
Abstract
Duchenne muscular dystrophy (DMD) is caused by dystrophin gene mutations. Restoration of dystrophin by exon skipping was demonstrated with the phosphorodiamidate morpholino oligomers (PMO) class of splice-switching oligomers, in both mouse and dog disease models. The authors report the results of Good Laboratory Practice–compliant safety pharmacology and genotoxicity evaluations of AVI-4658, a PMO under clinical evaluation for DMD. In cynomolgus monkeys, no test article–related effects were seen on cardiovascular, respiratory, global neurological, renal, or liver parameters at the maximum feasible dose (320 mg/kg). Genotoxicity battery showed that AVI-4658 has no genotoxic potential at up to 5000 μg/mL in an in vitro mammalian chromosome aberration test and a bacterial reverse mutation assay. In the mouse bone marrow erythrocyte micronucleus test, a single intravenous injection up to 2000 mg/kg was generally well tolerated and resulted in no mutagenic potential. These results allowed initiation of systemic clinical trials in DMD patients in the United Kingdom.
Keywords
Duchenne muscular dystrophy (DMD) affects 1 in every 3500 male and, in rare cases, female newborns worldwide 1 and results from mutations of the dystrophin gene. Lack of dystrophin leads to reduced sarcolemmal stability with actin filament contraction and increased intracellular calcium influx followed by muscle fiber degeneration. The clinical effect of a disrupted reading frame in the dystrophin gene is dramatic and lethal. 1,2 In DMD patients, the first symptoms involve the lower limbs and appear between the third and fifth year. These boys develop hypertrophic calves, show difficulty in running and climbing stairs, run on their tiptoes, and frequently fall. Muscle weakness progresses to the shoulder girdle upper arm and trunk muscles and loss of ambulation before the age of 12 years. Histological changes are readily apparent with light microscopy analysis of cross sections from patient muscle biopsies. They involve variation in fiber size with atrophic and hypertrophic fibers, degeneration and regeneration of the muscle fibers, infiltration of inflammatory cells and fibrosis, and characteristic central location of the nuclei within muscle cells. The fiber membrane destabilization results in leakage of the enzyme creatine kinase (CK), resulting in very high serum CK levels (20 000 to 50 000 U/L compared with 80 to 250 U/L in unaffected individuals). These levels decline as the patients get older, and the overall muscle mass decreases progressively. One-third of all affected boys are mentally impaired, and learning difficulties are common. Due primarily to the loss of muscle strength and integrity, DMD patients usually die in their 20s from cardiorespiratory failure. 1,2
Despite extensive effort, no effective disease-modifying therapy for DMD is yet available. However, a delay of the onset of disease manifestations and an improvement in quality of life can be achieved by drugs that decelerate progression of the DMD pathology. These include glucocorticoids, 3 most commonly prednisolone and deflazocort, which can improve muscle strength and delay loss of ambulation by up to 2 to 3 years. 1 The mechanisms of their beneficial effect are not well understood but likely include anti-inflammatory activity, which may prevent the additional damage caused by the infiltration of mononuclear cells into the muscle upon necrosis. 4 However, the benefits of glucocorticoid therapy come at a price of frequent side effects, which can include obesity, spine deformities, bone loss, and growth retardation. 5,6 To date, the most effective treatment for prolonging the life of DMD boys has been assisted ventilation with portable ventilators. Ventilation has been shown to increase the average life expectancy of DMD boys from 19 to 24 years. All of the above treatments are palliative and do not address the underlying cause of the disease: loss of dystrophin expression. No current treatment reverses or arrests the progression of DMD.
In addition to DMD, a milder, allelic form of muscular dystrophy called Becker muscular dystrophy (BMD) exists. 1,2 In BMD, unlike DMD, the reading frame is not disrupted, and an internally truncated yet functional dystrophin protein is produced. Most BMD patients remain ambulant for life and have a near-normal life expectancy. Work by Kole and others proposed the use of antisense oligonucleotides to modulate dystrophin mRNA splicing and convert out-of-frame DMD mutations into the nearest in-frame BMD-like mutation, to produce an internally deleted Becker-like functional dystrophin protein. 7-10 The mechanism of splice-switching oligomer (SSO) modulation of dystrophin pre-mRNA splicing involves hybridization to specific motifs involved in splicing and exon recognition in the pre-mRNA. This prevents normal spliceosome assembly and results in skipping of the target exon in the mature RNA transcript. 11,12 In the case of out-of-frame dystrophin gene deletions, selective removal of specific flanking exons should result in in-frame mRNA transcripts that may be translated into an internally deleted, BMD-like, and functionally active dystrophin protein. 9,10
Unmodified DNA and RNA oligonucleotides have poor in vivo stability and therefore are ineffective as drugs. The most common chemical modification used in early-generation oligonucleotides to improve the stability and pharmacokinetics was the introduction of the phosphorothioate linkage in place of the natural phosphodiester linkage. These phosphorothioate compounds have been used extensively in both preclinical and clinical evaluations, and the dose-limiting toxicities and adverse effects are well established. 13 Initial evaluations in primates led to mortality following intravenous bolus injections of phosphorothioate oligonucleotides at doses as low as 10 mg/kg, 14 while other effects included lethargy, central hypotension, and reduced cardiac output. Other notable toxic effects associated with phosphorothioate oligonucleotides include complement activation and prolonged coagulation times. 15 The latter, specifically linked to the phosphorothioate component of the oligonucleotides, is therefore considered a class effect, 16 as were observed hepatotoxic effects. 17 These dose-limiting toxicities are most likely due to high Cmax achieved with bolus injections. Studies subsequent to those in which mortality was observed have typically been limited to lower doses and increased infusion times to reduce the toxic effects. 13
To be effective in modulating splicing, SSOs must not activate RNA cleavage by RNase H, which would destroy the pre-mRNA before splicing can occur. 18 Phosphorodiamidate morpholino oligomers (PMOs) productively compete with the splicing factors for target sequences in pre-mRNA during splicing, and in addition, their stability and in vivo uptake and bioavailability are improved compared with natural oligonucleotides. PMOs have standard nucleic acid bases attached to the morpholino-phosphoroamidate backbone (Figure 1 ), which, unlike other sugar-phosphate backbone oligonucleotides, is uncharged. 19 PMOs are very resistant to enzymatic degradation in vivo, providing unparalleled stability and somewhat different biodistribution than other oligonucleotides. Other modifications that can be used as SSOs include previous-generation chemistries such as the 2′-O–substituted 2′-O-methyl phosphorothioate (2′OMe), and locked nucleic acids/phosphorothioate. 20
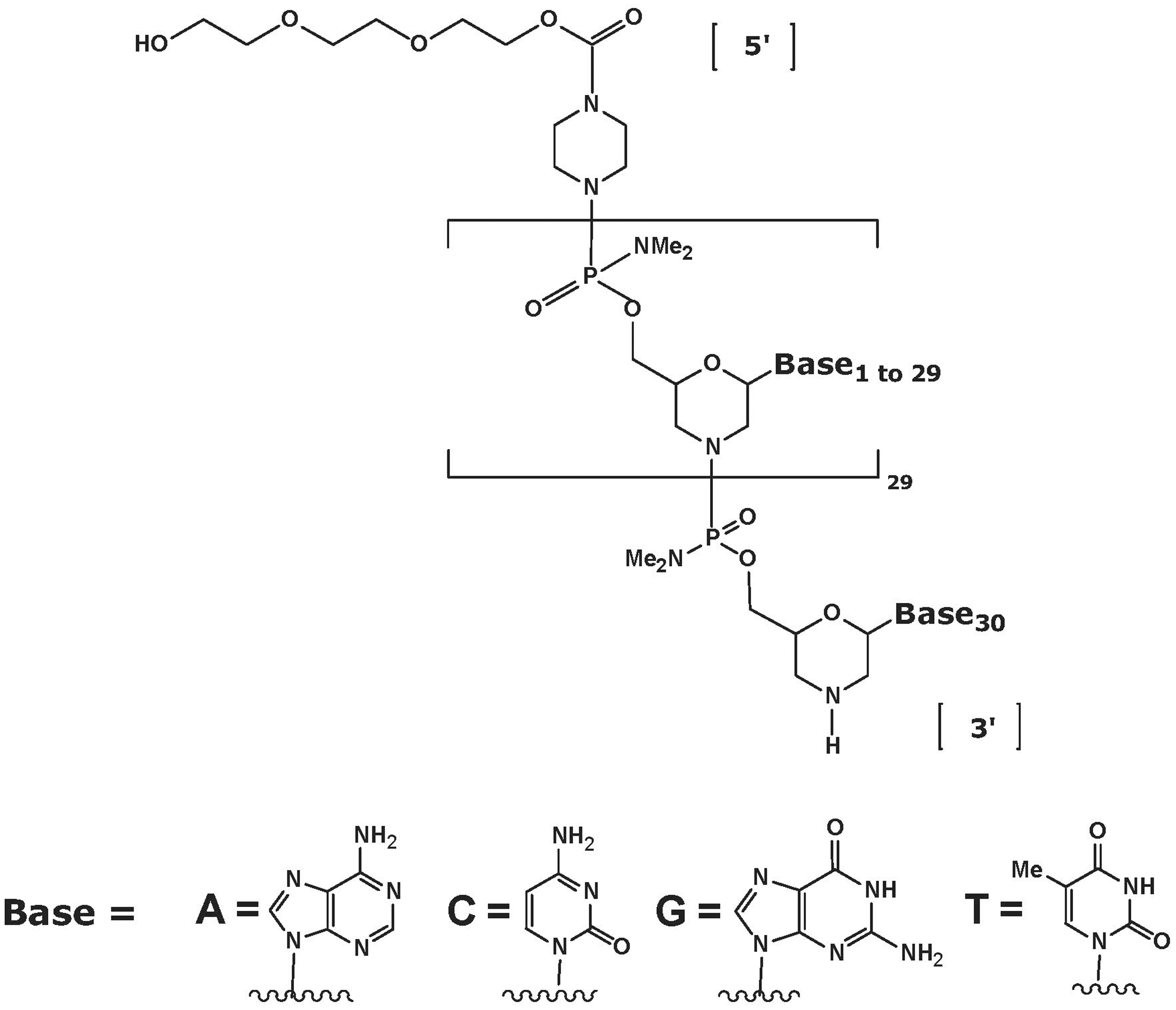
Structure of phosphorodiamidate morpholino oligomers.
