
Abstract
A battery of toxicological studies was conducted to investigate the genotoxicity and repeated-dose oral toxicity of creatyl-
Keywords
Introduction
Creatine (N-[aminoiminomethyl]-N-methyl glycine) and leucine occur naturally in foods such as meats (beef, chicken, and pork), seafood, and dairy products, with leucine also being found in some grains, soybeans, nuts, beans, seaweed, and sesame. 1 –3 In humans, 95% of creatine is found in skeletal muscle, with the remaining 5% found in the heart, brain, testes, retina, and other tissues. 4,5 Creatine is known for its use in fitness supplements and is an important contributing compound in cellular energy production systems, specifically that of creatine kinase/phosphocreatine. 4,6 Leucine is a branched chain amino acid involved in the regulation of skeletal muscle protein synthesis as well as influencing other physiological processes such as enabling adenosine triphosphate generation, insulin secretion from the pancreatic β-islet cells, and activation of cell signaling pathways. 2,7
Creatine and leucine are not known to naturally form bonds with one another endogenously in humans or in nature. Creatyl-
Materials and Methods
Chemicals
All chemical reagents, solvents, pharmaceuticals, and other chemicals used in the studies were of analytical or pharmaceutical grade. Agar bacteriological and nutrient broth number 2 were purchased from Oxoid Ltd (Hampshire, England); minimal glucose agar (MGA) plates were sourced from Merck Life Science GmbH (Eppelheim, Germany); biotin,
Test Article
Creatyl-
The homogeneity, concentration, and stability of the test article in ultrapure water (American Society for Testing and Materials type 1: prepared in the laboratory) were determined in a separate validated assay conducted according to Organization for Economic Co-operation and Development (OECD) Principles Good Laboratory Practice (GLP), C(97)186/Final, 12 Guidance for Industry Q2(R1) Validation of Analytical Procedures: Text and Methodology (November 2005 International Council for Harmonization [ICH]), 13 Santé et Consommateurs (Directorate General Health and Consumers; European Commission) Guideline 3030/99rev.4, July 11, 2000 (annex II [part A, section 4] and annex III [part A, section 5] of Directive 91/414/EEC), 14 and Harmonized Guidelines for Single-Laboratory Validation of Methods of Analysis (2002 International Union of Pure Applied Chemistry). 15
The test article was stable up to 250 mg/mL at room temperature for 1 day and in a refrigerator (5°C ± 3°C) for 3 days. Because the 90-day study employed repeated dosing, the test solutions were prepared and kept refrigerated no longer than 3 days before administration, and in addition to the up-front verification of vehicle suitability, analytical control of the formulations was performed 3 times (prior to initiation and again at weeks 7 and 12) during the study.
In Vitro Studies
Bacterial reverse mutation test
The bacterial reverse mutation test was conducted to investigate the potential mutagenic activity of CLL by measuring its ability to induce reverse mutations at selected loci of Salmonella typhimurium strains TA98, TA100, TA1535, and TA1537 and Escherichia coli strain WP2 uvrA (Moltox, Inc) in the presence and absence of activated rat liver S9. The study was performed following methods previously described by Ames et al, 16 Maron and Ames, 17 Kier et al, 18 and Venitt and Parry 19 and according to the following internationally accepted guidelines and recommendations: OECD Guideline 471 (July 21, 1997), 20 ICH Guidance S2(R1) (2012), 21 Commission Regulation (EC) (No 440/2008 B13/14, May 30, 2008), 22 the Environmental Protection Agency (EPA) Health Effects Test Guidelines (Office of Prevention, Pesticides, and Toxic Substances [OPPTS] 870.5100, EPA 712-C-98-247; 1998), 23 and OECD GLP, C(97)186/Final. 12
Ultrapure water was chosen as the test article vehicle for the bacterial reverse mutation test based on a preliminary solubility test. The preliminary solubility test was conducted by examining test article mixtures of varying concentrations suspended in ultrapure water and was followed by a preliminary range finding test (cytotoxicity test) utilizing the plate incorporation method and conducted in triplicate. Six concentrations, 5,000, 1,600, 500, 160, 50, and 16 μg/plate, of the test article were selected for the initial and confirmatory tests based on the preliminary test results. Test solutions were freshly prepared with ultrapure water at the beginning of the experiments by suspending varying amounts of CLL in ultrapure water to achieve concentrations of 50, 16, 5, 1.6, 0.5, and 0.16 mg/mL for administration of 100 μL of the test solutions per plate. Strain-specific positive controls were selected according to the guidelines and prepared for the experiments.
Positive controls without metabolic activation were NPD (4 µg/plate) for TA98, SAZ (2 µg/plate) for TA100 and TA1535, 9AA (50 µg/plate) for TA1537, and MMS (2 µL/plate) for E coli WP2 uvrA. The positive control for all strains for experiments with metabolic activation was 2AA (2 and 50 µg/plate for all S typhimurium strains and E coli WP2 uvrA, respectively). Dimethyl sulfoxide served as the vehicle and negative control for NPD, 9AA, and 2AA. Ultrapure water served as the vehicle and negative control for MMS and SAZ. S9 mix sensitivity, reliability, and promutagen activation potential (ethidium bromide, cyclophosphamide, benzo(a)pyrene, and 2-AA) were verified by the supplier.
A standard plate incorporation procedure was used for the initial mutation test. Tester strains were exposed to the test article, at each concentration, and positive, negative, and untreated controls, both with and without metabolic activation, were poured onto MGA plates and incubated for 48 hours at 37°C. The confirmatory mutation test was conducted using a 20-minute preincubation procedure prior to plating. Plates were again incubated for 48 hours at 37°C. All experiments were conducted in triplicate.
