
Abstract
Vorinostat (SAHA, Zolinza), a histone deacetylase inhibitor, is assessed in nonclinical studies to support its approval for cutaneous T-cell lymphoma. Vorinostat is weakly mutagenic in the Ames assay; is clastogenic in rodent (ie, CHO) cells but not in normal human lymphocytes; and is weakly positive in an in vivo mouse micronucleus assay. No effects are observed on potassium ion currents in the hERG assay up to 300 μM (safety margin ~300-fold the ~1 μM serum concentration associated with the 400 mg/d maximum recommended human dose. No rat respiratory or central nervous system effects are found at 150 mg/kg (>2-fold maximum recommended human dose). No cardiovascular effects, including effects on QTc interval, are observed after a single oral dose (150 mg/kg) in dogs. Vorinostat is orally dosed daily in rats (controls, 20, 50, or 150 mg/kg/d) and dogs (controls, 60, 80, or 100/125/160 mg/kg/d) for 26 weeks with a 4-week recovery. Rat vorinostat-related adverse findings are decreased food consumption, weight loss, and hematologic changes; a no observed adverse effects level is not established. In dogs, adverse effects are primarily gastrointestinal; the no observed adverse effects level is 60 mg/kg/d (~6-fold maximum recommended human dose). Toxicities are reversible and can be monitored in the clinic.
Histone deacetylases (HDACs) are a family of enzymes that catalyze the removal of acetyl groups from nucleosomal histones. Along with the histone acetyltransferases (HATs), a separate family of enzymes that add acetyl groups, HDACs help regulate chromatin structure and gene expression by varying the acetylation of lysine residues at the N-terminal of histones contained within nucleosomes. In addition, nonhistone proteins have been identified as substrates of HDACs. Disruptions of HDAC and HAT functions are associated with cancer; the molecular events underlying the effects of HDACs and their inhibition have recently been reviewed. 1 The classification and localization of the various mammalian (human) HDACs have been described previously. 2 Members of class I HDACs, including HDAC1, HDAC2, and HDAC3, are expressed ubiquitously in human tissues and various cell lines. 3 Drugs have been shown to inhibit HDAC activity. 4,5 The first HDAC inhibitor to be approved for marketing by the US Food and Drug Administration was vorinostat (suberoylanilide hydroxamic acid [SAHA], Zolinza), and it was approved for the treatment of cutaneous manifestations in patients with cutaneous T-cell lymphoma (CTCL) who have progressive, persistent, or recurrent disease on or following 2 systemic therapies 6,7 (http://www.accessdata.fda.gov/drugsatfda_nda/2006/021991s000_Zolinza_PharmR.pdf). Vorinostat inhibits HDAC activity by binding to a zinc ion in the catalytic domain of both class I (HDAC1, HDAC2, HDAC3, and to a lesser extent HDAC8) and class II HDACs (HDAC6) in the nanomolar range (30-779 nM). Vorinostat induced cell death in transformed cells by selectively altering gene expression and the function of proteins associated with caspase-dependent and -independent death. 8 The development of vorinostat, its current applications, and its future perspectives have been recently reviewed. 9 To support its clinical evaluation and approval, vorinostat was assessed in genotoxicity, safety pharmacology, and chronic repeated-dose toxicity studies in rats and dogs, and results are summarized here. Developmental toxicity studies in rats and rabbits, and the female and male fertility studies, have been described elsewhere. 10,11
Methods
Drug and Dose Formulation
Vorinostat used in the preclinical studies was synthesized by Organichem (Rensselaer, NY), and specifications were verified by Merck & Co. The purity of the drug was 99.6%. Dose formulations of vorinostat for the animals dosed by the oral route were prepared as suspensions in a vehicle of 1.0% (wt/vol) carboxymethylcellulose sodium (Sigma-Aldrich, St. Louis, Mo) and 0.5% (vol/vol) polysorbate 80 (Crodia, Mill Hall, Pa) in deionized water at daily dose volumes of 5 mL/kg (safety pharmacology studies, including cardiovascular, respiratory, and functional observational battery studies) or 10 mL/kg (26-week repeated-dose toxicity study) to each rat. The formulations were prepared daily and stirred continuously during use. During the specified dosing intervals, animals were administered vehicle or drug formulation orally once daily. For the dog studies, the animals were dosed using capsules. The capsules contained a blend of vorinostat and the excipients, a formulation similar to that currently approved for human use in the United States. Concentrations, homogeneity, and stability of dosing solutions and stability of the test article used in the capsules were evaluated with validated assays to confirm that targeted dose levels were achieved.
Animal Care and Use and Regulatory Compliance
The studies were conducted at Merck Research Laboratories (West Point, Pa) or at Covance Research Laboratories (Madison, Wis). All studies were carried out under Good Laboratory Practice regulations for preclinical laboratory studies, unless the studies were exploratory. All animal housing and care procedure were in compliance with the Federal Animal Welfare Act and the Institute for Laboratory Animal Resources. All procedures carried out on the animals were reviewed and approved by the Institutional Animal Care and Use Committee. The animal facilities were fully accredited with the Association for Assessment and Accreditation of Laboratory Animal Care International.
Genotoxicity
These studies were done as a comprehensive assessment of genotoxicity, consistent with OECD regulatory guidelines 12-14 and as previously described. 15-17 Specific protocols are described below.
Microbial mutagenesis
Vorinostat was tested in 2 laboratories using a modified pre-incubation Ames test for reversion at the his locus. 18,19 Vorinostat was tested at concentrations from 30 or 100 up to 5000 μg per plate in triplicate with and without a liver activation system (S-9) prepared from Arochlor 1254 or phenobarbital/β-naphthoflavone-treated male Sprague-Dawley rats. Pre-incubation was at 30°C (laboratory 1) or 37°C (laboratory 2) for 30 minutes. Both laboratories tested Salmonella strains TA98, TA100, and TA15135 and Escherichia coli WP2 uvrA. One laboratory also tested Salmonella TA1537 and TA1538, whereas the other used TA97a.
5-Fluorouracil forward mutation assay in Salmonella
This assay is based on induced 5-fluorouracil (FU) resistance in the Salmonella TA100 derived strain FU100. Assay protocol, mutational target, and spectra have been described previously. 20,21 Briefly, exponentially growing FU100 were treated with controls or vorinostat (up to 5000 μg/mL) with and without S-9 activation for 2 hours at 37°C with shaking. Cultures were then diluted 10-fold and cytotoxicity was assessed (by optical density) after 3-hour and 21-hour recovery incubation at 37°C. Cultures plated in 384-well plates were then incubated for 3 days in medium containing FU and the pH indicator bromocresol purple. “Positive wells” were yellow because of growth of FU-resistant colonies and were enumerated using a SpectraMax spectrophotometer. The assay is positive when there is an increase in mutant frequency that is 4 standard deviations or more above the historical control mean in at least 2 cultures, without excessive toxicity (≥5% survival).
Chromosome aberrations in human lymphocytes in vitro
Ficoll-Hypaque purified human blood lymphocytes were cultured for 2 days prior to treatment in RPMI medium containing 20% fetal bovine serum, L-glutamine, antibiotics, and 2% phytohemaglutinin M (PHA-M). Duplicate cultures were treated with controls or vorinostat dissolved in DMSO (final concentration 1%) with or without S-9 activation for 3 hours and harvested 22 hours from the beginning of treatment. Incubation with S-9 was in serum-free medium containing cofactors and S-9 from Arochlor 1254 induced male Sprague-Dawley rats. An additional set of cultures was treated for 22 hours without S-9. Colcemid was added 2 hours before cultures were fixed, and stained slides were scored under code numbers such that the reader did not know which slides were from controls and which from test substance–treated cultures. The high doses selected for aberration scoring were based on mitotic suppression. Two hundred metaphase cells containing 44-48 chromosomes were typically scored for aberrations, usually 100 cells from each duplicate culture. The Fisher exact test was used for statistical analysis.
Chromosome aberrations in Chinese hamster ovary (CHO) cells in vitro
Cultures were incubated with controls or vorinostat dissolved in DMSO (final concentration 1%) with or without S-9 activation for 3 hours and harvested 20 hours from the beginning of treatment. Incubation with S-9 was carried out in serum-free medium containing cofactors and S-9 from phenobarbital/β-naphthoflavone-induced male Sprague-Dawley rats. An additional set of cultures was incubated continuously for 20 hours without S-9. Colcemid was added 1 to 3 hours before cells were fixed, and stained slides were scored under code. The top doses selected for scoring were based on suppression of cell growth. For aberration analysis, 200 metaphase cells containing 19 to 23 chromosomes were scored for each treatment unless aberration frequencies were high. Noted, but not included in aberration totals, were the following: gaps (achromatic regions less than or equal to the width of a chromatid, or larger lesions with visible connecting material across the gap), polyploid and endoreduplicated cells, and pulverized and double-minute chromosomes. Cells with 10 or more aberrations were classified as severely damaged cells and scored as 1 aberrant cell but as 10 aberrations when calculating total frequency. Statistical analysis of the response included pairwise comparisons with a Dunnett adjustment for multiple comparisons. In a second test, parallel sets of cultures, designated for flow cytometric analysis to assess DNA synthesis inhibition (below), received 0.1 μM 5-bromodeoxyuridine (BrdUrd; Sigma-Aldrich, St. Louis, Mo) for the last 30 minutes of the 3-hour incubation with vorinostat without S-9. Vorinostat was also tested for aberrations in a second laboratory that used treatments for 5 hours with or without S-9 and harvested 23 hours from the beginning of treatment.
Flow cytometry for detecting DNA synthesis inhibition
Assessment of DNA synthesis was done as previously described. 17 Immediately after treatment and BrdUrd labeling, cell pellets were fixed and stained for BrdUrd detection using an anti-BrdUrd monoclonal antibody and FITC-labeled secondary antibody to mouse anti-BrdUrd and total DNA using propidium iodide (PI). Analysis was performed using an EPICS Elite ESP flow cytometer (Coulter Electronics, Hialeah, Fla) equipped with an Innova 90-6 argon-ion laser (Coherent, Palo Alto, Calif) at 488 nm to excite the 2 fluorochromes (FITC and PI). Information from 104 cells was collected for each sample. Inhibition of DNA synthesis was measured as suppression of BrdUrd uptake. The relative quantity of BrdUrd incorporated per S-phase cell was calculated from the difference in the mean fluorescent intensity (channel number) between the BrdUrd-positive cell population (S-phase cells) and the BrdUrd-negative population (G1, G2, and M cells).
Mouse micronucleus test
Six-week-old male Crl:CD1(ICR) mice were dosed once, orally, with vehicle or vorinostat at 500, 1000, and 2000 mg/kg (1500, 3000, and 6000 mg/m2, respectively). The high dose is a limit dose for the micronucleus test, 14 and only males were tested because there was no evidence for a sex difference in toxicity in a range-finding study. Vorinostat was suspended in 1% (wt/vol) carboxymethylcellulose sodium/0.5% (vol/vol) Tween 80 in deionized water. The dosing volume was 20 mL/kg. Bone marrow cells, enriched for erythrocytes as previously described, 22 were collected from groups of 5 to 7 mice at 24 and 48 hours after dosing. Two thousand polychromatic erythrocytes (PCEs) were scored under code for micronuclei (MN) from each of 5 mice per dose group. The frequencies of PCE and mature, normochromatic erythrocytes (NCEs) were also recorded among 1000 erythrocytes per mouse. Statistical analysis included pairwise comparisons with an adjustment for multiplicity of testing and a trend test.