AVI-4658 is a PMO drug with the general structure described in Figure 1, with the base sequence CTC CAA CAT CAA GGA AGA TGG CAT TTC TAG. It is designed to skip exon 51 of human dystrophin and thus restore dystrophin expression in DMD patients having certain mutations. 21 AVI-4658 targets the pre-mRNA transcripts of the dystrophin gene, causing exon 51 to be skipped from the mature, spliced mRNA. In cells from DMD patients with deletions in exons 50, 52, 52-63, 45-50, 48-50, or 49-50, exon skipping restored or is expected to restore the reading frame and produce an internally truncated, BMD-like form of dystrophin. Here, we report the results of a safety pharmacology evaluation of the PMO AVI-4658 in cynomolgus monkeys following intravenous and subcutaneous administration at doses up to the maximum feasible dose of 320 mg/kg. We also report the results of a standard genotoxicity battery evaluation using AVI-4658 at concentrations up to 5000 μg/mL in an in vitro mammalian cell chromosome aberrations test, up to 5000 μg/plate in a bacterial reverse mutation assay, and up to 2000 mk/kg as a single intravenous administration in a mouse micronucleus assay.
Materials and Methods
The safety pharmacology evaluation was performed by MDS Pharma Services (Lyon, France). The testing facility is Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) accredited, and the study plan was reviewed by the ethical committee, according to the following animal health and welfare guidelines: guide for the care and use of laboratory animals, NRC, 1996, Decree no. 2001-464 regarding the experiments with laboratory animals described in the Journal Officiel de la République Française on May 29, 2001, Decree no. 2001-486 relating to the protection of animals used in scientific experiments described in the Journal Officiel de la République Française on June 6, 2001. The study was conducted according to the following: guideline on safety pharmacology studies for human pharmaceuticals (November 8, 2000, issued as CPMP/ICH/539/00–ICH S7A, published in the Federal Register, vol 66, no. 135, July 13, 2001, pp 36791-36792) and guideline on nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals (May 12, 2005, issued as CPMP/ICH/423/02–ICH S7B, published in the Federal Register, vol 70, no. 202, October 20, 2005, pp 61133-61134). All phases of this study performed at the testing facility were conducted in compliance with the following Good Laboratory Practice (GLP) regulations: OECD Principles of Good Laboratory Practice concerning mutual acceptance of data in the assessment of chemicals, dated November 26, 1997, (C[97] 186 Final), “Principles of Good Laboratory Practice” described in the French Official Journal on March 23, 2000, Organization for Economic Co-operation and Development (OECD) GLP consensus document (the application of the OECD principles of GLP to the organization and management of multisites studies, ENV/JM/MONO [2002]9, June 25, 2002).
The genotoxicity battery was performed by BioReliance (Rockville, MD). This study was conducted in compliance with the most recent version of the US Food and Drug Administration GLP regulations, 21 CFR part 58, and the OECD Principles of Good Laboratory Practice, C(97)186/Final, and in compliance with the testing guidelines ICH S2A (1996), ICH S2B (1997), and OECD 474 (1998). The number of mice and the procedures and experimental design used for this study have been reviewed and were approved by the BioReliance Institutional Animal Care and Use Committee 8 and 10. All procedures involving mice performed at BioReliance follow the specifications recommended in The Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington, DC, 1996). The mice were housed in an AAALAC-accredited facility.
Safety Pharmacology Evaluation of AVI-4658
Animals and animal husbandry
Six male cynomolgus monkeys (Macaca fascicularis) were used in this study, with a weight range of 2.7 to 2.9 kg and an age range of 2 to 3 years. Animals were housed in 1 room for the study in an air-conditioned building with a target temperature of 22°C ± 2°C, relative humidity >40%, a minimum 10 air changes per hour, and 12 hours light (artificial)/12 hours dark. Animals were housed singly in stainless steel mesh cages. Animals were fed expanded complete commercial primate diet at approximately 100 g diet/animal per day. In addition, animals received fruit or vegetable daily (apple, banana, or carrot). Certificates of analysis for the diet and drinking water are maintained in the archives of the testing facility, which conducted the tests according to current animal welfare guidelines. The normal dark cycle was interrupted on occasions (for up to 45 minutes) to allow completion of technical procedures. These differences were not considered to have affected the outcome of the study.
Procedures for telemetry and venous catheter implantation
Animals were fasted for at least 15 hours before surgical procedures. For implantation of the telemetry device, each animal was premedicated with a subcutaneous injection of glycopyrrolate (Robinul V, Vétoquinol SA; 0.01 mg/kg) and then anesthetized with an intramuscular injection of ketamine (Imalgène 500, Mérial SAS; 15 mg/kg) and xylazine hydrochloride (Rompun 2%, Bayer AG; 0.7 mg/kg). In addition, local oropharyngeal anesthesia was provided with a spray of lidocaine chlorhydrate (Xylocaine 5 % Nébuliseur, AstraZeneca). The hair on the abdomen, the inguinal area, and the thorax was clipped. During surgery, the level of anesthesia was maintained with gaseous anesthetic (1% to 5% isoflurane in oxygen, AErrane, Laboratoire Baxter). An antibiotic prophylaxis by intramuscular injection with long-acting amoxycilline (Clamoxyl LA, Pfizer Italia SRL) at 30 mg/kg was given 48 hours before surgery. The transmitter body was implanted into the abdominal cavity under aseptic conditions. The pressure catheter (polyurethane tubing that extends out of the device body) was inserted into the lower abdominal aorta via the femoral artery and the bipotential leads then placed. Animals received antibiotic prophylaxis by intramuscular injection with long-acting amoxycilline (Clamoxyl LA, Pfizer; 30 mg/kg) and an analgesic prophylaxis intramuscular injection of tolfenamic acid (4% Tolfédine, Vétoquinol; 4 mg/kg), once right after implantation and then repeated 4 times at 48-hour intervals. The surgical wounds were disinfected with iodine (Vétédine, Vétoquinol) for 7 days. After surgery, animals were allowed to recover for approximately 3 weeks before the first treatment. Prior to intravenous dosing, the animals were implanted with a venous catheter, under anesthesia similar to that above. The catheter was attached to the delivery system via a tether and a swivel joint.
Safety examinations
Arterial blood pressure, heart rate, electrocardiogram, respiratory parameters, global neurological activity, and renal and liver functions were examined following 3 separate subcutaneous administrations and 1 single intravenous administration of AVI-4658 in the conscious male cynomolgus monkey. All observations, including respiratory parameters, were collected from the freely moving animal. AVI-4658 was administered by the subcutaneous route (phase 1) at 0 (vehicle), 40, 160, and 320 mg/kg on days 0, 7, 14, and 21 and by the intravenous route (phase 2) at 0 (vehicle) and 320 mg/kg on days 38 and 45 using a dosing volume of 2.67 mL/kg with an infusion rate of 1 mL/min (Table 1). Subcutaneous and intravenous dosing were examined to support both routes for potential clinical administration. The high dose (320 mg/kg) was used to support up to 100 mg/kg clinically, based on allometric scaling. For intravenous dosing, 320 mg/kg was the maximum feasible dose based on dose volume and solubility of the compound. The low dose for phase 1 was selected based on the expected therapeutic dose (human equivalent dose of approximately 10 mg/kg). For phase 1, animals were randomized in a Latin square design. For both phases, there were at least 6 days between each testing session. Each animal served as its own control.