Colony numbers were determined by manually counting; from this, mean values, standard deviations, and mutation rates were calculated. A result was considered positive if: a dose-related increase in revertant colonies occurred, and/or a reproducible biologically relevant positive response for at least 1 dose group occurred in at least 1 strain with or without metabolic activation.
An increase was considered biologically relevant if: a reproducible increase in revertants at least twice as high as the reversion rate of negative controls occurred in strains TA98, TA100, and/or E coli WP2 uvrA and/or a reproducible increase in revertants at least 3 times higher than the reversion rate of negative controls occurred in strains TA1535 and/or TA1537.
A result was considered negative if neither of the above criteria were met.
In vitro mammalian chromosomal aberration test
The in vitro mammalian chromosomal aberration test was performed to determine whether CLL or its metabolite(s) could induce structural chromosomal aberrations. It was performed in accordance with internationally accepted guidelines: OECD 473 (2014), 24 EPA Health Effects Test Guidelines OPPTS 870.5375, 25 and OECD GLP, C(97)186/Final. 12
Dulbecco modified Eagle medium was used as the solvent for the test article due to its compatibility with the test system (V79 male Chinese hamster lung cells; European Collection of Cell Cultures, Salisbury, United Kingdom) and the availability of historical control data. The stock solution was prepared by dissolving CLL in DME medium to achieve a concentration of 50 mg/mL. Test solutions were freshly prepared at the beginning of each experiment by diluting stock solution with DME medium to achieve concentrations of 500, 1,000, and 2,000 µg/mL.
The positive control for use without metabolic activation was prepared by dissolving EMS in DME medium to achieve concentrations of 0.4 and 1.0 µg/mL, and the positive control for use with metabolic activation was prepared by dissolving cyclophosphamide in DME medium to achieve a concentration of 5.0 µg/mL. The DME medium served as the negative control. The use of EMS as a positive control is a deviation from the guideline and EMS was chosen as a positive control because it is a known, widely used mutagen and clastogen and the test facility has a broad historical database.
A cytotoxicity assay was conducted as a pretest for the purpose of selecting concentrations for the main test, experiments A and B. The pretest was conducted using the same procedures described below for the main test. Based on the cell counts, relative increase in cell counts (RICC), an indicator of cytotoxicity, was calculated. Results obtained were used for dose selection for experiments A and B.
For experiments A and B, the test article was prepared as described above at concentrations of 500, 1,000, and 2,000 µg/mL. In experiment A, V79 cultures (5 × 105 cells/group) were then exposed to the negative control, respective positive controls, or each test article concentration for a 3-hour period at 37°C with and without S9 metabolic activation (15 mg/mL). Following the exposure period, the cells were washed with DME medium and growth medium was added. Sampling was made 20 hours after the start of treatment (approximately 1.5 normal cell cycles from the beginning of treatment).
Experiment B was conducted as described for experiment A, except that the exposure period without metabolic activation was 20 hours (exposure time with metabolic activation remained 3 hours), and sampling was made after 20 and 28 hours for groups treated without metabolic activation and after 28 hours for groups treated with metabolic activation.
The pH and osmolality of the negative control and test article treatment solutions of all concentrations were measured in all 3 experiments (pretest, experiment A, and experiment B). All individual test article and negative and positive control experiments were carried out in duplicate and concurrent measures of cytotoxicity were also set up and assessed.
Chromosomes were prepared for analysis by treatment with colchicine (0.2 μg/mL) for 2.5 hours followed by harvesting, swelling with 0.075 M KCl, and washing in fixative for approximately 10 minutes before dropping onto slides, air drying, and staining with 5% Giemsa for scoring.
Three hundred well-spread metaphase cells containing 22 ± 2 chromosomes from each experimental group were evaluated for structural aberrations (slides were coded and scored blind) and were equally divided among the duplicates (150 metaphases/slide). Chromatid and chromosome type aberrations (gaps, deletions, and exchanges) were recorded separately. Polyploid and endoreduplicated cells were also scored. Nomenclature and classification of chromosomal aberrations were based on publications by International System for Human Cytogenic Nomenclature (1985) 26 and Savage (1975) 27 and Savage (1983). 28
There was a deviation to the OPPTS guideline regarding choice of the study top dose as 2,000 μg/mL. The dose was chosen according to updated guideline, OECD 473 (2014). The deviation did not influence the quality or integrity of the study.
Animal Studies
In vivo mammalian micronucleus test
The in vivo mammalian micronucleus test was conducted to investigate the genotoxic potential of CLL to cause the formation of micronuclei in erythrocytes of the bone marrow of treated animals. The study was conducted in compliance with OECD 474 (2014), 29 US EPA OPPTS 870.5395, 30 and OECD GLP C(97)186/Final. 12 The Institutional Animal Care and Use Committee of Toxi-Coop Zrt permitted conduction of the study.
The test article was formulated in aqua ad injectabilia (water for injection, vehicle; NATURLAND KFT, Budapest, Hungary) to achieve concentrations of 200, 100, and 50 mg/mL. Test article formulations were prepared fresh and administered within 2 hours at a dose volume of 10 mL/kg bw. Cyclophosphamide served as the positive control and was dissolved in aqua ad injectabilia to achieve a concentration of 6.0 mg/mL for administration of the standard dosing volume of 10 mL/kg bw. Aqua ad injectabilia served as the negative control.