Safety Pharmacology
Evaluation of hERG current using patch clamp analysis
The whole-cell patch clamp recording technique was used to study potassium currents in CHO-K1 cells heterologously expressing the hERG potassium channel, following published procedures for data acquisition and analysis, 23,24 with the following modifications: cells were dissociated with 0.05% trypsin plus 0.53 mM EDTA (GIBCO-BRL) or 0.48 mM EDTA 4 mM Na (Versene, GIBCO-BRL Life Technologies); data acquisition and analysis were performed using pCLAMP 8 or eCLAMP software (Axon Instruments, Molecular Devices, Sunnyvale, Calif). In vitro formulations of vorinostat, and 2 other HDAC inhibitors, LAQ824 and LBH589, were prepared from DMSO stock solutions diluted in HEPES buffered solution (132 mM NaCl, 4 mM KCl, 1.2 mM MgC12, 10 mM HEPES, 11.1 mM glucose, pH 7.35, 0.1% DMSO final). LAQ824 and LBH589 were synthesized by Merck Research Laboratories (Boston, Mass).
Evaluation of cardiovascular, functional observational battery and respiratory effects
The cardiovascular study was conducted in conscious dogs previously implanted with Konigsberg solid-state pressure transducers with a biopotential lead for the measurement of arterial blood pressure and electrocardiogram (ECG), as previously described. 25,26 Doses selected were the same as the doses for the 26-week repeated-dose toxicity study in dogs. Data were transmitted via radiotelemetry and recorded by CA Recorder Systems (D.I.S.S. LLC, Pinckney, Mich). Arterial blood pressure, heart rate, PR interval, QRS interval, and QT interval were recorded and/or calculated from the acquired data. QT interval was also reported as the rate-corrected QT interval using Fridericia’s rate correction formula. 27 Data were collected for 24 hours or more prior to dosing and 24 hours or more after dosing for vehicle. Data were collected for 2 hours or more prior to dosing and 24 hours or more after dosing for 20, 50, and 150 mg/kg (400, 1000, and 3000 mg/m2, respectively). Maximum serum concentration (Cmax) was approximately 2 hours post dose. Data were collected as 15-minute mean values and reported as mean 15-minute values ± standard error of the mean.
Rats were tested in the oral functional observational battery study, as part of the safety pharmacology core battery, 28,29 approximately 30 minutes post dose by observers blinded to the dose condition of each animal. Vorinostat was administered as an oral suspension at doses of 20, 50, and 150 mg/kg (120, 300, 900 mg/m2), doses the same as those used in the rat 26-week repeated-dose toxicity study. During this time, a white noise generator at approximately 70 dB was activated and remained on for the duration of the study. The following observations/tests were performed: home cage observations, hand-held observations (such as lacrimation, salivation, piloerection), open-field observations (such as posture, gait, locomotion, number of rears, physical signs), and stimulus activity responses (pupil response, pinna response, response to noise, approach, touch, and tail pinch) and measurements (forelimb and hindlimb grip strengths, body temperature, foot splay, body weight).
Vorinostat was administered as a single dose to male Sprague-Dawley rats by oral gavage to determine its effects on the respiratory system, using whole-body plethysmography. Respiratory rate, tidal volume, minute ventilation, and PenH (an index of airway resistance) were measured following administration of 20, 50, or 150 mg/kg vorinostat or vehicle (Buxco BioSystem XA Data Acquisition System, Wilmington, NC).
General Toxicology
The toxicity and toxicokinetics (TK) of vorinostat in rats and dogs were examined in chronic 26-week repeated-dose toxicity studies with 4-week recovery periods. For the rat studies there were necropsies after 13 and 26 weeks of dosing and after 4 weeks of recovery. Thirty or 40 Sprague-Dawley rats (Charles River Laboratories, Portage, Mich) per sex per group were used in the chronic rat study, with the animals at 8 weeks of age at study start. The beagle dogs ranged in age from 7 to 12 months at study start for the repeated-dose toxicity study (Covance Laboratories, Madison, Wis), and group sizes were 3 to 6 per sex per group. The doses for the rat repeated-dose toxicity study were 20, 50, or 150 mg/kg/d (120, 300, or 900 mg/m2/d, respectively), based on the 4-week repeated-dose toxicity study with the NOAEL of 20 mg/kg/d. For the 26-week repeated-dose toxicity study in beagle dogs, doses were 20 or 60 mg/kg (400 or 1200 mg/m2/d, respectively) in the low- and mid-dose groups, with an escalating high dose of 80/100/125/160 mg/kg (3200 mg/m2/d) on days 16, 30, and 97, respectively. Evaluations conducted in both species included clinical signs, food consumption, body weight, serum chemistry, hematology, plasma coagulation, urinalysis, flow cytometric immunophenotyping of rat peripheral blood, gross pathology, ophthalmologic examinations, organ weights, and microscopic histopathology. In addition, serum samples were collected for determination of vorinostat and its 2 major metabolite levels (O-glucuronide of vorinostat and 4-anilino-4-oxobutanoic acid) in rats and dogs; electrocardiographic evaluations were conducted only in dogs.
Clinical Observations
Mortality observations were recorded twice daily and clinical observations of physical appearance and behavior were recorded daily during predose, dosing, and the dose-free recovery period. Body weight changes were recorded weekly for rats and twice weekly for dogs in the dosing phase, and these measurements were recorded weekly in the recovery period. Food consumption (estimated) was recorded weekly for the rats and measured and recorded daily for the dogs during the predose, dosing, and dose-free recovery phases of the studies.
Electrocardiography
Electrocardiogram tracings and heart rates were recorded on all dogs on 4 occasions: prior to initiation of treatment, during week 13, at the conclusion of the dosing period, and during the last week of the recovery period. Data were recorded twice per day (before dosing and approximately 2 hours post dose) and once per day during pretest and recovery periods. All animals were acclimated to the procedures and equipment in training sessions conducted twice in the week prior to each scheduled recording. The ECG determinations included heart rate and PR, QRS, QT, QTc, and RR intervals measured using 10 leads. In addition, a qualitative assessment of the trace for rhythm and other abnormalities was performed. QTc was calculated using Van de Water’s equation. 30
Ophthalmology
Ophthalmic examinations were performed on all rats and dogs once prior to initiation of the dosing, during the final week of dosing, and during the final week of the recovery period. An additional examination was performed at week 13. The pupils were dilated with a mydriatic agent and the eyes were examined with an indirect ophthalmoscope.
Clinical Pathology
Blood samples from a jugular vein of dogs or rats were collected twice for clinical pathology analyses during the predose period; weeks 4, 13, and 25 during the dosing period; and week 4 of the postdose recovery period. Standard hematologic, coagulation, and clinical chemistry parameters were measured. The anticoagulants for the hematology and coagulation samples were potassium EDTA and sodium citrate, respectively. Urine was collected for urinalysis and urine chemistry tests once prior to initiation of treatment and in weeks 4 and 13, at the end of dosing, and once during the recovery period. Prior to each scheduled sampling for clinical pathology, animals were fasted for at least 6 hours. Urine specimens were collected for at least 6 hours on wet ice before blood collection.
Creatine kinase (total and isoenzymes) was measured in blood samples collected from the jugular vein prior to dosing, during weeks 13 and 25, and once during week 4 of the recovery period. Samples were collected without an anticoagulant, stored at room temperature, and allowed to clot. Samples were centrifuged and the serum was harvested, stored in a freezer at –60°C to –80°C, and analyzed by LabCorp-Preclinical (San Diego, Calif).
Peripheral Blood Immunophenotyping
In the 26-week rat study, blood samples were collected from all interim sacrifice animals on day 28 and at the interim sacrifice and from animals at the end of the recovery period. FAST Systems (Rockville, Md) analyzed whole-blood specimens for lymphocyte phenotypes by flow cytometry using a Beckman-Coulter EOICS XL MCL flow cytometer. The absolute cell counts were determined as was the percentage of total lymphocytes expressing a particular phenotype.
Histopathology
Animals were fasted overnight, anesthetized with sodium pentobarbital, and euthanized by exsanguination. Terminal body weights were recorded prior to exsanguination. The necropsy performed on each animal included examination of the external features; the abdominal, thoracic, and cranial cavities; organs; and tissues. Tissues were preserved in 10% neutral-buffered formalin, embedded in paraffin, sectioned, stained with hematoxylin and eosin, and examined by light microscopy. Tissue slides from each animal in the control and high-dose groups and each animal that died or was sacrificed at an unscheduled interval were examined microscopically.
Statistics
One-way analysis of variance (ANOVA) was used in the repeated-dose toxicity studies. If the ANOVA was significant (P < .05), then the Dunnett t test was used for controls versus treated group comparisons for body weight, body weight change, food consumption, clinical pathology, and organ weight change.
Toxicokinetics (TK)
The TK of vorinostat was determined on day 7 and during the last week of dosing in the oral 26-week repeated-dose rat study. Samples were collected from the first set of 3 animals per sex per group predose at approximately 1 and 12 hours post dose. Samples were collected from a second set of 3 animals per sex per group approximately 15 minutes and 1.5 and 18 hours post dose. Samples were collected from a third set of animals 30 minutes and 3 and 24 hours post dose. Samples were collected from a fourth set of animals 45 minutes and 6 hours post dose. Additional time points were collected on week 13 prior to dosing and 0.5 hours after dosing. Animals were not fasted and the blood was collected from a jugular vein. Blood samples were held at room temperature for up to 1 hour and allowed to clot. Serum samples were analyzed for vorinostat and its 2 major inactive metabolites O-glucuronide of vorinostat and 4-anilino-4-oxobutanoic acid by MDS Pharma Services (Blainville, Quebec, Canada) using a validated LC/MS/MS assay. The analytical range was 5.00 to 5000 ng/mL. Toxicokinetic parameters were analyzed using WinNonLin 4.0 (MDS Pharma Services, Montreal, Quebec, Canada).
In the 26-week repeated-dose toxicity study in dogs, blood was collected on days 7 and 56 and on 1 day during weeks 17 and 25. All animals were bled at the following time points: 0 (prior to dosing), approximately 30 minutes post dose, and 1, 1.5, 2, 3, 4, 8, 12, and 24 hours post dose. The blood samples were handled and analyzed as described above.
Results
Weak Mutagenicity Was Detected in the Ames Assay But Not in the 5-FU Test
The Ames assays gave weakly positive results in TA100 and TA97a (Table 1). Trends toward positive results were also seen in other strains. The results were considered borderline, because in addition to being weak and at high concentrations (3333 and 5000 μg/plate), they were not always reproducible within a laboratory (eg, TA1535 had a positive point in 1 experiment that was not reproducible in 2 subsequent experiments) or across laboratories (different strains met the positive criteria in the 2 laboratories). The increases were not dependent upon S-9 metabolic activation, indicating that metabolism (eg, by P450s) is not required for mutagenicity. In the 5-FU forward mutation test, vorinostat was negative at up to 5000 μg/mL, where survival at the top concentrations was 41% and 31%, with and without S-9, respectively (data not shown).
