemic exposure and rapid half-life of this compound. The Cmax of BT-11 in plasma of rats and dogs ranged from 19.3 to 210 ng/mL at doses ranging from 250 to 1,000 mg/kg. A possible sex difference in TK in dogs was suggested in that increasing doses increased AUC’s in male but did not in females for this species when determined after the first dose. However, the dose-dependent increase in plasma concentrations seen with male dogs on day 1 was not observed in samples collected on day 89. With a half-life of approximately 3 hours, orally dosed BT-11 has a low overall systemic exposure that does not proportionally scale with dosing increases in the tested animal species, which likely indicates that the gut absorptive capacity is saturable at high doses. The short half-life allows for minimal accumulation of drug in plasma, which indicates systemic exposure will not increase with extended dosing regimens. In addition to its low overall exposure, absorbed BT-11 is highly plasma bound, suggesting a low percentage of active drug in the body. BT-11 has a high metabolic stability with half-life over 1 hour in both microsomes and hepatocytes. Plasma protein binding and metabolic assays were conducted in rat, dog, and human cells with little to no observed differences, suggesting that rats and dogs are adequate models for the systemic processing of BT-11 postabsorption. The information on pharmacokinetics of BT-11 presented here and previously 15 suggest that orally administered BT-11 is unlikely to accumulate.
No target organ system was identified for BT-11 in general toxicity or safety pharmacology studies. In 500 mg/kg dosed female dogs, an increase in liver weight relative to brain weight and absolute heart weight was noted. However, the increases in organ weights were not significant relative to fasted body weight and no macroscopic or microscopic findings in either organ were recorded. No similar changes were observed in rats or dogs of other groups even within the higher, 1,000 mg/kg, group. While there was a slight increase in alkaline phosphatase enzyme, no increase in ALT or AST was observed. No significant change in creatine phosphokinase was observed that would correlate to a heart weight change and there were no findings noted in the cardiovascular safety pharmacology study. Further, the lack of difference in systemic exposure within the 500 mg/kg dosed female dogs relative to other groups, overall low systemic exposure, and low systemic bioactivity provide additional justification for a low risk of these findings impacting use in humans. Overall, the weight of evidence is indicative of a low risk of hepatic and cardiac toxicities.
Based on the observed TK of BT-11, the HPBL in vitro assay for genotoxicity tested concentrations of BT-11 roughly 1,000-fold higher than those reached in plasma. This suggests that the measured systemic concentrations of BT-11 poses low risk in terms of genotoxicity potential. A common concern for immunomodulatory therapies for IBD and other autoimmune diseases is a broad immunosuppression that opens the risk of increased infection and incidence of cancer. Based on currently measured parameters that demonstrate low overall plasma concentrations and high plasma protein binding, BT-11 is likely to have limited to no effects on the non-GI immune system. In 90-day repeat-dose studies, BT-11 at doses up to 1,000 mg/kg/d had no significant effects on hematology parameters, including subpopulations of white blood cells, and did not impact overall white blood cell counts or influence the appearance or size of lymphoid organs. Although currently untested, these results combined with the current knowledge on BT-11 and the LANCL2 pathway provide a preliminary indication that an increased risk of serious infections or cancer may not apply to BT-11.
The anti-TNF biologic, adalimumab, has been tested in cynomolgus monkeys at doses up to 214.8 mg/kg/wk (relative to a therapeutic dose of 40 mg every other week) in which adverse immune effects including decrease cellularity of B cells in splenic follicles were observed at 32 mg/kg/wk, and thymic involution was observed at 82.9 mg/kg/wk. Importantly, these adverse immune effects have carried forward into the clinic, resulting in increased risk of tuberculosis, black box warnings on opportunistic and rare infections, and a 2- to 4-fold increase in malignancies. Nonclinically, the genotoxicity studies in mice for adalimumab have questionable relevance due to the decreased affinity of adalimumab to its target in rodents. Adalimumab did not undergo carcinogenicity studies prior to approval likely due in part to the uncertainly surrounding the predictive power of these studies for large molecules. In contrast, mesalamine, a 5-ASA therapeutic, has proven to provide a low risk of side effects within patients with IBD. Nonclinically, mesalamine was observed to be nonmutagenic in Ames and micronucleus tests. Observations were made in 13-week oral toxicity studies at high doses, including renal lesions at 2,400 mg/kg in mice, 1,150 mg/kg in rats, and 250 mg/kg in cynomolgus monkeys. Mesalamine, and other 5-ASA therapeutics, has only mild efficacy at doses of 1.2 to 4.8 g/d, thus has a smaller nonclinical safety margin above therapeutic doses relative to the anticipated human efficacious dose of BT-11. Meanwhile, tofacitinib was observed to have significant immune effects in rats and monkeys, including bone marrow depletion, atrophy of lymphoid organs, and increased infections, including infection-related deaths. Histopathological changes were observed in lymphoid tissues at 10 mg/kg/d in rats. In monkeys, the 50 mg/kg/d dose caused 100% mortality in male monkeys and doses as low as 2 mg/kg, which results in an AUC only 7.7-fold higher than the human therapeutic dose, causing significant decreases in red blood cell counts.
In animal models of IBD, the effective oral dose of BT-11 is 8 mg/kg, once daily 2 for up to 8 weeks. In vivo safety pharmacology and 90-day repeat-dose toxicity studies support an NOAEL >1,000 mg/kg, providing a dosing multiple that is approximately 100-fold higher than the therapeutic dose (8 mg/kg). To observe mucosal healing in phase II human clinical studies, treatment periods of 12 weeks are generally required. The relatively short half-life (<5 hours in rats) and lack of accumulation in dogs suggest little risk of additional safety concerns at dosing periods extending past this time frame as well, though additional long-term studies will be needed to confirm this assertion. Future studies on drug–drug interactions and reproductive toxicology will further enhance knowledge on the safety profile of BT-11.
From previous results and those presented here, BT-11 is a promising oral investigational therapeutic for IBD that has limited ability to be absorbed systemically from the GI tract. BT-11 induces and maintains homeostasis of the gut immune system through activation of immunometabolic effects of the LANCL2 pathway to reduce inflammation and dysregulated immune responses that lead to tissue damage in IBD. No demonstrated safety concerns on common target organ systems or genotoxicity were demonstrated. Furthermore, BT-11 potentially could have a better safety profile than current IBD therapeutics, biologics, corticosteroids, and broad immunosuppressants that increase the risk of infection and cancer due to their broad distribution throughout the body and overall mechanism of action.
Footnotes
Authors’ Note
Studies were conducted at Experimur, MPI Research Inc, and Bioreliance Laboratories.
Author Contributions
Leber, A contributed to conception and design; acquisition, analysis, and interpretation; drafted manuscript; and critically revised manuscript. Hontecillas, R contributed to conception and design, analysis and interpretation, and critically revised manuscript. Zoccoli-Rodriguez, V contributed to acquisition and analysis and critically revised manuscript. Ehrich, M and Davis, J contributed to design, analysis and interpretation, and critically revised manuscript. Chauhan, J. contributed to conception and design, acquisition, and critically revised manuscript. Bassaganya-Riere, J. contributed to conception and design, interpretation, drafted manuscript, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: JBR, RH, AL, VZR, and JC are employees of Landos Biopharma. JBR is also a shareholder of Landos.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.