Eight-week-old, specific pathogen-free Crl:NMRI BR mice, weighing 30.8 to 34.1 g, were utilized for the study. They were acclimatized for 8 days and housed in groups (2 animals/cage in the pretest and in the high-dose group of the main test and 5 animals/cage in the other groups of the main test) in type I polypropylene/polycarbonate cages with laboratory bedding at 22°C ± 3°C, 40% to 70% relative humidity, and a 12-hour light–dark cycle. All animals received ssniff SM R/M-Z + H pellet diet (Soest, Germany) and potable water ad libitum.
A non-GLP preliminary toxicity test was performed to determine the appropriate maximum dose level for the main test and whether there were large differences in toxicity between male and female mice. A single dose of CLL was administered by gavage to 2 male and female Crl:NMRI BR mice at a concentration of 2,000 mg/kg bw. Animals were observed at regular intervals for 3 days for toxic signs and mortalities.
In the main test, male Crl:NMRI BR mice were randomly divided into groups of 5 (low- and mid-dose groups and positive control group) or 10 (negative control group and high-dose group) animals. A single dose of CLL was administered by gavage at test concentrations of 0 (negative control), 500, 1,000, and 2,000 mg/kg bw (the high dose is the limit dose for mammalian erythrocyte micronucleus tests and was selected based on the results of the preliminary acute toxicity test). The positive control, cyclophosphamide 60 mg/kg bw, was administered by intraperitoneal injection.
All animals were observed immediately after dosing and at regular intervals until sacrifice (by cervical dislocation) for mortality, visible signs of toxicity, or other reactions to treatment. At 24 hours after treatment, half of the mice from the negative control and high-dose groups and all the mice from the low-dose, mid-dose, and positive control groups were sacrificed. At 48 hours posttreatment, the remaining animals in the negative control and high-dose group were sacrificed. Bone marrow was obtained from 2 exposed femurs of the mice at each time point immediately after sacrificing. Four thousand polychromatic erythrocytes (PCEs) were scored per animal for frequency of micronuclei, with 1 slide from each animal being scored blind (original animal numbers covered with a blinding code number). The frequency of micronucleated cells was expressed as percentage of micronucleated cells based on the first 4,000 PCEs counted in the optic field. The proportion of PCE among total erythrocytes was determined for each animal by counting a total of at least 500 erythrocytes.
Ninety-day repeated-dose oral toxicity studies in rats
The 90-day study was conducted in order to evaluate the possible health hazards, including toxic effects and target organs, of repeated exposure to CLL in male and female rats and to determine the no observed adverse effect level (NOAEL). The study was conducted in compliance with OECD 408 (1998) 31 and OECD GLP, C(97)186/Final. 12 Care and use of study animals was in compliance with laboratory standard operating procedures under the permission of the laboratory’s Institutional Animal Care and Use Committee, as well as in accordance with the National Research Council Guide for Care and Use of Laboratory Animals and in compliance with the principles of the Hungarian Act 2011 CLVIII (modification of Hungarian Act 1998 XXVIII) regulating animal protection.
Specific pathogen-free Hsd.Han Wistar rats, 38 to 46 days old and weighing 143 to 165 g (males) and 109 to 132 g (females), at the start of the study, were randomly divided according to stratification by body weight into 4 groups (3 dose levels and 1 control group) of 20 animals (10 per sex per dose). Animals were acclimatized for 6 days. Animals were housed individually in type III polypropylene/polycarbonate cages in a room with 12-hour light–dark cycles, more than 10 air exchanges per hour, via central air conditioner, a temperature of 22°C ± 3°C, and a relative humidity of 30% to 70%. Animals received ssniff SM R/M-Z + H complete diet for rats and mice and potable tap water ad libitum except overnight food deprivation before blood sampling.
Doses were chosen based on an unpublished OECD 407 compliant 14-day repeated-dose oral toxicity study in Hsd.Han Wistar rats. There were no toxic changes in the examined parameters of the 14-day oral administration; therefore, a maximum tolerated dose could not be established and an NOAEL was estimated as the high dose of 5,000 mg/kg bw/d. As such, for the 90-day study, the test article was formulated in distilled water (Aqua purificata, Parma, Hungary) to achieve concentrations of 62.5, 125, and 250 mg/mL, for administration of 5,000 mg/kg bw/d as the high dose, and provide a 2-fold descending interval for the lower doses. Therefore, 10 rats/sex/group received, by gavage, once daily, doses of 0 (vehicle-control), 1,250, 2,500, and 5,000 mg/kg bw/d of the test article administered at a constant dosing volume of 20 mL/kg bw.
All animals were observed twice daily for signs of morbidity and mortality. General cage-side observations for clinical signs were made twice during the acclimation period and once daily after administration of the test article. Detailed observations were conducted weekly while handling the animal on days that the animals were weighed and food consumption measurements were taken. Using a modification of the procedure described by Irwin, 32 functional observations of sensory reactivity to various stimuli and assessments of grip strength and motor activity were made in the last exposure week.
Measurement of individual body weights was performed once during the acclimation, on day 0 (prior to the start of the study), twice weekly during weeks 1 to 4, once weekly during weeks 5 to 13, and immediately prior to sacrifice. Individual body weight changes were calculated. Food consumption was measured and feed efficiency calculated once weekly. Ophthalmological examination was performed on all rats during the acclimation period and during the last week of the study, on day 84.