Study Design
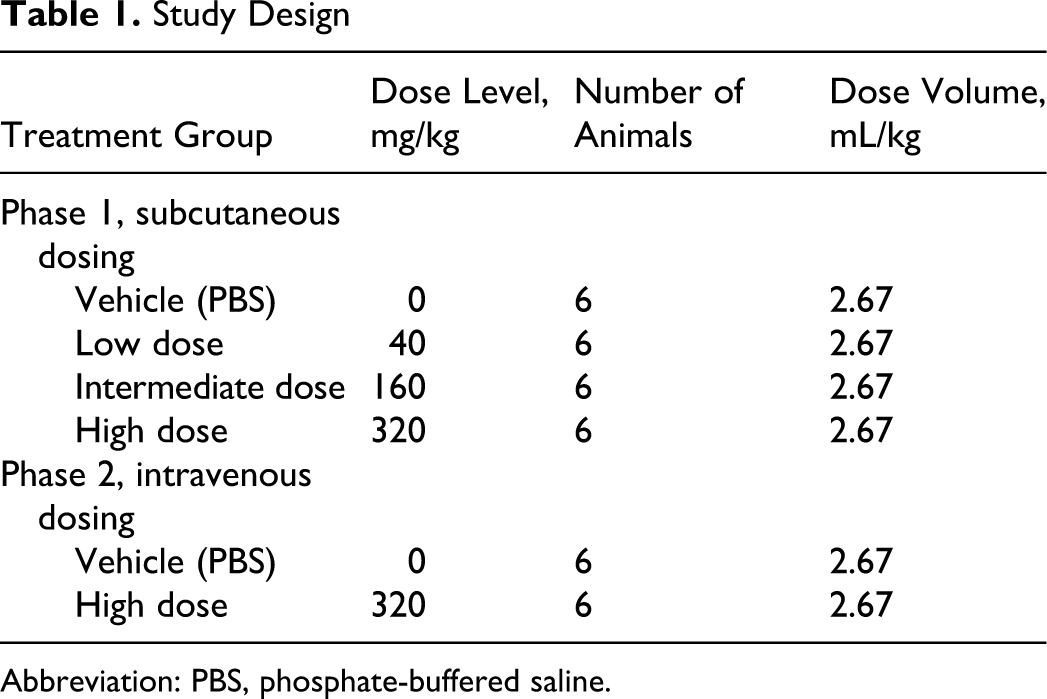
Abbreviation: PBS, phosphate-buffered saline.
Telemetry signals (body temperature, cardiovascular and respiratory parameters) were recorded. Time points were selected to correspond to the times of maximum exposure to the drug: phase 1: on days 0, 7, 14, and 21, starting at least 1.5 hours preadministration and for 21 hours postadministration phase 2: on days 38 and 45, starting at least 1.5 hours preadministration and for 24 hours postadministration
The telemetric system used consisted of an implantable TL11M3-D70-CCTP device (DSI, St Paul, MN), an RMC-1 receiver located on the top of each cage, a DEM data exchange matrix that centralizes the signals from all animals, an APR-1 ambient pressure reference that allows a barometric correction, and a microcomputer with acquisition card. For collecting and analyzing hemodynamic, cardiac, and respiratory parameters, Notocord-hem software (Notocord Systems SA, Croissy-sur-Seine, France) was used.
For cardiovascular analyses, the value for each parameter (systolic blood pressure, mean arterial blood pressure, diastolic blood pressure, and heart rate) was the mean of the values recorded for 5 minutes around the time point (selected times ±2.5 minutes). The value for each interval or complex (PR, RR, QT, or QRS) was the mean of the 10 best quality electrocardiogram (ECG) traces at the time point (selected time ±2.5 minutes). The value for respiration rate (f), inspiratory time (TI), expiratory time (TE), and AUCEMG was the mean of 10 values obtained around the time point (selected time ±2.5 minutes). Note that the signals recorded at the predefined time points were occasionally disturbed. In this case, data recorded a few minutes before or a few minutes after the theoretical time point were used instead. On a few occasions, electromyogram (EMG) parameter values were calculated from fewer than 10 values. In another few cases, sustained disturbances of the signal did not allow an accurate evaluation of any data at the predefined time point (detailed in the raw data). The results were expressed as mean ± standard error of the mean (SEM).
For respiration parameters, inspiratory time (TI, milliseconds) was defined as the duration of the diaphragmatic EMG burst. Expiratory time (TE, milliseconds) was defined as the time elapsed between 2 diaphragmatic EMG bursts, and respiration rate (f, breath/min) was calculated from the averaged respiratory cycle duration (TI + TE). The AUC of the rectified diaphragmatic EMG burst (AUCEMG) is that for which variation in amplitude has been shown to be correlated in humans and animals with variation of the tidal volume. 22-24 For this evaluation, the raw EMG signal is filtered and rectified. Moreover, AUCEMG is normalized such that AUCEMG values are expressed as a percentage change from the pretest value.
For neurological evaluations, all animals were examined at pretest and approximately 4 hours and 8 hours after treatment. Evaluated parameters included level of consciousness, motor function, and eye movements. Normal versus abnormal results were recorded and graded for each major parameter at each time point. All results for all animals were normal and noted as follows: level of consciousness: –1 = alert (normal); motor function: –1 = normal; eye movements: –1 = normal fixation and following of stimulus. Hepatic function evaluation, hematology, and other clinical chemistry analyses were performed on day –4 and day 24. Renal function evaluation and urine analysis were performed on day –3 and day 25.
Genotoxicity Evaluation of AVI-4658
Bacterial reverse mutation assay
The tester strains used were the Salmonella typhimurium histidine auxotrophs TA98, TA100, TA1535, and TA1537 as described by Ames et al 25 and Escherichia coli WP2 uvrA as described by Green and Muriel. 26 Salmonella tester strains were received from Dr Bruce Ames' designated distributor, Discovery Partners International (San Diego, CA). The E coli tester strain was received from the National Collection of Industrial and Marine Bacteria (Aberdeen, Scotland). Overnight cultures were prepared by inoculating from the appropriate master plate or from the appropriate frozen permanent stock into a vessel containing ~50 mL of culture medium. To ensure that cultures were harvested in the late log phase, the length of incubation was controlled and monitored. Aroclor 1254-induced rat liver S9 was used as the metabolic activation system. The S9 was prepared from male Sprague-Dawley rats induced with a single intraperitoneal injection of Aroclor 1254, 500 mg/kg, 5 days prior to sacrifice. The S9 was prepared by and purchased from MolTox (Boone, NC). Upon arrival at BioReliance, the S9 was stored at –60°C or colder until used. Each bulk preparation of S9 was assayed for its ability to metabolize at least 2 promutagens to forms mutagenic to S typhimurium TA100. The S9 mix was prepared immediately before its use and contained 10% S9, 5 mM glucose-6-phosphate, 4 mM β-nicotinamide-adenine dinucleotide phosphate, 8 mM MgCl2, and 33 mM KCl in a 100 mM phosphate buffer at pH 7.4. The sham S9 mixture (sham mix), containing 100 mM phosphate buffer at pH 7.4, was prepared immediately before its use. To confirm the sterility of the S9 and sham mixes, a 0.5-mL aliquot of each was plated on selective agar.
In the initial toxicity-mutation assay, the maximum dose of AVI-4658 tested was 5000 μg per plate; this dose was achieved using a concentration of 50 mg/mL and a 100-μL plating aliquot. The dose levels tested were 1.5, 5.0, 15, 50, 150, 500, 1500, and 5000 μg per plate. The test article formed soluble and clear solutions in sterile water for injection from 0.015 to 50 mg/mL. Neither precipitate nor background lawn toxicity was observed. No positive mutagenic responses were observed with any of the tester strains in either the presence or absence of S9 activation.
Based on the findings of this initial toxicity-mutation assay, the maximum dose plated in the confirmatory mutagenicity assay was 5000 μg per plate. For the confirmatory mutagenicity assay, the dose levels tested were 50, 150, 500, 1500, and 5000 μg per plate.
On the day of its use, minimal top agar, containing 0.8 % agar (W/V) and 0.5 % NaCl (W/V), was melted and supplemented with L-histidine, D-biotin, and L-tryptophan solution to a final concentration of 50 μM each. Top agar not used with S9 or sham mix was supplemented with 25 mL of water for each 100 mL of minimal top agar. For the preparation of media and reagents, all references to water imply sterile, deionized water produced by the Milli-Q Reagent Water System. Bottom agar was Vogel-Bonner minimal medium E (Vogel and Bonner, 1956) containing 1.5 % (W/V) agar. Nutrient bottom agar was Vogel-Bonner minimal medium E containing 1.5 % (W/V) agar and supplemented with 2.5 % (W/V) Oxoid Nutrient Broth No. 2 (dry powder). Nutrient Broth was Vogel-Bonner salt solution supplemented with 2.5% (W/V) Oxoid Nutrient Broth No. 2 (dry powder). Each plate was labeled with a code system that identified the test article, test phase, dose level, tester strain, and activation, as described in detail in BioReliance’s Standard Operating Procedures. One-half (0.5) milliliter of S9 or sham mix, 100 μL of tester strain, and 100 μL of vehicle or test article dilution were added to 2.0 mL of molten selective top agar at 45°C ± 2°C. After vortexing, the mixture was overlaid onto the surface of 25 mL of minimal bottom agar. When plating the positive controls, the test article aliquot was replaced by a 50-μL aliquot of appropriate positive control. After the overlay had solidified, the plates were inverted and incubated for approximately 48 to 72 hours at 37°C ± 2°C. Plates that were not counted immediately following the incubation period were stored at 2°C to 8°C until colony counting could be conducted.
The condition of the bacterial background lawn was evaluated for evidence of test article toxicity by using a dissecting microscope. Precipitate was evaluated after the incubation period by visual examination without magnification.
Revertant colonies for a given tester strain and activation condition, except for positive controls, were counted either entirely by automated colony counter or entirely by hand unless the plate-exhibited toxicity.