Mutagenicity Results for Vorinostat in the Pre-Incubation Ames Test
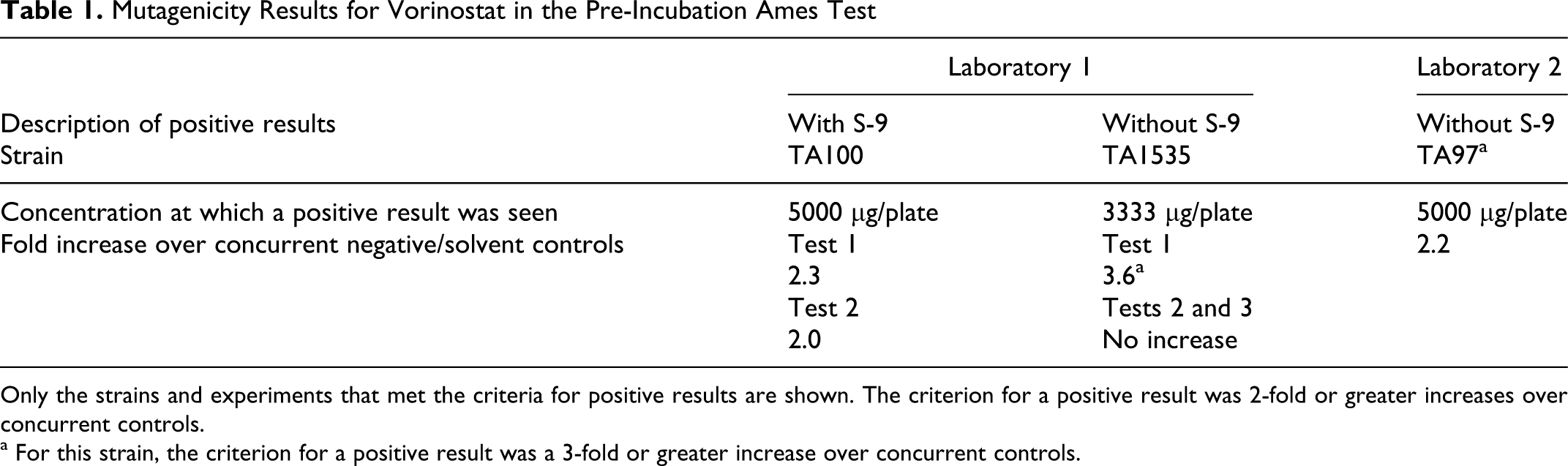
Only the strains and experiments that met the criteria for positive results are shown. The criterion for a positive result was 2-fold or greater increases over concurrent controls.
a For this strain, the criterion for a positive result was a 3-fold or greater increase over concurrent controls.
Chromosome Aberrations Were Induced in CHO Cells But Not in Human Lymphocytes
Vorinostat did not induce aberrations in purified lymphocytes up to concentrations limited by cytotoxicity assessed as mitotic suppression (data not shown). At the top scored doses, 3.8 mM after 3-hour treatments with and without S-9 and 0.15 mM after the 22-hour treatment, mitotic indices were reduced by 51%, 53%, and 56% compared with solvent controls, respectively.
In contrast, vorinostat induced a marked increase in aberrations at 20 hours in CHO cells treated for 3 hours (Figure 1). With S-9, there were up to 22% aberrant cells (at 700 μM) compared with 1% in concurrent solvent controls. Without S-9 there were 18% aberrant cells at 350 μM and 24% at 600 μM. The increases were associated with moderate to marked cytotoxicity (cell growth reduced to about 72% to 46% of controls) and included both chromosome breaks and exchanges. At higher concentrations, severely damaged cells and cells with multiple aberrations were noted. These results were replicated in an independent experiment, and after 20 hours of treatment, no increases in aberrations were seen up to 150 μM, where cell growth was 51% of controls. The increases in aberrations were also seen in a third assay in CHO cells in a different laboratory, where increases to about 28% cells with aberrations (compared with 5% in controls) were seen at about 380 μM after 5-hour treatments with and without S-9. To investigate whether the aberrations resulted from an indirect mechanism, we examined evidence for suppression of DNA synthesis.
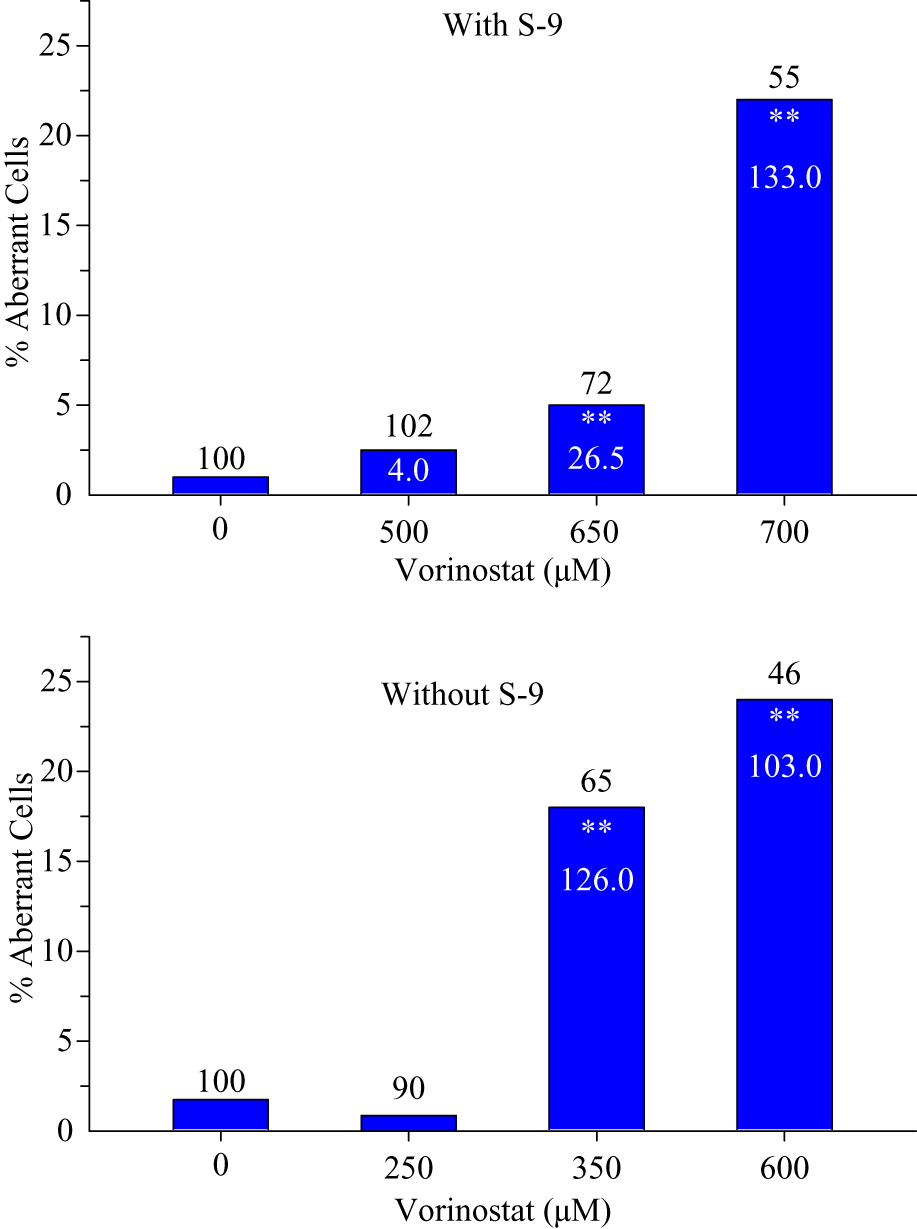
Percentages of cells with chromosomal aberrations in CHO cultures treated for 3 hours with vorinostat and harvested at 20 hours from the beginning of treatment: Numbers on tops of columns are population doublings (cytotoxicity) taken at harvest, as percentages of controls. Numbers inside columns are frequency of aberrations per 100 cells, as a cell may have more than one aberration. **Statistically significant increase over concurrent control (P ≤ .01).
Marked Suppression of DNA Synthesis Was Associated With Aberration Induction in CHO Cells
BrdUrd uptake was markedly suppressed during the 3-hour treatment without S9, to 17% and 9% of controls at 400 and 500 μM, respectively, effects similar to the positive control DNA polymerase inhibitor, aphidicolin (Figure 2). Marked aberration induction and cytotoxicity were seen in a concurrent assay.
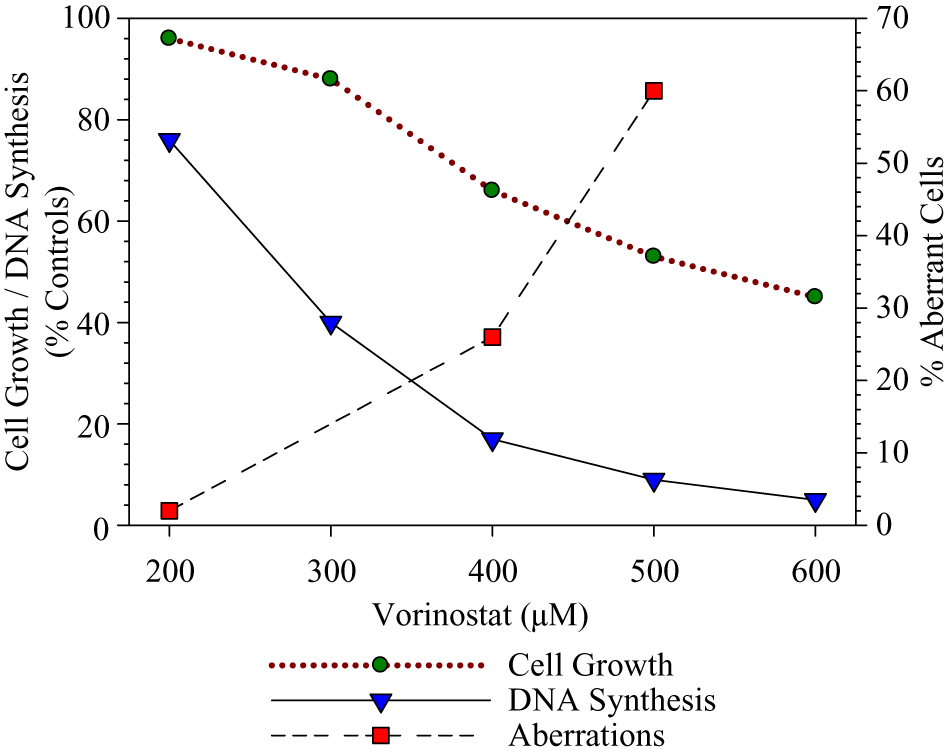
DNA synthesis inhibition associated with aberration induction and cytotoxicity in CHO cells treated for 3 hours with vorinostat without S9 activation. Data shown for DNA synthesis are means from 2 determinations per point (10 000 cells analyzed per determination).
The In Vivo Micronucleus Test Gave Equivocal Results With Vorinostat in Mouse Bone Marrow
There were small but statistically significant increases in micronucleated PCEs compared with the vehicle controls at all 3 dose levels (500, 1000, and 2000 mg/kg) at the 24-hour harvest and at 1000 mg/kg and 2000 mg/kg at the 48-hour time point (Table 2). The highest levels in treated groups were 2.6 and 2.7 MN per 1000 PCEs compared with the concurrent control mean of 1.0. This result is considered equivocal because the levels of MN-PCE were within our historical vehicle control range for CD-1 mice (mean about 1.5 MN-PCE/1000 PCE, typical range [99th percentiles] 0.3-2.8). There was a slight suppression of the number of PCE as a proportion of total erythrocytes in the 48-hour 2000 mg/kg–treated group, indicating possible bone marrow toxicity.