After an overnight fast of approximately 16 hours, following termination of treatment, blood samples were collected from the retro-orbital venous plexus under Isofluran CP anesthesia. Three samples were taken from each animal for evaluation of hematological and clinical chemistry parameters. Gross pathological examinations and determinations of selected absolute organ weights and relative organ weights (compared to body weight and brain weight) were conducted on all animals. Complete histopathological examinations were conducted on the preserved organs and tissues of all animals of the control and high-dose groups. Any gross lesions observed at necropsy also underwent histological examination.
Statistical Analyses
Statistical analyses were conducted using SPSS PC+ software (SPSS, Inc, Chicago, Illinois). Adhered to guidelines, the bacterial reverse mutation test results were interpreted based on the criterion of biological relevance; thus, no statistical analysis was performed. For the mammalian chromosomal aberration test, the Fisher exact test and the χ2 test were utilized to evaluate the number of aberrations and the number of cells with aberrations (with and without gaps). The number of aberrations in the treatment and positive control groups was compared to the concurrent negative control, and all groups were compared to the laboratory historical controls. Statistical analysis for the mammalian micronucleus test was conducted using the Kruskal-Wallis nonparametric 1-way analysis of variance (ANOVA) test.
For the 90-day study, Bartlett homogeneity of variance test was used to assess heterogeneity of variance between groups and was followed by a 1-way ANOVA if no significant heterogeneity was detected. If the result was positive, Duncan multiple range test was used to assess the significance of intergroup differences. Where significant heterogeneity was detected, the normal distribution of data was examined by the Kolmogorov-Smirnov test; the Kruskal-Wallis nonparametric 1-way ANOVA, followed by the Mann-Whitney U test for intergroup comparisons of positive results, was used in the case of a non-normal distribution. Statistical significance was assigned when P values were <.05. Male and female rats were evaluated separately.
Results
Bacterial Reverse Mutation Test