In vitro mammalian chromosome aberration test
Chinese hamster ovary (CHO-K1) cells (repository no. CCL 61) were obtained from American Type Culture Collection (Manassas, VA). The use of CHO cells has been demonstrated to be an effective method of detection of chemical clastogens. 27 Aroclor 1254–induced rat liver S9 was used as the metabolic activation system as above. The S9 was thawed and mixed with a cofactor pool to contain 2 mM magnesium chloride, 6 mM potassium chloride, 1 mM glucose-6-phosphate, 1 mM nicotinamide adenine dinucleotide phosphate, and 20 μL S9 per milliliter medium (McCoy’s 5A serum-free medium supplemented with 100 U penicillin/mL, 100 μg streptomycin/mL, 2 mM L-glutamine, and 2.5 μg/mL amphotericin B).
A preliminary toxicity assay was performed for the purpose of selecting dose levels for the chromosome aberration assay and consisted of an evaluation of test article effect on cell growth. 28 CHO cells were seeded at approximately 5 × 105 cells/25 cm2 flask and were incubated (37°C ± 1°C in a humidified atmosphere of 5% ± 1% CO2 in air). Treatment was carried out by refeeding the flasks with 4.5 mL complete medium (McCoy’s 5A medium supplemented with 10% fetal bovine serum [FBS], 100 U penicillin/mL, 100 μg streptomycin/mL, 2 mM L-glutamine, and 2.5 μg/mL amphotericin B) for the nonactivated study or S9 reaction mixture (3.5 mL serum-free medium plus 1 mL of S9/cofactor pool) for the activated study, to which was added 500 μL dosing solution of test article in vehicle or vehicle alone. Therefore, the final concentrations of serum-free medium components were diluted by 30% and S9 cofactor pool by 80%. The cells were exposed to vehicle alone and to 1 of 9 final concentrations (0.5 to 5000 μg/mL) of the test article for 4 hours in both the presence and absence of S9 activation and for 20 hours continuously in the absence of S9 activation. After the 4-hour exposure period, the treatment medium was removed, the cells washed with calcium and magnesium-free phosphate buffered saline (CMF-PBS), and refed with 5 mL complete medium. At 20 hours after the initiation of treatment, the cells were harvested by trypsinization and counted using a Coulter counter. Cell viability was determined by trypan blue dye exclusion.
The chromosome aberration assay was performed using standard procedures. 29 The CHO cells were seeded and treated as above. In the absence of both test article precipitation in the treatment medium and at least 50% toxicity, the highest dose level evaluated was 5000 μg/mL. After the exposure period, the treatment medium was removed, the cells washed with CMF-PBS, and refed with complete medium. Two hours prior to the scheduled cell harvest, Colcemid was added to duplicate flasks for each treatment condition at a final concentration of 0.1 μg/mL, and the flasks were returned to the incubator until cell collection. A concurrent toxicity test was conducted in both the nonactivated and the S9-activated test systems. Two hours after the addition of Colcemid, metaphase cells were harvested for both the nonactivated and S9-activated studies by trypsinization. Cells were collected and chromosome sample slides prepared and analyzed as described previously. 30 Slides were coded using random numbers by an individual not involved with the scoring process. Mitomycin C was used as the positive control in the nonactivated study at final concentrations of 0.1 and 0.2 μg/mL. Cyclophosphamide was used as the positive control in the S9-activated study at final concentrations of 10 and 20 μg/mL.
Mouse bone marrow micronucleus test
ICR mice were obtained from Harlan (Frederick, MD). At the time of use, the mice were 6 to 8 weeks old.
The mice were observed each day for signs of illness and other conditions of poor health. All mice were judged to be healthy prior to utilization in the study.
The micronucleus study was conducted using established and validated procedures. 31 Following an initial dose-range–finding study to determine the high dose, mice in the definitive micronucleus study were assigned to 7 experimental groups (5 of which were killed at 24 hours and 2 groups that were extended to 48 hours) of 5 males and 5 females each. The study design is shown in Table 2.
Mouse Bone Marrow Micronucleus Test Design
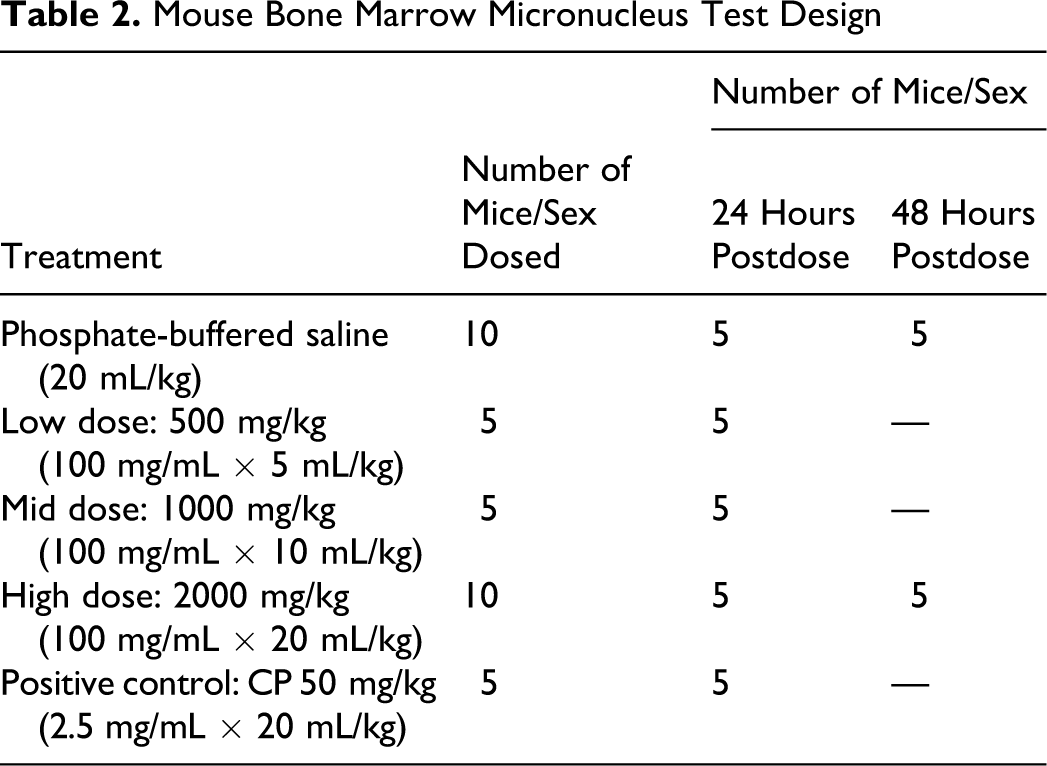
AVI-4658 was administered at a dose volume of up to 20 mL/kg by a single intravenous injection into the lateral tail vein by using 1 mL disposable polypropylene syringes with hypodermic needles (27G). At the scheduled bone marrow collection time, 5 mice per sex per treatment were euthanized by CO2 asphyxiation. Immediately following euthanasia, the femurs were exposed, cut just above the knee, and the bone marrow was aspirated into a syringe containing FBS. The bone marrow cells were transferred to a labeled centrifuge tube containing approximately 1 mL FBS. The tubes were identified by labels containing the study, group, and animal numbers. The bone marrow cells were pelleted by centrifugation at approximately 100g for 5 minutes, and the supernatant was drawn off, leaving a small amount of serum with the remaining cell pellet. The cells were resuspended, and a small drop of bone marrow suspension was spread onto a clean glass slide. Two slides were prepared from each mouse and fixed in methanol. One set of slides was stained with May-Gruenwald-Giemsa stain and permanently mounted and used in microscopic evaluation. Using a light microscope and a medium magnification (400×), an area of acceptable quality was selected such that the cells were well spread and stained.
Using oil immersion (1000×), the following cell populations and cellular components were evaluated and enumerated: polychromatic erythrocytes (PCEs), normochromatic erythrocytes (NCEs), and micronuclei. Two thousand PCEs per mouse were scored for the presence of micronuclei, resulting in evaluation of a total of 10 000 PCEs per each treatment group. To control for bias, bone marrow slides were coded using a random number table by an individual not involved with the scoring process. The number of NCEs and micronucleated NCEs (MNCEs) in the field of 1000 total erythrocytes (ECs) was determined for each animal to determine the proportion of polychromatic erythrocytes to total erythrocytes (PCEs/ECs). Micronuclei are round, darkly staining nuclear (chromosome) fragments with a sharp contour and diameters usually from 1/20 to 1/5 of an erythrocyte. Micronuclei may occur in PCEs (MPCEs) or NCEs (MNCEs). The proportion of PCEs/ECs was also recorded per 1000 ECs per each animal.
Statistical significance was determined using the Kastenbaum-Bowman tables, which are based on the binomial distribution. 32 All analyses were performed separately for each sex and sampling time. To quantify the proliferation state of the bone marrow as an indicator of bone marrow toxicity, the proportion of PCEs/ECs was determined for each mouse and treatment group. The proportion of PCEs/ECs in test article–treated animals should not be less than 20% of the control value. The test article would have been considered to induce a positive response if a dose-responsive increase in the incidence of MPCEs is observed and 1 or more doses are statistically elevated relative to the vehicle control (P < .05, Kastenbaum-Bowman tables) at any sampling time.
Results
Safety Pharmacology Evaluation of AVI-4658
AVI-4658 administration did not affect the health status and body weights of animals throughout the study period. Furthermore, no injection site reactions were detected following any of the subcutaneous or intravenous administrations.