Vorinostat Results in the Micronucleus Assay in Mouse Bone Marrow Polychromatic Erythrocytes
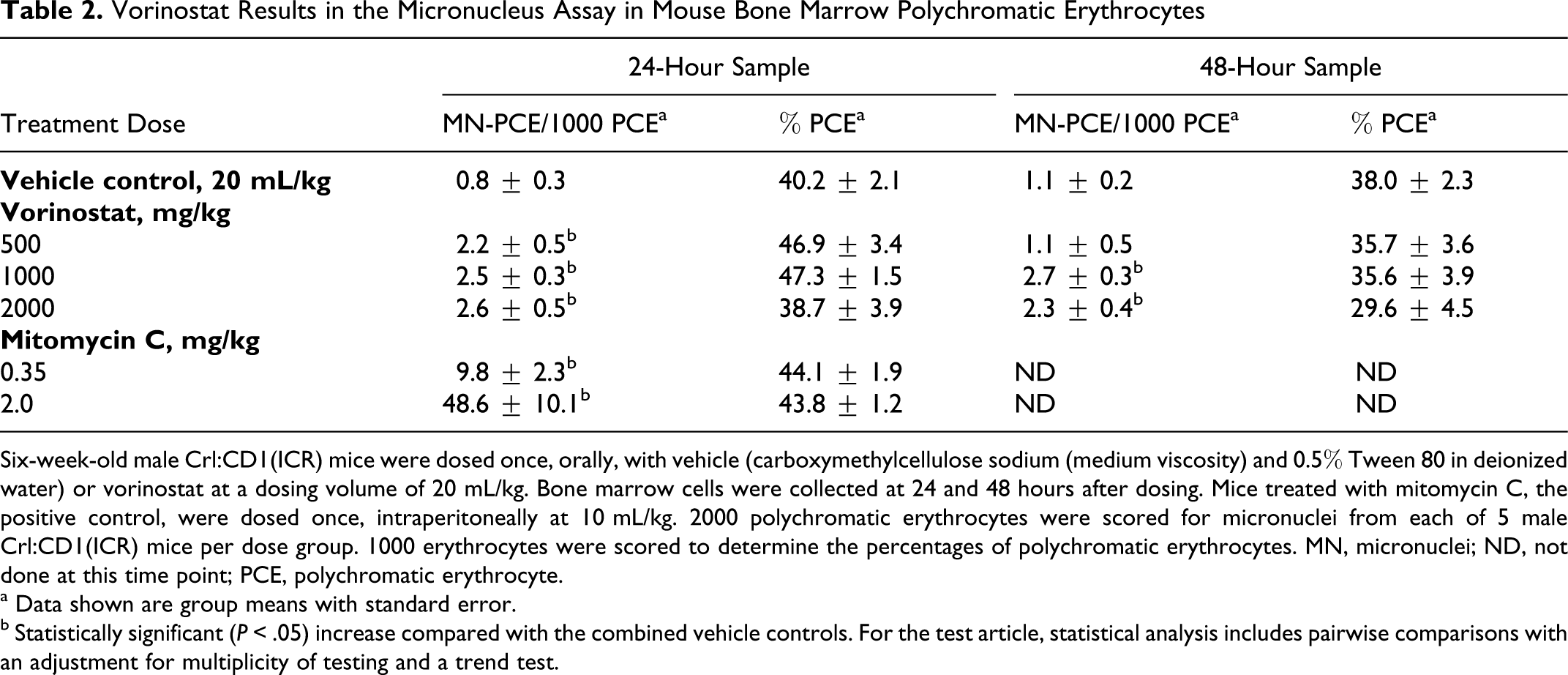
Six-week-old male Crl:CD1(ICR) mice were dosed once, orally, with vehicle (carboxymethylcellulose sodium (medium viscosity) and 0.5% Tween 80 in deionized water) or vorinostat at a dosing volume of 20 mL/kg. Bone marrow cells were collected at 24 and 48 hours after dosing. Mice treated with mitomycin C, the positive control, were dosed once, intraperitoneally at 10 mL/kg. 2000 polychromatic erythrocytes were scored for micronuclei from each of 5 male Crl:CD1(ICR) mice per dose group. 1000 erythrocytes were scored to determine the percentages of polychromatic erythrocytes. MN, micronuclei; ND, not done at this time point; PCE, polychromatic erythrocyte.
a Data shown are group means with standard error.
b Statistically significant (P < .05) increase compared with the combined vehicle controls. For the test article, statistical analysis includes pairwise comparisons with an adjustment for multiplicity of testing and a trend test.
Safety Pharmacology Studies Provided Margins Against Adverse Cardiovascular, Respiratory, and Neurological Effects
Vorinostat was tested for effects on potassium currents mediated by hERG channels heterologously expressed in CHO-K1 cells using standard whole-cell voltage clamp techniques. Vorinostat had little or no effect on potassium current at concentrations of 300 μM. Vorinostat produced a decrease of 8% in potassium current at a maximal testable concentration of 300 μM, with an estimated IC50 value greater than 300 μM. The small effect at 300 μM, indistinguishable from the effect of vehicle alone, represents a safety margin ~300-fold compared with the peak serum concentration (approximately 1 μM) associated with a maximum recommended human dose (MRHD) of 400 mg/d. In contrast, 2 other HDAC inhibitors, LAQ824 and LBH589, inhibited hERG with IC50 values of 2.5 and 3.1 μM, respectively (Figure 3).
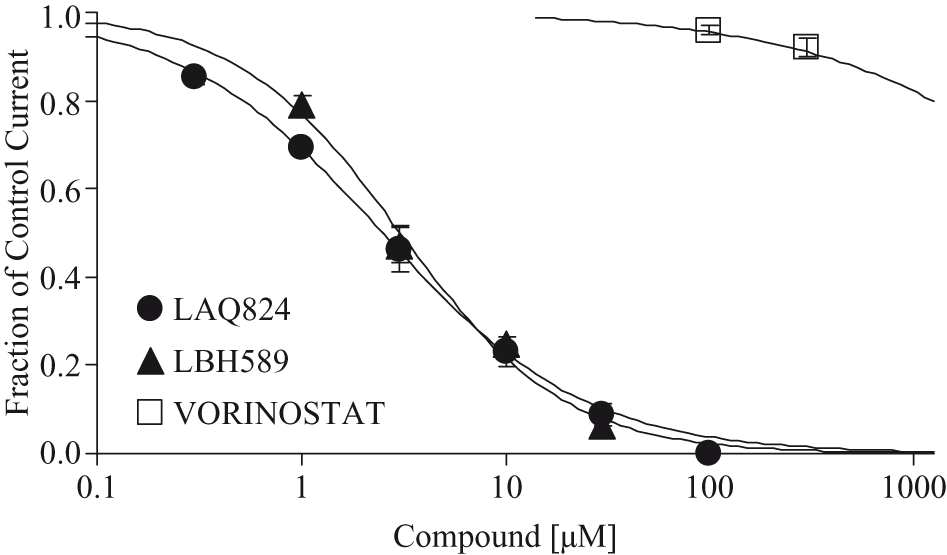
Inhibitory effects of hydroxamates LAQ824, LBH589, and vorinostat on hERG channel potassium currents. Concentration dependence of hERG channel inhibition by LAQ824 (filled circles), LBH589 (filled triangles), and vorinostat (open squares) is quantified as the peak tail current amplitude measured at steady state for each concentration of test agent and normalized to the control tail current amplitude for each cell. LAQ824, LBH589, and vorinostat inhibited the hERG channel with IC50 values of 2.5, 3.1 and > 3 µM, respectively. Plotted data are the mean ± SEM (n ≥ 3, ≥ 3, and ≥ 8, respectively).
In vivo safety pharmacology studies included respiratory (whole-body plethysmography) and functional observation battery (FOB) studies in rats and an oral cardiovascular study in telemetered dogs. No treatment-related effects of vorinostat in either study conducted on rats were observed up to the highest tested dose of 150 mg/kg/d (900 mg/m2).
In telemetered dogs, no cardiovascular effects, including QT/QTc interval prolongation, were observed following oral administration of vorinostat at doses up to 150 mg/kg (3000 mg/m2), with an exposure of 0.963 ± 0.349 μM 2 hours post dose. This represents a multiple of approximately 1 compared with the peak human concentration.
Additional respiratory and cardiovascular assessments were incorporated into the oral 26-week toxicology studies of vorinostat in rats and dogs, with no observed treatment-related effects.
Findings in Repeated-Dose Toxicity Study in Rat Were Primarily Weight Loss, Decreased Food Consumption, and Hematologic Effects
The most notable treatment-related changes after 26 weeks of dosing included reduced body weight and food consumption in both females and males following administration of vorinostat at doses of more than 50 mg/kg/d (Figures 4 and 5). A dose-dependent decrease in body weight gain was observed for males and females given 50 or 150 mg/kg/d. Mean body weight gains from weeks 1 to 14 and from weeks 1 to 27 were significantly less than controls for both sexes at either 50 or 150 mg/kg/d. From weeks 1 to 27, the mean body weight gain for males was 96%, 87%, or 64% for 20, 50, or 150 mg/kg/d, respectively, of the mean body weight gained by control males. From weeks 1 to 27, the mean body weight gain for females was 102%, 83%, or 70% at doses of 20, 50, or 150 mg/kg/d, respectively, compared with the mean body weight gained by control females. Males still weighed significantly less than control males (76%) at the end of the recovery period. Females were not significantly different than control females after 2 weeks of recovery; at the end of the recovery period, their mean body weight was 91% of controls.
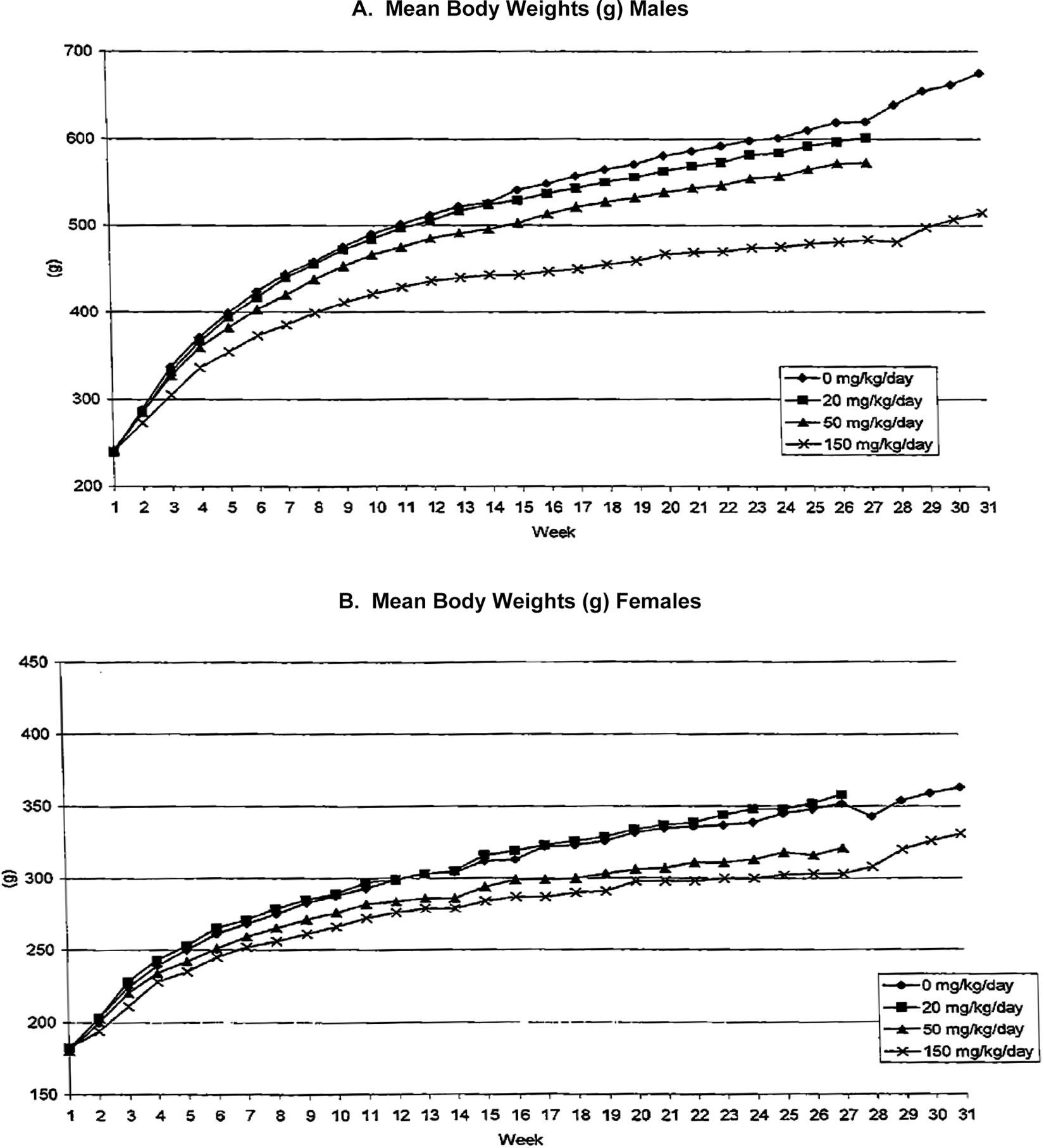
Mean body weight for male (A) and female (B) rats.