Clear solutions were obtained in the preliminary solubility test when the test article was dissolved in ultrapure water at a concentration of 50 mg/mL and when 100 µL of the aforementioned test solution was mixed with 500 µL of phosphate buffer and 2 mL of top agar. In the preliminary concentration range-finding test, no precipitation or cytotoxicity was observed at concentrations ranging from 5,000 to 5 µg/plate with and without S9 in tester strains TA98 and TA100, and revertant colony numbers were as expected with the negative and positive controls (data not shown). Therefore, the initial and confirmatory mutation tests were conducted with ultrapure water as the test article vehicle at concentrations of 5,000, 1,600, 500, 160, 50, and 16 μg/plate.
In main tests, all tester strains exhibited the necessary specific phenotypic characteristics, culture titers were in the order of 109 cell/mL, no precipitation of the test article was observed, the concurrent negative vehicle controls remained within the range of the corresponding historical control data, and the concurrent positive controls induced the expected increases (at least 3-fold compared to corresponding negative controls) in revertant colony numbers. Numbers of revertant colonies observed in untreated controls were similar to observations in the negative controls. In the initial mutation test, revertant numbers with metabolic activation in tester strains TA98 at 5,000 µg/plate and E coli WP2uvrA at all concentrations except 50 µg/plate fell below the corresponding actual historical negative control range established in the testing laboratory; however, the background lawn development was not affected in any case, the lower values were not extreme outliers, and a dose-related inhibitory tendency was not additionally noticed. Finally, no concentration-related or biologically relevant increases in revertant colonies were observed in any tester strain at any test article concentration with or without S9 mix in the initial or confirmatory mutation tests (see Tables 1 and 2).
Bacterial Reverse Mutation Test, Initial Mutation Test.a,b
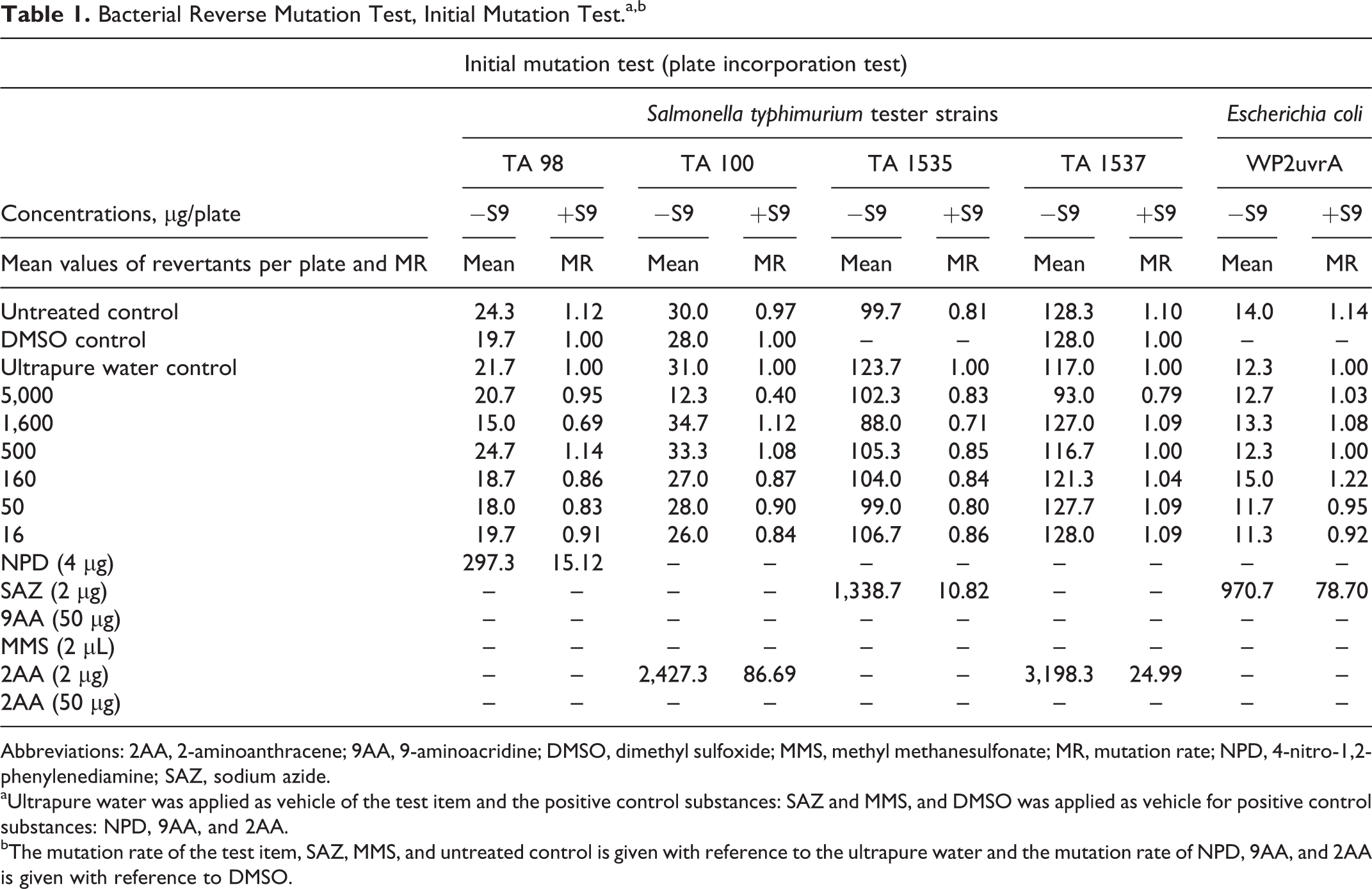
Abbreviations: 2AA, 2-aminoanthracene; 9AA, 9-aminoacridine; DMSO, dimethyl sulfoxide; MMS, methyl methanesulfonate; MR, mutation rate; NPD, 4-nitro-1,2-phenylenediamine; SAZ, sodium azide.
aUltrapure water was applied as vehicle of the test item and the positive control substances: SAZ and MMS, and DMSO was applied as vehicle for positive control substances: NPD, 9AA, and 2AA.
bThe mutation rate of the test item, SAZ, MMS, and untreated control is given with reference to the ultrapure water and the mutation rate of NPD, 9AA, and 2AA is given with reference to DMSO.
Bacterial Reverse Mutation Test and Confirmatory Mutation Test.a,b
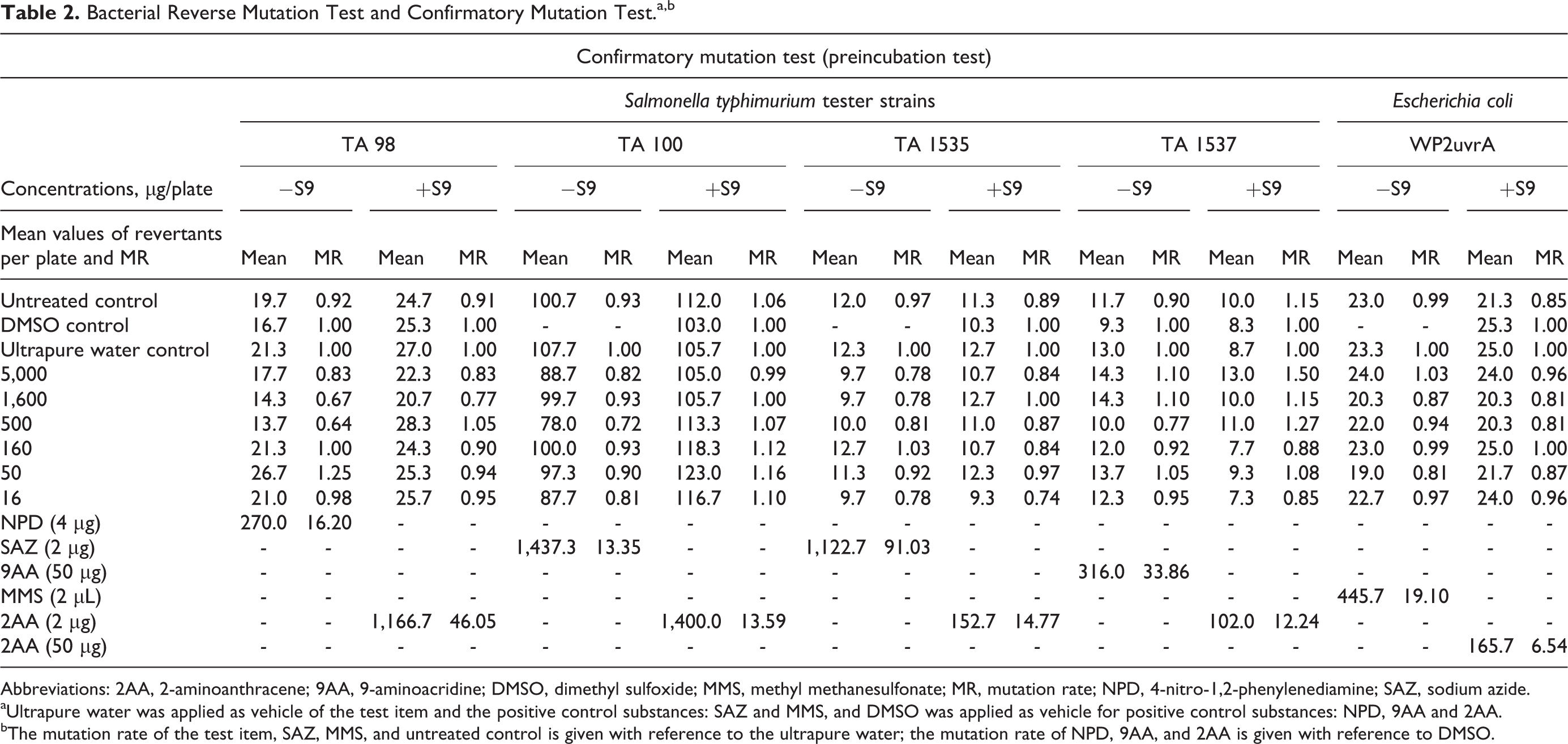
Abbreviations: 2AA, 2-aminoanthracene; 9AA, 9-aminoacridine; DMSO, dimethyl sulfoxide; MMS, methyl methanesulfonate; MR, mutation rate; NPD, 4-nitro-1,2-phenylenediamine; SAZ, sodium azide.