Various cardiovascular parameters were measured. AVI-4658 injected intravenously at a high dose of 320 mg/kg had no effect on either arterial, systolic, or diastolic blood pressure; heart rate, QT and QTc interval durations (Figure 2 ); or RR and PR intervals and QRS complex durations (data not shown). Similarly, subcutaneous administration had no effect on any of these parameters (data not shown). These data show that single, high-dose administrations of the AVI-4658 PMO compound do not alter key heart function parameters.
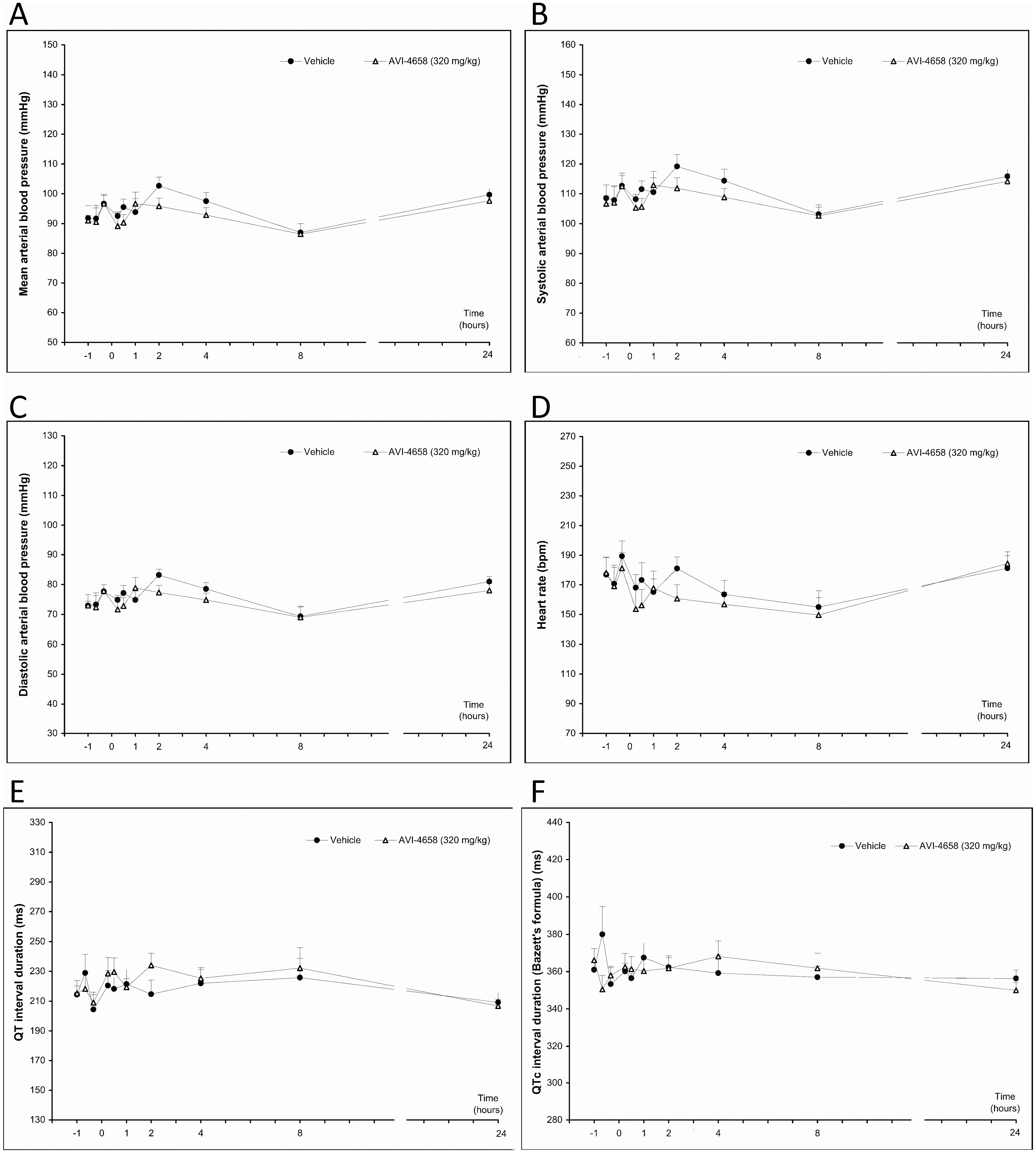
Evaluation of cardiac parameters following intravenous injections of AVI-4658 at 320 mg/kg or placebo in same animals. Parameters were measured up to 24 hours following injection, including (A) mean arterial blood pressure, (B) systolic blood pressure, (C) diastolic blood pressure, (D) heart rate, (E) QT interval, and (F) QTc interval.
Intravenous and subcutaneous injections had no effect on the respiratory rate, inspiratory rate, expiratory rate, or AUCEMG (Figure 3 ; subcutaneous data not shown); AUCEMG is a marker of tidal volume compared with the pretest values. These data demonstrated the pulmonary safety of AVI-4658, even at high doses. No neurological changes, including level of consciousness, motor function, and eye movements, were detected following any dose administration of AVI-4658.
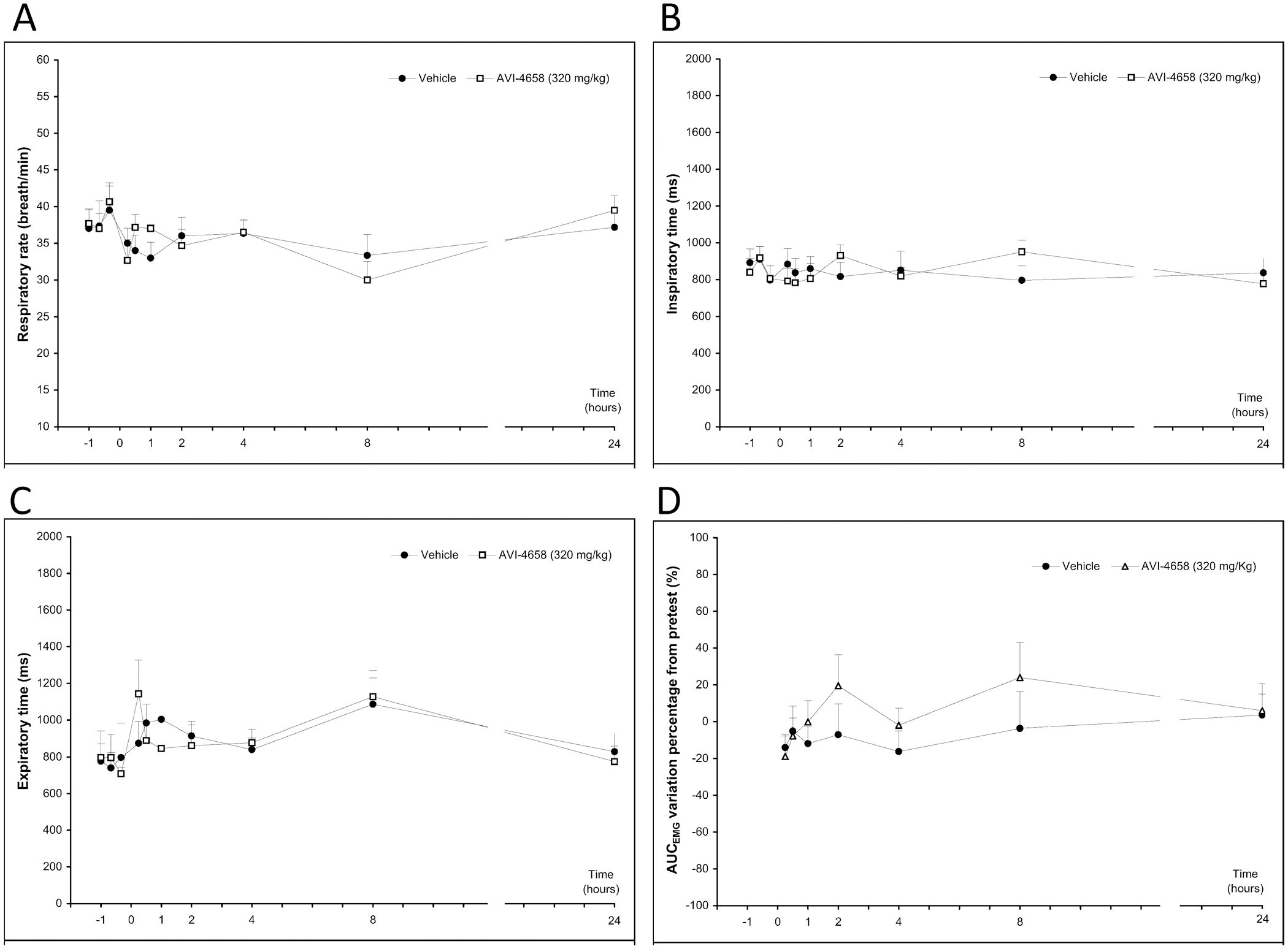
Evaluation of respiratory parameters following intravenous injections of AVI-4658 at 320 mg/kg or placebo in same animals. Parameters were measured up to 24 hours following injection, including (A) respiratory rate, (B) inspiratory rate, (C) expiratory time, and (D) AUCEMG (a marker of tidal volume).