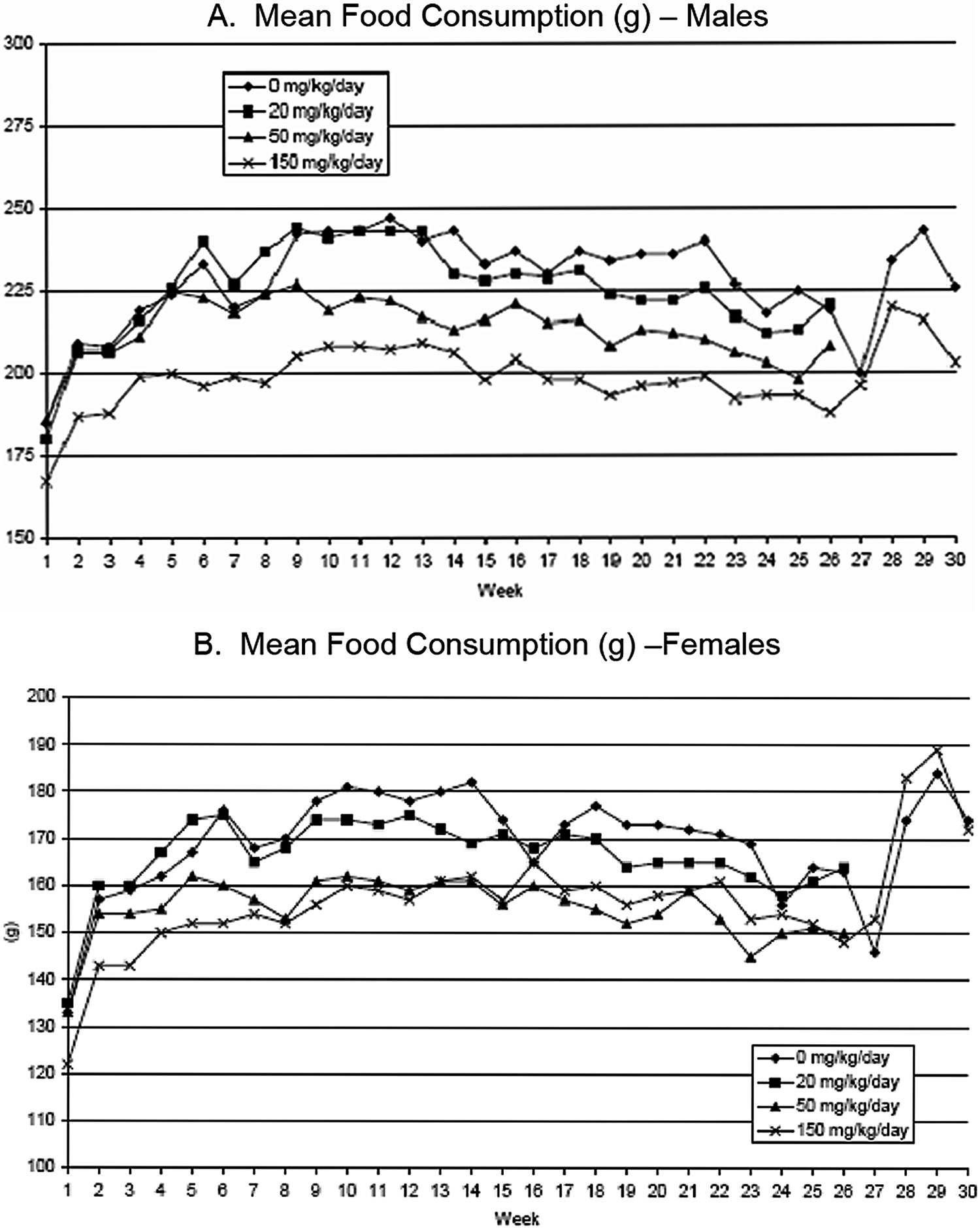
Mean food consumption for male (A) and female (B) rats.
Mean food consumption (Figure 5A) was reduced in males at 50 and 150 mg/kg/d throughout the treatment period, compared with control males. At 20 mg/kg/d in males, there was decreased mean food consumption in 1 or more weeks during the treatment period. In females at 50 and 150 mg/kg/d, mean food consumption was reduced for most weekly intervals during the treatment period. In recovery, male mean food consumption was less than control males in the last 2 weeks of treatment, but not from the initial first 2 weeks of treatment. Females were not different compared with control females during the recovery period (Figure 5B).
The most prominent adverse effects of toxicologic importance were the effects of vorinostat on white blood cell (WBC) counts, globulin, and absolute reticulocyte counts. Lower WBC counts (primarily lymphocytes, including all B- and T-cell subtypes from immunophenotyping) and higher absolute reticulocyte counts (apparently in response to extravascular hemolysis) were observed at all doses in at least 1 sex at more than 1 time interval. (Effects of vorinostat have been observed on the lymphoid system in previous studies. 31 ) Animals administered vorinostat at doses of 150 mg/kg/d (3.6 times a clinical dose of 400 mg/d) were more affected (Table 3). At 150 mg/kg/d the increased absolute reticulocyte count (+154% and +132%, males and females, respectively) is consistent with decreased red blood cell count (–12% and –10% males and females, respectively), hemoglobin (–10% and –9% males and females, respectively), and hematocrit (–5%). Platelet counts for the males were decreased throughout the study (–19% at week 4 and –26% at week 14) (data not shown) and mildly lower at 27 weeks (–20%). In females, the lower platelet count was only observed at the interim clinical pathology interval (–20% at week 14) and not at 27 weeks.
Vorinostat Effects, by Daily Dose, on Hematology Parameters After 26 Weeks of Dosing in Rats
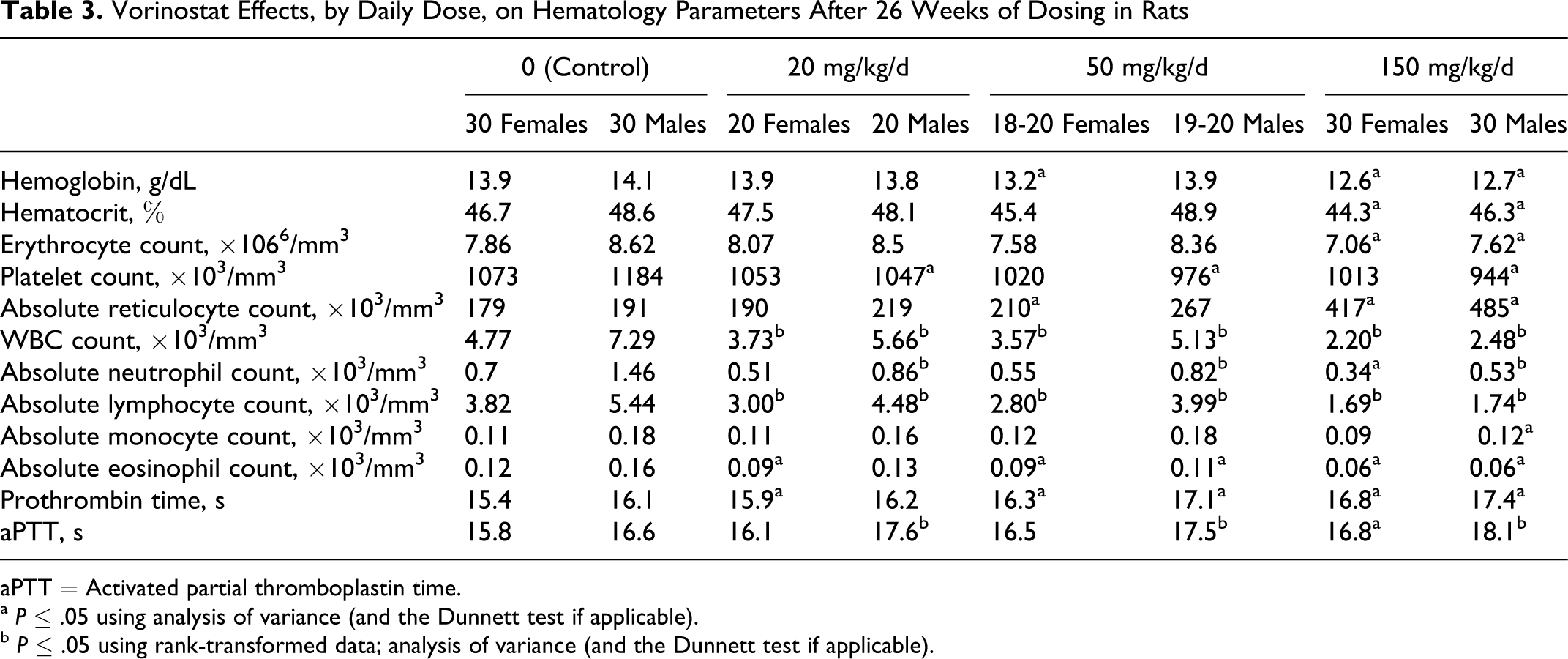
aPTT = Activated partial thromboplastin time.
a P ≤ .05 using analysis of variance (and the Dunnett test if applicable).
b P ≤ .05 using rank-transformed data; analysis of variance (and the Dunnett test if applicable).
The effects on circulating lymphocyte numbers (Table 3) were consistent with lymphoid depletion in the spleen and thymus (see below). The effect on absolute reticulocyte counts was consistent with a regenerative response to loss of red blood cells (red blood cell count, hemoglobin, and hematocrit), which were lower at the 150 mg/kg/d dose and the observation of splenic pigment, suggesting that the loss was likely due to extravascular hemolysis (see below).
With the exceptions of globulin and total protein levels, there were no dose-dependent, treatment-related findings in the serum biochemistry or urine parameters measured (Table 4). Although other parameters were statistically significantly different than controls and are delineated in the tables, these values were within historical control ranges, did not exhibit a dose-dependent response, and/or were not considered to be toxicologically significant. There were no treatment-related differences observed for total creatine kinase or subtypes, including creatine kinase isoenzyme MM (data not shown).