aUltrapure water was applied as vehicle of the test item and the positive control substances: SAZ and MMS, and DMSO was applied as vehicle for positive control substances: NPD, 9AA and 2AA.
bThe mutation rate of the test item, SAZ, MMS, and untreated control is given with reference to the ultrapure water; the mutation rate of NPD, 9AA, and 2AA is given with reference to DMSO.
In Vitro Mammalian Chromosomal Aberration Test
Creatyl-
In experiments A and B, there were no statistically significant differences between the test article treatment and vehicle or historical negative controls, with or without metabolic activation, and no dose–response relationship was observed (see Table 3). There was no increase in the rate of polyploidy or endoreduplicated metaphases after treatment with the differing concentrations of CLL. There were no significant differences in pH and osmolality between the test article treatment and control groups.
Summary of Chromosomal Aberration Test Results.
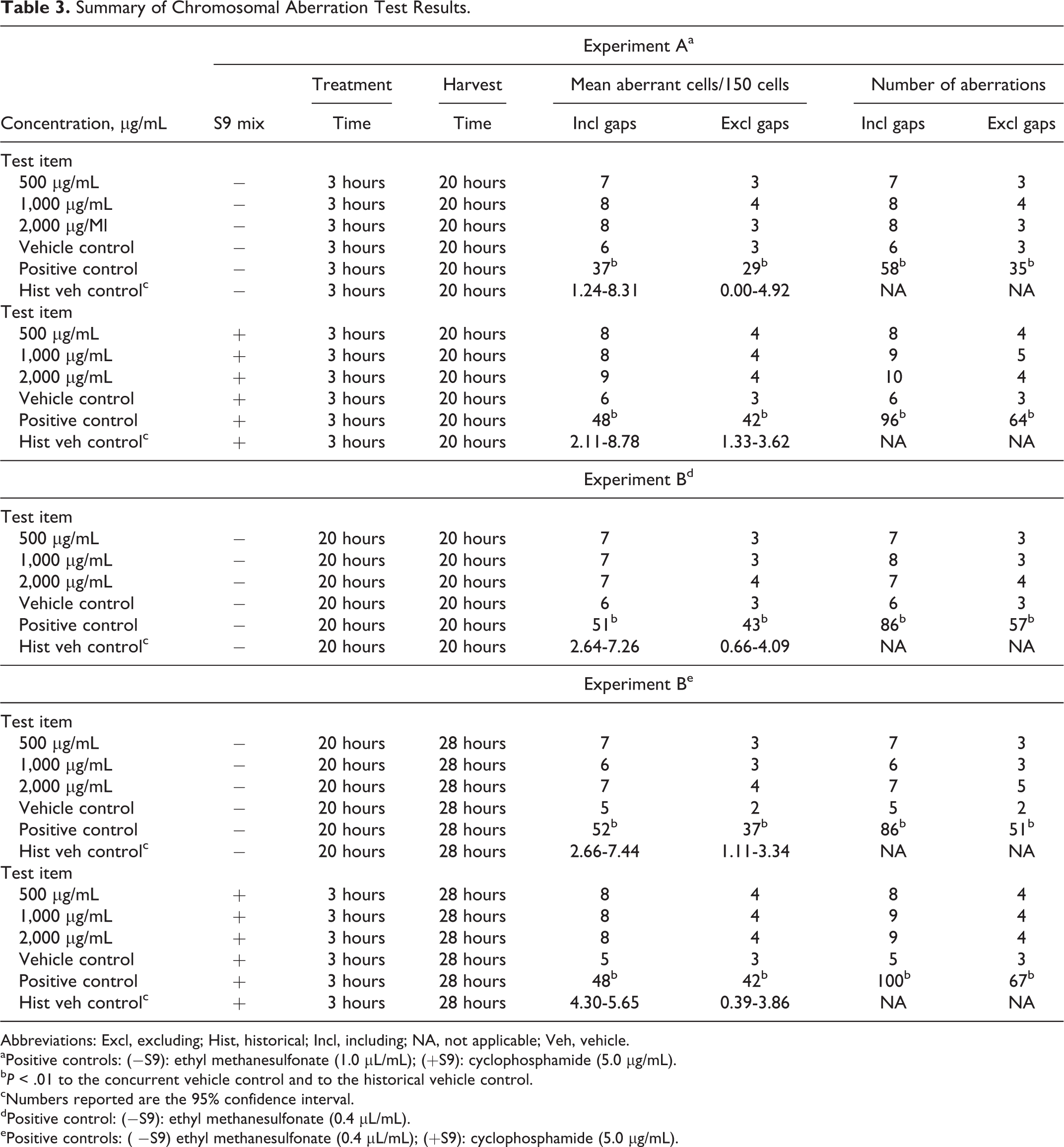
Abbreviations: Excl, excluding; Hist, historical; Incl, including; NA, not applicable; Veh, vehicle.