In addition to the evaluations listed above, the cynomolgus monkeys were evaluated for markers of renal and hepatic function; a panel of urinalysis, hematology, and serum chemistry tests were performed prior to and following the subcutaneous administration phase. The data show that AVI-4658 administration did not affect blood urea nitrogen, urine or serum creatinine, alanine aminotransferase, aspartate aminotranferase, and serum albumin (Figure 4A-D ). The glomerular filtration rate and free water clearance were not affected (Figure 4E), nor were key hematological parameters, including activated partial thromboplastin time and prothrombin time, affected (Figure 4F). Additional urine, hematology, and serum chemistry parameters also showed no alterations as a result of AVI-4658 administration.
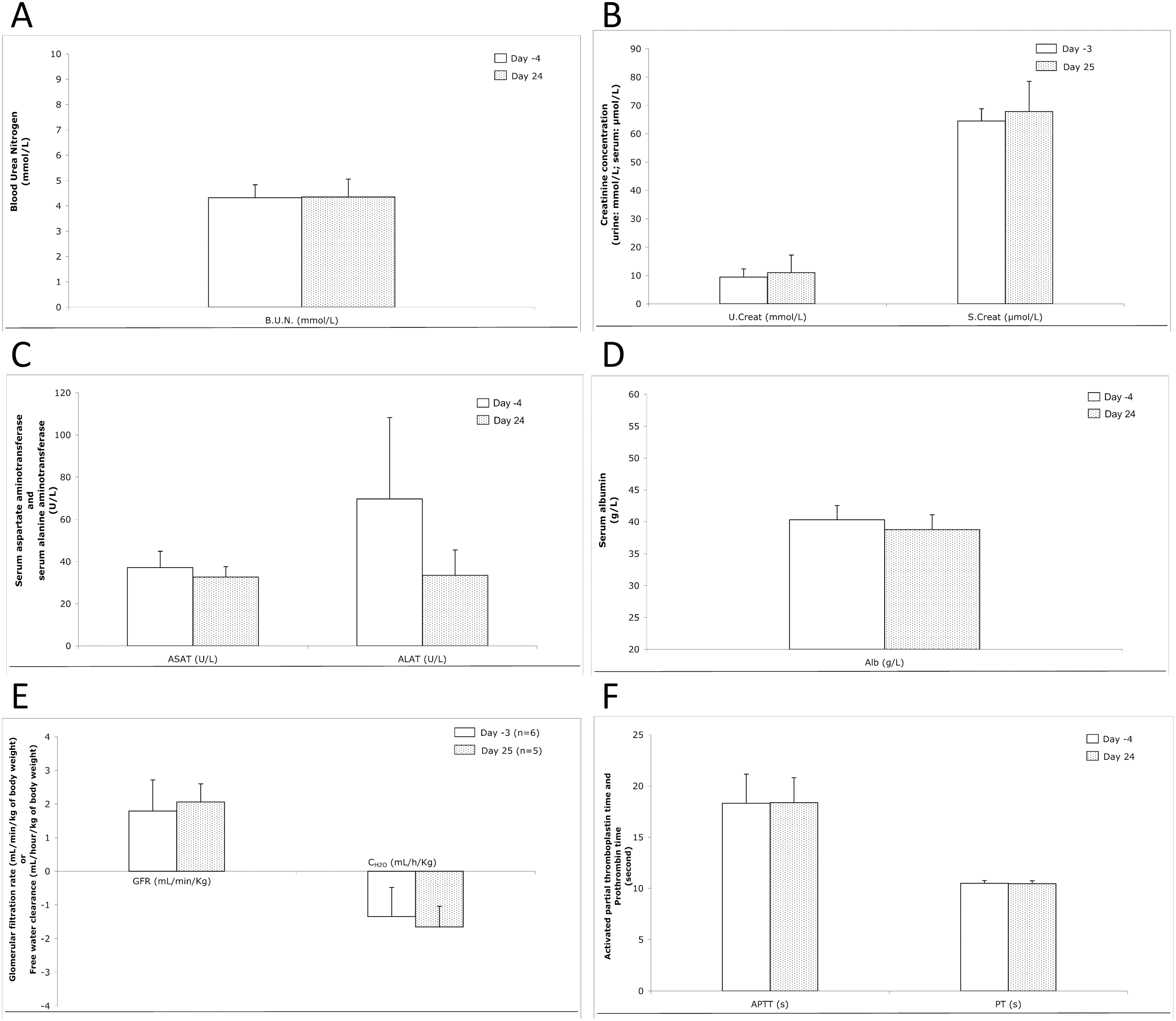
Evaluation of hepatic and renal function as well as serum chemistry and hematology following subcutaneous injection of AVI-4658 up to 320 mg/kg. Parameters were measured up to 24 hours following injection, including (A) blood urea nitrogen (BUN), (B) urine and serum creatinine, (C) aspartate aminotranferase (AST) and alanine aminotransferase (ALT), (D) serum albumin, (E) glomerular filtration rate (GFR) and water clearance, and (F) activated partial thromboplastin time (APTT) and prothrombin time (PT).
Genotoxicity Evaluation of AVI-4658
Bacterial reverse mutation assay
AVI-4658 was tested in the bacterial reverse mutation assay using S typhimurium tester strains TA98, TA100, TA1535, and TA1537 and E coli tester strain WP2 uvrA in the presence and absence of Aroclor-induced rat liver S9. In the initial toxicity-mutation assay, doses from 1.5, 5.0, 15, 50, 150, 500, 1500, and 5000 μg per plate were tested. In the initial toxicity-mutation assay, no positive mutagenic response, precipitate, or background lawn toxicity were observed (data not shown). Based on the findings of the initial toxicity-mutation assay, the maximum dose plated in the confirmatory mutagenicity assay was 5000 μg per plate. In this assay, no positive mutagenic response was observed at the dose levels tested of 50, 150, 500, 1500, and 5000 μg per plate. For all test strains, the numbers of revertant colonies on plates treated with AVI-4658 at any dose were similar to those in the vehicle control, both in the presence and absence of metabolic activation by rat liver S9 (Table 3). The positive control used in each tester strain induced significantly higher numbers of revertants than either the vehicle control or any of the AVI-4658–treated groups. Neither precipitate nor appreciable toxicity was observed at any dose level.
Bacterial Mutation Assay
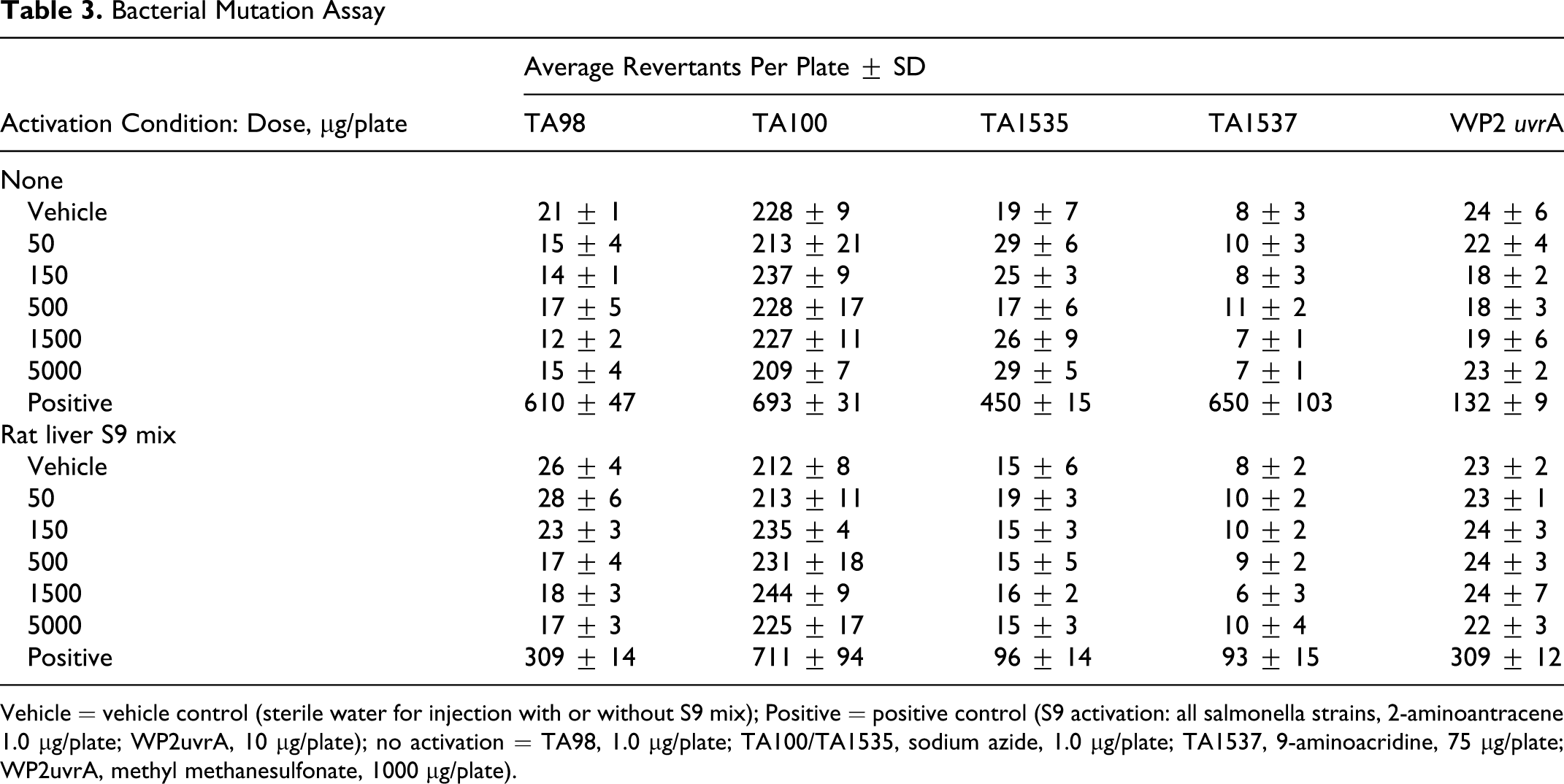
Vehicle = vehicle control (sterile water for injection with or without S9 mix); Positive = positive control (S9 activation: all salmonella strains, 2-aminoantracene 1.0 μg/plate; WP2uvrA, 10 μg/plate); no activation = TA98, 1.0 μg/plate; TA100/TA1535, sodium azide, 1.0 μg/plate; TA1537, 9-aminoacridine, 75 μg/plate; WP2uvrA, methyl methanesulfonate, 1000 μg/plate).