Vorinostat Effects, by Daily Dose, on Serum Biochemistry Parameters After 26 Weeks of Dosing in Rats
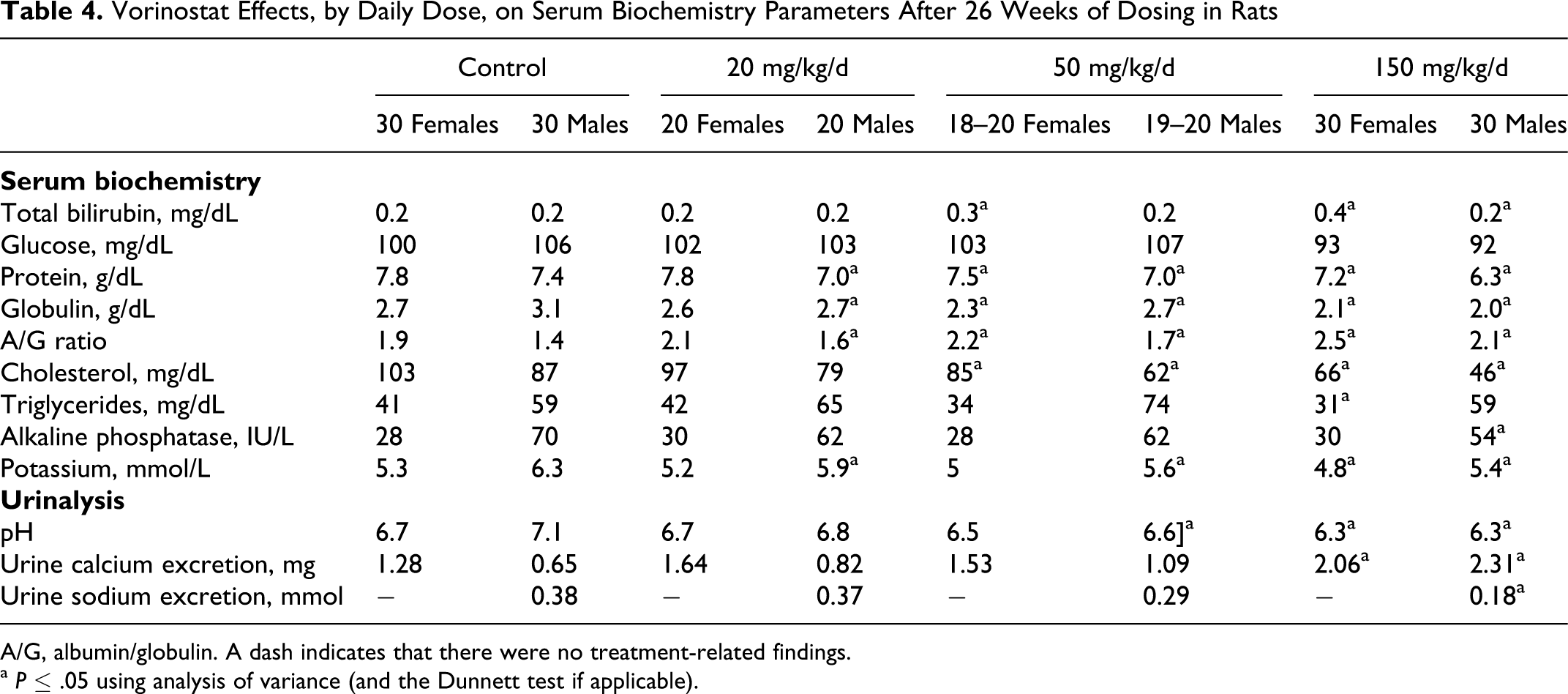
A/G, albumin/globulin. A dash indicates that there were no treatment-related findings.
a P ≤ .05 using analysis of variance (and the Dunnett test if applicable).
At the end of the 4-week recovery period, all the serum clinical chemistry parameters for the rats dosed at 150 mg/kg/d were reversible or trended toward reversibility (Table 5). Of greatest toxicologic importance, the absolute lymphocyte count and globulin level (reduced by 25% and 16% respectively, compared with controls) were improved compared with the differences at week 27 (reduced 68% and 35%, respectively). Absolute lymphocyte counts, helper T-lymphocytes, suppressor/cytotoxic T cells, and B cells trended toward reversibility in males and were completely reversible in females. Other parameters that did not completely reverse by the end of the recovery period in males only included lower platelet count, white blood cell count, total protein, globulin, and albumin to globulin ratio.
Vorinostat Effects Remaining in Male Rats After a 4-Week Recovery Period

A/G, albumin/globulin; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; WBC, white blood cell.
Data presented as percentage changes from concurrent controls at that time (except number evaluated and body weight).
Only change in female rats (n = 10 per group): alkaline phosphate (IU/L) was +29 in controls and +38 at 150 mg/kg/d.
a For all values in this column, P ≤ .05, 2-sample t test.
b Four-week recovery with controls and 150 mg/kg/d treated group only.
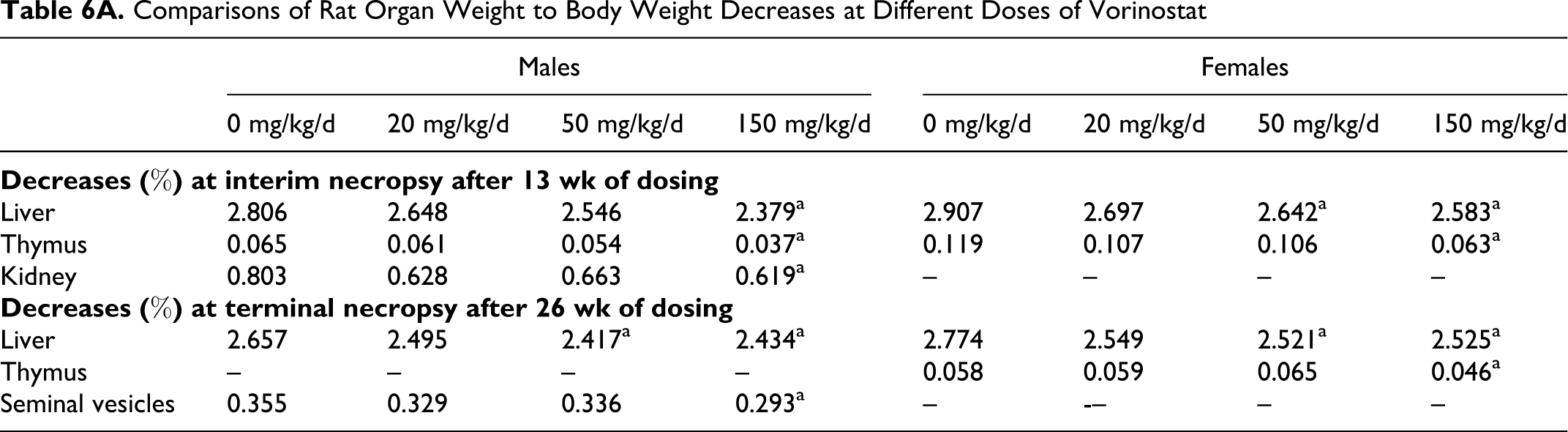
Pathology analyses demonstrated treatment-related decreased thymus weight, splenic and thymic lymphoid depletion, and histopathologic observations of bone marrow erythroid hyperplasia/myeloid hypoplasia. At the interim sacrifice after 13 weeks of dosing, treatment-related decreases in mean absolute and relative liver, kidney, and thymus weights, relative to body weights, were observed (Table 6). Only the animals dosed at 150 mg/kg/d showed reduced thymus weights that correlated with the microscopic observation of lymphoid depletion. The other tissues decreased in weight did not show any histomorphologic correlations. Animals dosed at 50 or 150 mg/kg/d had femur and sternum bone marrow erythroid hyperplasia/myeloid hypoplasia. The low-dose animals had minimal effects. At the terminal sacrifice, mean absolute and relative thymus, liver, and seminal vesicle weights, relative to body weights, were decreased (Table 6). Vorinostat treatment did demonstrate organ weight changes in specific organs at both the interim and terminal necropsies, as shown in Table 6B, with organ to brain ratios significantly decreased compared with concurrent controls. Again, only the decreased thymus weights corresponded with histomorphologic changes.
Comparisons of Rat Organ Weight to Body Weight Decreases at Different Doses of Vorinostat
B. Rat Organ to Brain Weight Ratio With 150 mg/kg/d Vorinostat
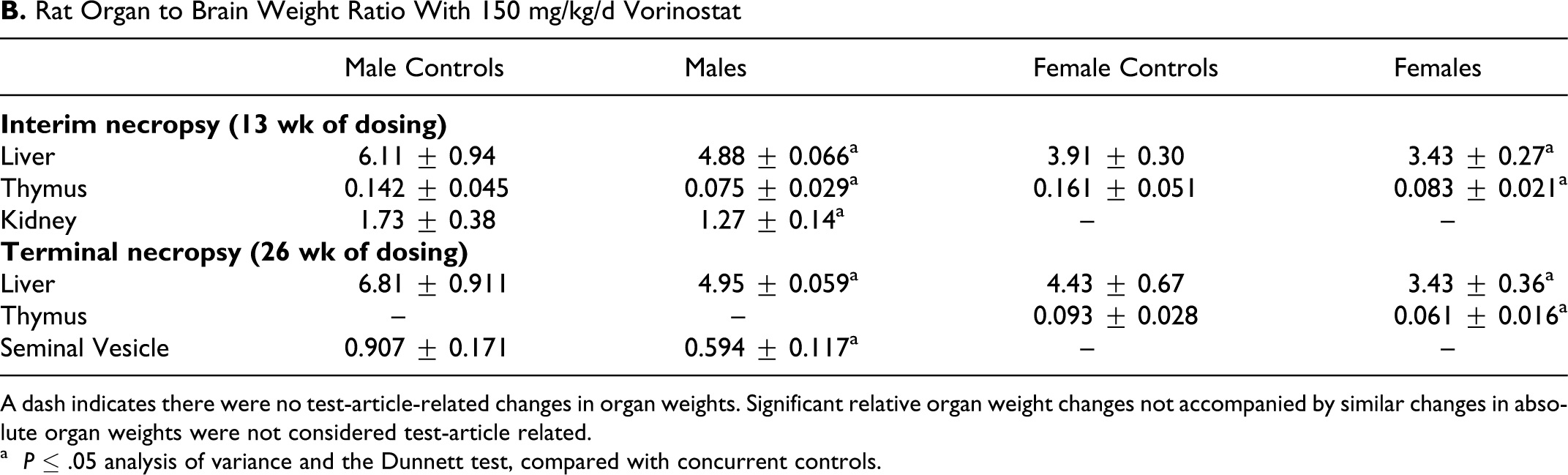
A dash indicates there were no test-article-related changes in organ weights. Significant relative organ weight changes not accompanied by similar changes in absolute organ weights were not considered test-article related.
a P ≤ .05 analysis of variance and the Dunnett test, compared with concurrent controls.
Animals dosed at 150 mg/kg/d had minimal thymic and splenic lymphoid depletion (Table 7). Animals at all doses had minimal bone marrow erythroid hyperplasia/myeloid hypoplasia of the femur and sternum. There appeared to be a qualitative loss of myeloid tissue, because decreased numbers of granulocyte precursors were present. Animals at all dose levels had minimally to slightly increased splenic pigment, a treatment-related finding secondary to extravascular hemolysis. There was no evidence of intravascular hemolysis or hemoglobinuria, and the splenic pigment indicated extravascular phagocytosis of erythrocytes.