aPositive controls: (−S9): ethyl methanesulfonate (1.0 μL/mL); (+S9): cyclophosphamide (5.0 μg/mL).
b P < .01 to the concurrent vehicle control and to the historical vehicle control.
cNumbers reported are the 95% confidence interval.
dPositive control: (−S9): ethyl methanesulfonate (0.4 μL/mL).
ePositive controls: ( −S9) ethyl methanesulfonate (0.4 μL/mL); (+S9): cyclophosphamide (5.0 μg/mL).
In the concurrent negative control group, the percentage of cells with structural aberrations without gaps was less than 5%, confirming the suitability of the cell line used. The number of aberrations found in the solvent controls was in the range of historical control data. The concurrent positive controls caused the expected statistically significant increases of cells with structural chromosome aberrations as compared to solvent controls and were compatible with historical positive control data; thus, the study was considered valid.
In Vivo Mammalian Micronucleus Test
No mortality, signs of toxicity, or sex-specific effects were observed in the preliminary toxicity test; therefore, the micronucleus test was conducted at the doses described in males only. In the main study, no mortality, clinical signs of toxicity, or adverse reactions to treatment were observed in any animals during the study.
The single oral administration of 500, 1,000, and 2,000 mg/kg bw of CLL did not induce statistically significant increases in the frequency of micronucleated PCEs (MPCEs) in male mice at either 24 or 48 hours after the treatment compared to the concurrent negative control and historical negative control values (historical data not shown). The frequencies of MPCEs for the negative controls were within an acceptable range and compatible with the historical control data. The positive control showed a statistically significant increase in MPCE numbers compared to the concurrent and historical negative controls, demonstrating an acceptable sensitivity of the test. In the 2,000 mg/kg bw dose group, the number of PCEs was slightly decreased compared to the negative control group at the 24- and 48-hour sampling points, indicating evidence of exposure of the bone marrow to the test article (see Table 4).
Summary of Mammalian Micronucleus Test.a
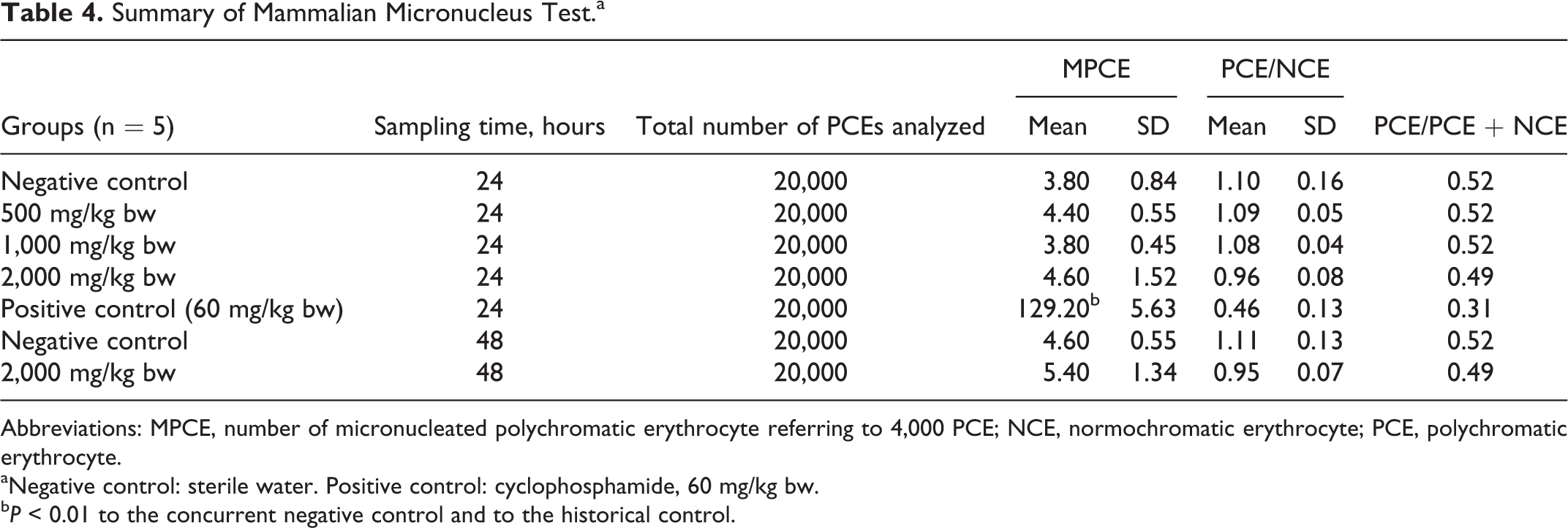
Abbreviations: MPCE, number of micronucleated polychromatic erythrocyte referring to 4,000 PCE; NCE, normochromatic erythrocyte; PCE, polychromatic erythrocyte.
aNegative control: sterile water. Positive control: cyclophosphamide, 60 mg/kg bw.
b P < 0.01 to the concurrent negative control and to the historical control.