Data sets for tester strains TA1535/TA1537 and TA98/TA100/WP2 uvrA were judged positive if the increase in mean revertants at the peak of the dose response was equal to or greater than 3.0 or 2.0 times the mean vehicle control value, respectively. In our tests, only the respective positive control treatments met the criteria for a positive increase in mean revertants compared with historical control data.
In vitro mammalian chromosome aberration test
AVI-4658 was tested in the chromosome aberration assay using CHO cells in both the absence and presence of an Aroclor-induced S9 activation system. Cytotoxicity was determined by the population doubling method. In the preliminary toxicity assay, the maximum dose tested was 5000 μg/mL, and no substantial toxicity (ie, at least 50% cell growth inhibition, relative to the vehicle control) was observed at any dose level (not shown). Based on these findings, the doses chosen for the chromosome aberration assay ranged from 1250 to 5000 μg/mL for all 3 treatment conditions.
In the chromosome aberration assay, the cells were treated for 4 and 20 hours in the nonactivated test system and for 4 hours in the S9-activated test system. All cells were harvested 20 hours after treatment initiation. The results (Table 4) show that the percentage of cells with structural or numerical aberrations in the test article–treated groups was not significantly increased relative to vehicle control at any dose level (P > .05, Fisher exact test). Based on the findings of this study, AVI-4658 was concluded to be negative for the induction of structural and numerical chromosome aberrations in CHO cells in both nonactivated and S9-activated test systems.
In Vitro Mammalian Chromosome Aberration Test
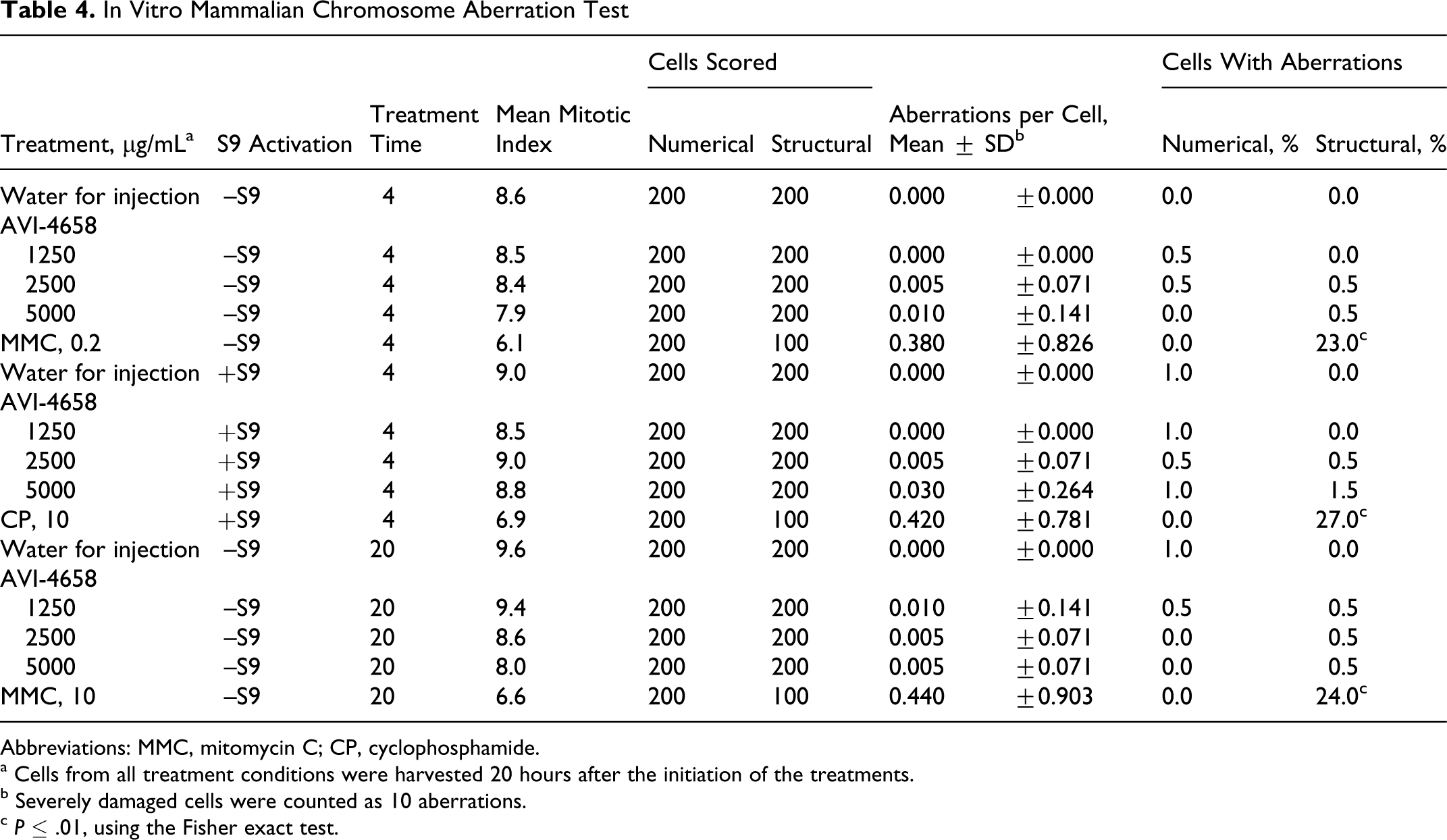
Abbreviations: MMC, mitomycin C; CP, cyclophosphamide.
a Cells from all treatment conditions were harvested 20 hours after the initiation of the treatments.
b Severely damaged cells were counted as 10 aberrations.
c P ≤ .01, using the Fisher exact test.
Mouse bone marrow micronucleus test
The mouse micronucleus assay was performed in 2 phases. In the first phase, the dose-range–finding study was designed to investigate the toxicity of the test article and to set dose levels for the definitive micronucleus study. The second phase, the definitive micronucleus study, was designed to evaluate the potential of the test article to increase the incidence of micronucleated polychromatic erythrocytes in the bone marrow of male and female ICR mice.
In the dose-range–finding study, groups of 5 male and 5 female mice were exposed to AVI-4658 up to 2000 mg/kg intravenously. The highest dose was achieved by administration of a stock formulation (100 mg/mL) at a volume of 20 mL/kg. No mortality occurred at 2000 mg/kg, and only piloerection was noted in all the animals within an hour of dose administration. In the absence of mortality, the highest dose for the definitive micronucleus study was set at 2000 mg/kg.
The definitive micronucleus study consisted of 7 groups, each containing 5 male and 5 female mice. Mice in 5 of these groups were treated either with the controls (vehicle or positive) or with AVI-4658 at 500, 1000, or 2000 mg/kg and were euthanized 24 hours after treatment (see the Materials and Methods section). The remaining 2 groups were dosed with either control or 2000 mg/kg and euthanized 48 hours after treatment. At the time of euthanasia, femoral bone marrow was collected, and bone marrow smears (slides) were prepared and stained with May-Gruenwald-Giemsa stain. Bone marrow cells (PCEs) were examined microscopically for the presence of micronuclei (MPCEs). A statistical analysis of data was performed using the Kastenbaum-Bowman tables (binomial distribution, P ≤ .05). The incidence of MPCEs and the ratio of PCEs/ECs served as indication of test article clastogenicity and cytotoxicity, respectively. The results are summarized in Table 5.