Rat Terminal Necropsy—Histomorphologic Findings after 26 Weeks of Vorinostat Dosing
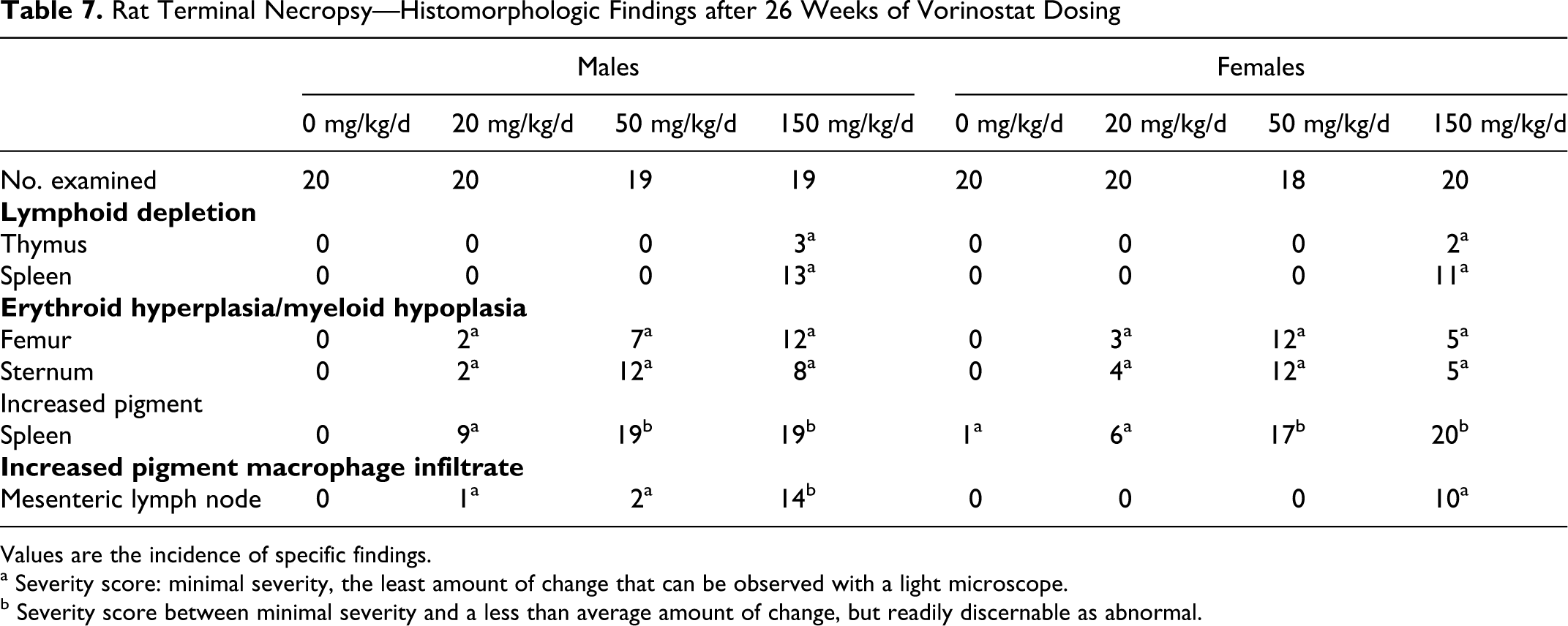
Values are the incidence of specific findings.
a Severity score: minimal severity, the least amount of change that can be observed with a light microscope.
b Severity score between minimal severity and a less than average amount of change, but readily discernable as abnormal.
In summary, the adverse vorinostat treatment–related changes included reduced body weight and food consumption, lower globulin and white blood cell counts, higher absolute reticulocyte counts, decreased thymus weight, and histopathologic findings in the thymus, bone marrow, and spleen. All of these effects were reversible or trended toward reversibility following 4 weeks of recovery, including thymic lymphoid depletion and femur/sternum erythroid hyperplasia/myeloid hypoplasia. Other treatment-related effects were not considered toxicologically significant. Based on the fact that 1 or more of these treatment-related findings (lower white blood cell counts and globulin and increased absolute reticulocyte count) were observed in all dose groups at more than 1 interval, a no observed adverse effect level (NOAEL) was not established for this study.
Treatment-Related Findings in Repeated-Dose Toxicity Studies in Dogs Were Primarily Gastrointestinal
In an oral exploratory repeated-dose escalation study in female and male beagle dogs, which was used to determine the dose levels for the 26-week GLP oral repeated-dose toxicity and toxicokinetic study, the feeding schedule affected the ability of the dogs to tolerate vorinostat. When food was offered in the first several hours after vorinostat administration at doses of 60 mg/kg/d (1200 mg/m2/d) or 80 mg/kg/d (1600 mg/m2/d), food consumption was low and animals tended to lose body weight. In the dogs with continuous access to food overnight 5 to 7 hours after dosing, vorinostat was reasonably well tolerated when administered up to 160 mg/kg/d (3200 mg/m2/d) after animals first received lower dose levels: 80 mg/kg/d (1600 mg/m2/d), 100 mg/kg/d (2000 mg/m2/d), and 120 mg/kg/d (2400 mg/m2/d). A further increase to 200 mg/kg/d (4000 mg/m2/d) was not well tolerated, and signs of gastrointestinal (GI) toxicity were apparent. Naïve dogs administered 160 mg/kg/d (3200 mg/m2/d) from the initial dose developed GI toxicity, including reddening of the stomach, cecum, and colon within 13 days of dosing.
In the 26-week oral repeated-dose oral toxicity and toxicokinetic study in female and male beagle dogs with a 4-week recovery, no adverse effects were observed when vorinostat was administered at doses of 20 mg/kg/d and 60 mg/kg/d and animals were fed overnight. For the high-dose group, dosing with vorinostat began at 80 mg/kg/d and, in order to demonstrate treatment-related effects, was escalated in succession to 100 mg/kg/d, 125 mg/kg/d, and 160 mg/kg/d on study days 16, 30, and 97, respectively. The NOAEL of vorinostat was 60 mg/kg/d in this study.
All animals except 1 female survived until the end of the study. No treatment-related findings, at any dose, in the surviving animals were noted for the end points of mean body weight, mean food consumption, ophthalmologic abnormalities, electrocardiographic parameters, or blood pressure. The average body weights for control females and males at the start of the study were 9.6 and 11.5 kg, respectively, and at the end of dosing in the high-dose dogs the mean body weights were 9.5 and 11.6 kg in the females and males, respectively. Clinical pathology results were only observed in the high-dose female dogs, and the differences were very slight, were not abnormally low or considered adverse, and were reversible. These changes included mild decreases in red blood cell counts (–9%), hemoglobin (–12%) and hematocrit (–10%), and WBC counts (–29%) (primarily attributable to lower absolute neutrophil counts [–29%]) with a slight increase in platelets (+27%). Absolute reticulocyte counts were unaffected, and bone marrow histopathology indicated no obvious hematopoietic abnormalities in females that survived to necropsy.
Gastrointestinal toxicity, characterized by physical signs (inappetence and diarrhea) and macroscopic or microscopic findings attributed to vorinostat, was associated with the high-dose regimen (particularly at 160 mg/kg/d [3200 mg/m2/d]), including the unscheduled sacrificed dog. This female dog lost approximately 26% of her body weight (2.4 kg) between study weeks 22 and 24, accompanied by decreased food consumption. Findings consistent with dehydration in the moribund female included mild to moderate increases in red blood cell count, hemoglobin, hematocrit, white blood cells, albumin, globulin, and urea nitrogen. These were considered secondary to treatment. Multiple red areas in the mucosa of the stomach correlated with slight gastric erosions, which were considered treatment related. Other adverse treatment-related effects included slight crypt epithelial degeneration in the duodenum, jejunum, and ileum.
At terminal necropsy in the high-dose group, 1 male dog had macroscopic findings of red pinpoint foci in the gastric mucosa and linear red areas in mucosa of cecum, colon, and rectum, all of which were considered treatment related. Other findings, all of which were minimal to moderate, and were found in 1 or more high-dose dogs, with a higher prevalence in males. The findings included acute inflammation with erosion in the esophagus and stomach; acute inflammation, crypt cell necrosis, and villous blunting in the jejunum, ileum, and cecum; and acute inflammation and regeneration of crypt epithelium in the cecum and colon. There were no findings in the 20 or 60 mg/kg/d dose groups. After recovery, 1 high-dose female had treatment-related findings of hemorrhage in the cecum, minimal villous blunting, and crypt epithelium regeneration. All other microscopic lesions resolved in the other dogs. In general, GI toxicity was reversible with time.
Toxicokinetics in Rat and Dog Indicated Exposure of Vorinostat Less Than in Humans
The exposures of vorinostat were less at all doses in both rats and dogs than the mean exposure at the maximum recommended human dose of 400 mg/d (AUC0-t h was 1588 hċng/mL and Cmax ~320 ng/mL). For vorinostat, exposure margins (the ratio of animal to human vorinostat AUC values) were less than 1. The AUC0-t h in the pivotal rat 6-month repeated-dose toxicity study at 150 mg/kg/d (900 mg/m2/d) was 1197 hċng/mL and the Cmax was 449 ng/mL at 2 hours after dosing (sexes combined) (Table 8). The exposure in the 6-month dog repeated-dose toxicity study was less than that in rats (362 hċng/mL). The Cmax was 130 ng/mL at 2 hours after the 160 mg/kg/d (3200 mg/m2/d) dose (sexes combined). In rats, dogs, and humans, the same pharmacologically inactive metabolites O-glucuronide of vorinostat and 4-anilino-4-oxobutanoic acid have been identified. For O-glucuronide of vorinostat, the exposure of this metabolite was less than unity (based on the ratio between animal/human exposure) at the highest doses tested in either rat or dog. However, in the rat, the exposure to 4-anilino-4-oxobutanoic acid was 1.7 compared with human exposure, demonstrating that this metabolite has been adequately tested in a nonclinical species. These data suggest that humans appear to glucuronidate vorinostat to a greater extent than do rats or dogs.
Toxicokinetic Data in Rats and Dogs Administered Vorinostat
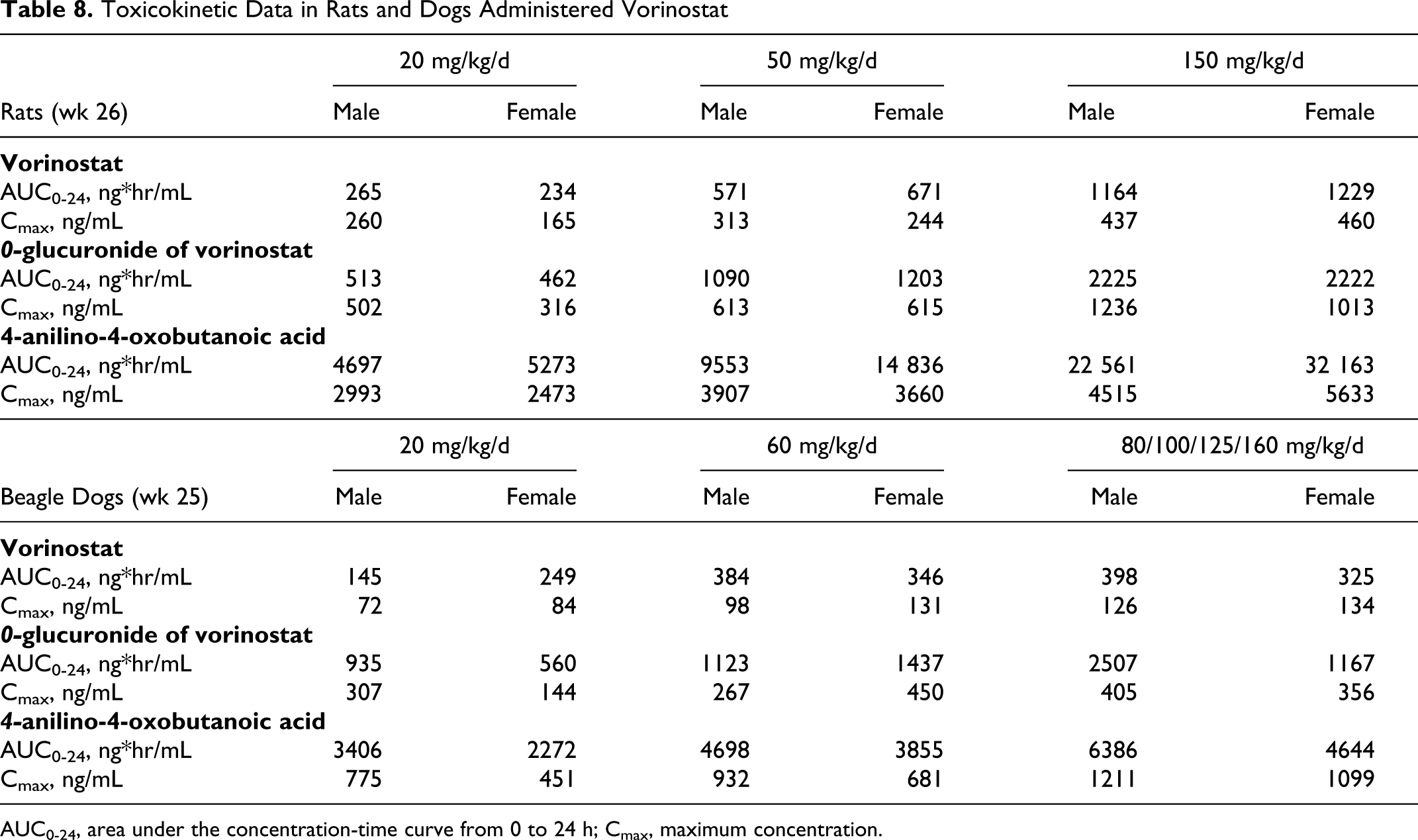
AUC0-24, area under the concentration-time curve from 0 to 24 h; Cmax, maximum concentration.