Micronucleus Test
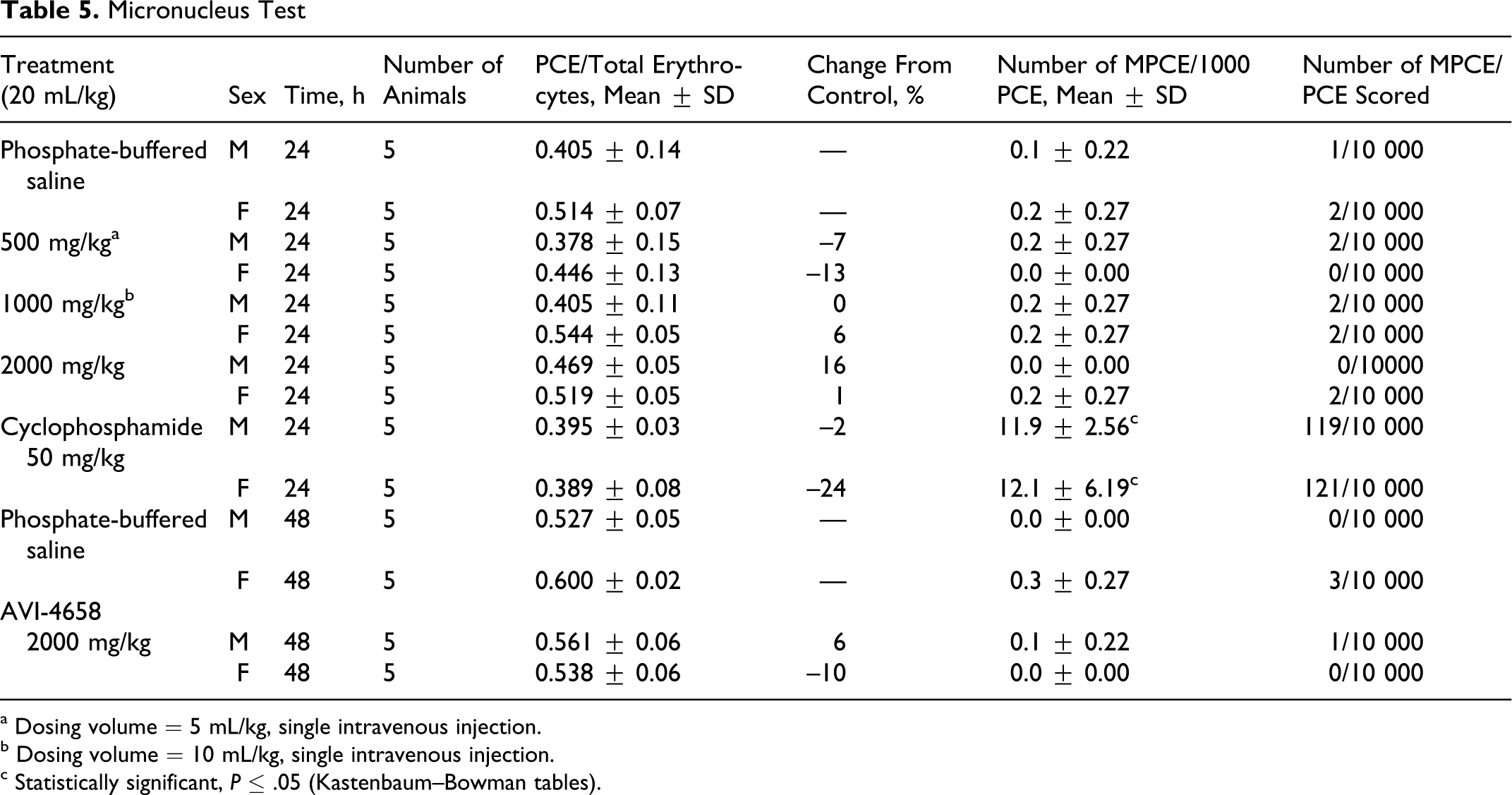
a Dosing volume = 5 mL/kg, single intravenous injection.
b Dosing volume = 10 mL/kg, single intravenous injection.
c Statistically significant, P ≤ .05 (Kastenbaum–Bowman tables).
Slight reductions in the PCEs/ECs ratio, up to 13%, were observed in some of the AVI-4658 groups at 24 or 48 hours after dose administration relative to the vehicle control groups. However, the effects were not dose dependent, and reductions of this magnitude suggest that the test article did not inhibit erythropoiesis. No statistically significant increase in the incidence of MPCEs in the AVI-4658–treated groups was seen relative to the respective vehicle control groups in male or female mice at 24 or 48 hours after dose administration (P > .05, Kastenbaum-Bowman tables).
Cyclophosphamide, the positive control, induced a statistically significant increase in the incidence of MPCEs (P ≤ .05, Kastenbaum-Bowman tables) in both male and female mice. The number of MPCEs in the vehicle control groups did not exceed the historical vehicle control range. Therefore, the controls used in this test were valid and showed that a single intravenous injection of AVI-4658 at doses up to and including 2000 mg/kg did not induce a significant increase in the incidence of MPCEs in bone marrow of male and female ICR mice.
Discussion
The therapeutic potential of DMD-applicable PMO SSOs has been demonstrated in vivo, in both the dystrophic mdx mouse model 33 and a canine model, CXMD beagle. 34 Importantly, in a head-to-head comparison between 2′OMe and PMO in the mdx mouse, PMOs were shown to be substantially more potent than the corresponding 2′OMe phosphorothioate compound following systemic administration at similar doses. 35
AVI-4658 is a PMO developed to restore dystrophin in certain subsets of DMD patients. In cynomolgus monkeys, following subcutaneous or intravenous administration, no test article–related effects were seen on arterial blood pressure, heart rate, ECG, respiratory parameters, global neurological activity, or renal and liver functions at the maximum feasible dose (320 mg/kg). No mutagenicity was observed in the bacterial reverse mutation assay, a CHO chromosome aberration assay, or a mouse bone marrow micronucleus assay.
Based on these data, a proof-of-concept clinical trial to evaluate the safety and efficacy of AVI-4658, delivered by intramuscular injection in DMD boys, was recently performed in the United Kingdom. 36 In this study, DMD patients, subdivided into 2 groups, received 0.09 mg (2 boys) or 0.9 mg (5 boys) of AVI-4658 in 900 μL of normal saline in one of their extensor digitorum brevis (EDB) muscles and 900 μL normal saline in the contralateral EDB. Both EDB muscles were biopsied either at 3 or 4 weeks following the injections. All safety evaluations showed no adverse events related to administration of AVI-4568. Furthermore, specific dose-dependent exon skipping was clearly demonstrated in the treated EDB muscles compared with the contralateral saline-injected muscles. Strong dystrophin production was clearly evident in the treated muscle in all individuals in the high-dose cohort. Dystrophin sarcolemmal localization suggests appropriate interaction with other members of the DGC complex.
AVI has now initiated a dose-ranging clinical study in the United Kingdom, in ambulant DMD patients in whom the safety and efficacy of repeated doses of AVI-4658, delivered intravenously, is being assessed. Enrollment is planned for up to 24 subjects in 6 cohorts. Each subject will be treated once weekly for 12 weeks and will undergo a muscle biopsy 2 weeks after the last administration. Subjects will receive AVI-4658 at 0.5, 1.0, 2.0, 4.0, 10.0, or 20.0 mg/kg per injection. Overall, this study will explore a wide (40-fold) dose range, up to relatively high doses, to thoroughly explore and understand the nature of the dose response, clinically, by assessment of expression of dystrophin and improvement of the condition in patients. The animal safety studies reported here were crucial in enabling such a study and in demonstrating the safety of AVI-4658 and the PMO class of compounds in general. Once a safe and effective dose has been established in the clinic, further confirmatory clinical studies are planned in both Europe and the United States.
Clinicians have recently tested a single 0.9-mg intramuscular dose of 2′OMe-based SSO (with a negatively charged backbone) targeted to exon 51 in 4 DMD patients. 37 As with the AVI-4658 intramuscular study, analysis of the biopsies from drug-injected tibialis anterior muscle showed evidence of accurate skipping of exon 51 and evidence of dystrophin protein expression. These same researchers are now performing an additional clinical study to investigate 2′OMe oligonucleotides following subcutaneous injections of up to 6 mg/kg for 5 weeks and recently reported dystrophin expression, although this was not quantified, with limited clinical benefit. 38 The reasons for not escalating the doses further in this study were not clear but may be related to concerns over the dose-limiting toxicities seen in preclinical studies with phosphorothioate compounds (reviewed in by Rayburn et al 13 ). Further preclinical results with the 2′OMe oligonucleotides are needed before determining whether the dose-limiting toxicities of phosphorothioates will inhibit their utility in the treatment of DMD or other exon-skipping amenable diseases, especially considering the lack of preclinical toxicity seen with the AVI-4658 PMO. To help inform decisions as to which of the 2 chemistries may better serve DMD boys and be appropriate for lifelong administration, the results contained in this article may be especially important if clinical studies for both chemistries are planned to run simultaneously.
The favorable safety profile of the uncharged PMOs has been remarkably consistent and predictable, as more than 460 patients have been safely dosed with PMO in clinical trials, for a variety of indications. This body of knowledge suggests that AVI–4658 will have the clinical safety profile characteristic of other PMO class drugs and is in direct contrast to phosphoroathioate compounds, which show class effect dose-limiting toxicities that can include mortality in preclinical testing. Importantly, the safety of PMO drugs will allow for a thorough exploration of the clinical dose response of AVI-4658, while clinical studies define the effective dose that may offer the first effective treatment of the lethal genetic childhood disease of DMD.
Footnotes
Acknowledgments
We would like to thank study directors O. Boucheix of MDS Pharma and V. O. Wagner, R. Gudi, and L. Krsmanovic of BioReliance for their efforts in performance of the safety pharmacology and bacterial reverse mutation assay, chromosome aberration test, and mouse micronucleus evaluation of AVI-4658, respectively. We would also like to thank R. Kole, S. Stadnicki, and P. Medeiros for their critical evaluation of this article.
The authors declared no potential conflicts of interests with respect to the authorship and/or publication of this article.
The authors declared that they received no funding with respect to the authorship and/or publication of this article.