Discussion
The results indicate a weak potential for genotoxicity, with weak positive results in the Ames assay for mutation in bacteria and the in vivo assay for micronuclei and an interesting cell-type difference in chromosome aberration induction. Positive results were observed at toxic concentrations in the transformed CHO cell line, and negative results were observed in normal human lymphocytes.
The reason for the small increase in mutation in the Ames test in Salmonella is not known, but mutation at the his locus requires specific interaction with DNA, so this mutation frequency may be related not to the pharmacological activity of vorinostat but possibly to its hydroxamic acid chemical structure. Certain derivatives of hydroxamic acids are known to be mutagenic. 32 Certainly this increase in revertants is small and occurs only at high concentrations, compared with many genotoxic cancer chemotherapeutic agents. For example, in the same assay cisplatin is clearly positive at 1 μg per plate 33 ; vorinostat gave a positive result only at 5000 μg/plate (approximately 8 mM) and an increase in mutation frequency that was only 2 to 3 times background. The forward mutation assay for 5-fluorouracil resistance was used to gain additional information on mutagenic potential in bacteria. The results of this assay correlate highly with the results of the Ames test for many classes of known mutagens, 20 and the negative results here also indicate that vorinostat is not a potential mutagen.
Induction of chromosome damage has been reported with other HDAC inhibitors, for example, trichostatin in human TK6 lymphoblasts 34 and apicidin in CHO cells. 35
Why the aberration induction was seen with the immortalized rodent CHO cell line but not in normal human lymphocytes is not clear. Our previous work demonstrated that chromosome damage produced in CHO cells by non-DNA reactive chemicals is often associated with marked toxicity and inhibition of DNA synthesis 17 ; such effects are likely to have a threshold and thus not occur at lower concentrations. The aberration induction by vorinostat in CHO cells is likely related to toxicity and the suppression of DNA synthesis and also to the ability of cells to continue to move through the cell cycle despite damage. This ability is known to be quite different in normal human cells compared with cultured rodent CHO cells that are immortalized, transformed, and tumorigenic and have mutated p53 gene function. 36 Rodent cells also have much less stringent cell cycle checkpoints compared with normal human cells. 37,38 Thus, rodent cells that are damaged may continue to cycle, whereas human cells are more likely to arrest in response to damage and in certain circumstances to undergo apoptosis. Because HDAC inhibitors affect cell cycle checkpoints, the different results in normal versus transformed cells are possibly related to the ability of transformed cells to bypass checkpoints, characteristics that likely result in the differential killing of tumor cells by HDAC inhibitors. 39,40
Although the in vivo micronucleus results are statistically significant, the effect is considered equivocal, because the micronucleus levels are within the historical control range, and a test for reproducibility has not been performed. Micronuclei can arise from chromosome breakage or from chromosome loss due to errors of chromosome segregation at mitosis. If the increases are weak but “real,” either process might result from HDAC inhibition, because structural chromosome breakage is induced by HDAC inhibitors in transformed cells. Potential for aneuploidy has been reported for HDAC inhibitors, for example, premature exit from mitosis (mitotic slippage) in HeLa cells, 41 and polyploidy in cancer cell lines, especially those lacking p21 or p53 function, but not in normal human or mouse embryonic fibroblasts. 42 Such mitotic abnormalities lead to cell death, 39,42 and because mitotic slippage and polyploidy are reported to occur in transformed but not normal cells, this does not seem a likely explanation for any micronucleus induction in mouse bone marrow from vorinostat-treated animals. Mitotic abnormalities have been seen in cultured normal (MRC-5) fibroblasts that entered mitosis with hyperacetylated histones due to treatment with trichostatin A. 43 Lagging chromosomes and chromatin bridges at cytokinesis were seen, implying potential for induction of aneuploidy and micronuclei if the cells do progress out of mitosis. Increases in micronuclei (also known as Howell Jolly bodies) are associated with rapid replication of red blood cells, after direct stimulation (eg, with erythropoietin) or in regenerative anemia. 44 Although hematologic disturbances are described here with vorinostat, it seems unlikely that such effects would occur in mice 24 hours after a single dose of the drug at the time micronuclei were scored.
The effects of 2 other hydroxamic acid HDAC inhibitors, LAQ824 and LBH589, on hERG channel potassium currents have been reported elsewhere, 45,46 with IC50 values of 10 μM and 3.9 μM, respectively. Small differences between our data and those previously reported may be attributed to differences in assay methodology and underscore the importance of systematic assessments, not only in hERG channel assays but also in preclinical electrocardiographic determinations and efficacy information in order to establish an accurate safety margin for the risk of delayed ventricular repolarization. Limited clinical cardiac safety information is available for the hydroxamates compared here. A preliminary report described dose-related increases in QTc of less than 20 milliseconds at doses up to 200 mg/m2 for LAQ824. One patient had QTc greater than 500 milliseconds: in cycle 1 after dosing at 200 mg/m2 and after dose reduction to 125 mg/m2 in a second cycle, a 10-second run of unsustained torsades de pointes. 45 A published report described dose-limiting QTc prolongations during clinical trials of LBH589, with tolerability at doses less than 11 mg/m2. A clear relationship between dose administered and reversible, asymptomatic QTc prolongation was noted, but it was not reflected in plasma pharmacokinetic variables. Investigators speculated that LBH589 could be initiating a physiologic process that prolongs QTc but has no temporal dependency on LBH589 plasma concentration or exposure. 46 Electrocardiographic data from reported clinical trials with vorinostat indicated no major QT abnormalities in electrocardiographic measurements up to doses of 300 mg/m2. 47 No QT abnormalities were observed in any of the preclinical studies. Recent data from a phase I study in patients with advanced cancer assessed the effect of a supratherapeutic dose (800 mg; 462 mg/m2) of vorinostat on the QTcF interval (QTc calculated using Fridericia’s equation). Administration of this single supratherapeutic dose of vorinostat did not prolong the QTcF interval. 48
In addition to clinical considerations, a larger issue raised by these recent studies is the potential for mechanism-based cardiac toxicities among HDAC inhibitors. 49 In this context, our data suggest little connection between potency as inhibitors of HDACs (nM) and potency affecting hERG (μM). hERG channel inhibition is just one potential mechanism of QT prolongation, and QT prolongation is not a straightforward correlate of ventricular fibrillation.
The definitive chronic toxicity studies in both rats and dogs achieved toxicologic end points, including weight loss, inappetence, diarrhea, and hematologic changes. Although many of these effects were consistent with the pharmacology of the drug and other cytotoxic agents (weight loss, inappetence, and hematologic effects), the severity of the vorinostat-related findings on absolute reticulocyte counts, red blood cell counts, hematocrit, and hemoglobin and the proliferative effects on lymphoid tissue in the rats were considered to be treatment-related adverse findings. The high dose of 150 mg/kg/d (900 mg/m2/d) was clearly more affected than the lower doses. The effects on circulating lymphocyte numbers and globulin were consistent with histopathologic findings in lymphoid tissue. The effect on absolute reticulocyte count is consistent with a regenerative response to the loss of red blood cells (red blood cell count, hemoglobin, and hematocrit were consistently lower in the high dose animals), and the observation of splenic pigment indicated the loss was likely due to extravascular hemolysis, consistent with the erythroid hyperplasia. Erythroid hyperplasia (increased numbers of erythroid precursors) and myeloid hypoplasia (reduced numbers of granulocytic precursors) were evident in the bone marrow histologic sections and in sacrifice bone marrow smears from the interim necropsy after 13 weeks of dosing. All clinical pathology effects at the high dose trended toward reversibility or completely reversed following the 4-week recovery.
The gastrointestinal vorinostat-related findings in the 26-week toxicity study in dogs included microscopic lesions throughout the intestinal tract and only in the high dose. These findings included inflammation in the esophagus and small intestine, with epithelium regeneration in the colon, all of which were reversible or trended toward reversibility following the 4-week recovery.
Although the mechanism underlying the antitumor activity of vorinostat is not yet entirely known, it is known that the action of this drug leads to changes in expression of specific genes via acetylation of histones and transcription factors as well as inhibiting mitosis. These effects cause growth arrest and cell death. 9,31 Vorinostat is active in inducing differentiation, cell growth arrest, or apoptosis by interfering with the cell cycle. Observations in the repeated-dose toxicity studies that proliferating tissues (hematopoietic, lymphoid, and gastrointestinal) are the ones predominantly affected in these studies, particularly at the high doses, are consistent with current understanding of the mechanism of action of this drug.
The apparent half-life of vorinostat in the repeated-dose toxicity studies was 1 to 2 hours for rats and to more than 5 hours in dogs after 25 weeks of daily oral dosing. The apparent half-life in preclinical species is thus appreciably longer than the terminal half-life of 12 minutes calculated after a single intravenous dose 50 and is consistent with the human half-life after oral administration (1.34 hours). In the oral repeated-dosing paradigm, the actual rate of elimination is offset by absorption in the GI tract. Although the half-life is relatively short, the duration of the accumulation of histone acetylation in blood peripheral mononuclear cells from patients treated with vorinostat is more than 8 hours and persists for 6 months with continued dosing, suggesting the sustained biological effect of the drug. 47
Expressed on the basis of body surface area, all doses, except the dose of 20 mg/kg/d (120 mg/m2/d) in the 26-week repeated-dose toxicity study in rats, were higher than the clinical dose of 400 mg (250 mg/m2/d) by 1.2 or more. Although the exposure margins of vorinostat and metabolites in dog and rat were less than the human exposures, both species were dosed at or above the MTD. Therefore, using higher dosages via the intended clinical oral route of administration to achieve higher exposures was not feasible.
Conclusions
We have extensively evaluated the preclinical safety of vorinostat, the first HDAC inhibitor approved by the FDA for a cancer indication. Vorinostat was positive in all genotoxicity assays performed except in the chromosomal aberration assay in normal human lymphocytes. Preclinical ECG assessments in the chronic dog study did not indicate that vorinostat had any cardiac safety issues. There does not appear to be a class effect of HDAC inhibitors and QTc prolongation. The findings in the 26-week repeated-dose toxicity study in rats demonstrated the presence of a regenerative anemia that involved extravascular phagocytosis of erythrocyte components and exhibited no evidence of intravascular hemolysis. The findings in the 26-week repeated-dose toxicity study in dogs demonstrated GI lesions that were reversible. The toxicities observed in these repeated-dose toxicity studies demonstrate that the nonclinical dosing regimen was satisfactory to characterize the potential toxicities associated with chronic vorinostat administration and did not reveal any adverse effects that could not be monitored in humans.
Footnotes
Acknowledgments
We thank Janet Gormley and Patricia Scripture for their contributions.