Ninety-Day Repeated-Dose Oral Toxicity Study in Rats
There were no mortalities in the control, 1,250, 2,500, or 5,000 mg/kg bw/d groups during the 90-day (males) and 91-day (females) treatment period. Daily cage-side observations, weekly detailed clinical observations, and the functional observation battery revealed no differences between test article groups and controls. The stool in the bedding material was softer than normal in all males and females in the mid- and high-dose groups from days 9 and 6, respectively, up to the end of the treatment period. There were no changes observed on ophthalmoscopic examination of any of the treatment or control groups.
Mean body weights were similar in the male and female control and treatment groups throughout the study. Some statistically significant differences in body weight gain with respect to controls were observed at various times for the male test groups and for the intermediate-dose female test group (see Table 5). The mean daily food consumption of males in the test article groups was similar to the controls during the entire treatment period. There were statistically significant differences in food consumption for females in the 2,500 and 5,000 mg/kg bw/d groups over 7 consecutive weeks during the study and in cumulative food consumption (see Table 6). Although mean feed efficiency was similar in the 1,250 mg/kg bw/d males and females and their respective controls, there were statistically significant changes in feed efficiency in males and females in the 2,500 and 5,000 mg/kg bw/d groups, sporadically throughout the study (see Table 7).
Body Weight Gain in Males and Females in a 90-Day Repeated-Dose Oral Toxicology Study.
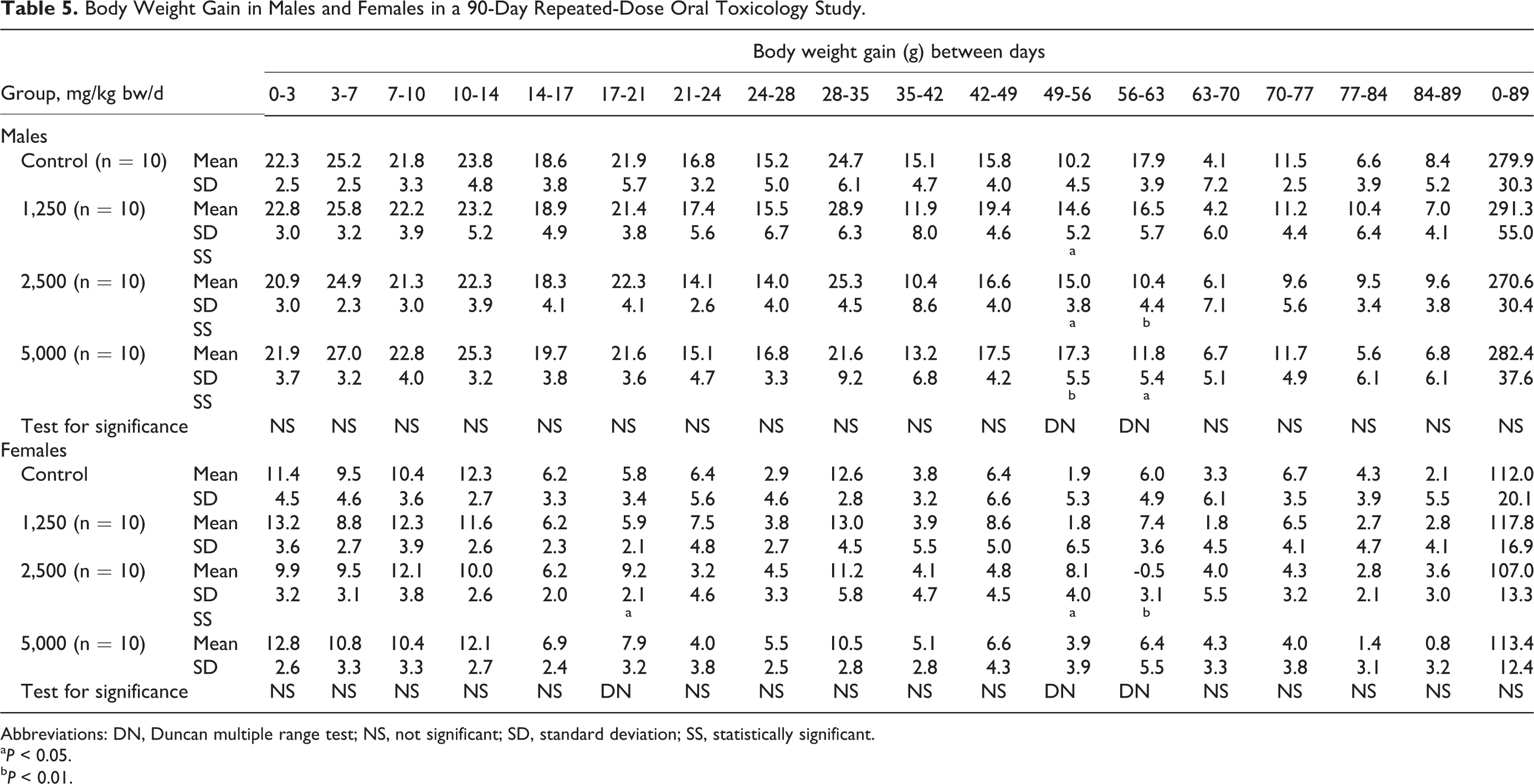
Abbreviations: DN, Duncan multiple range test; NS, not significant; SD, standard deviation; SS, statistically significant.
a P < 0.05.
b P < 0.01.
Daily Mean Food Consumption of Females.
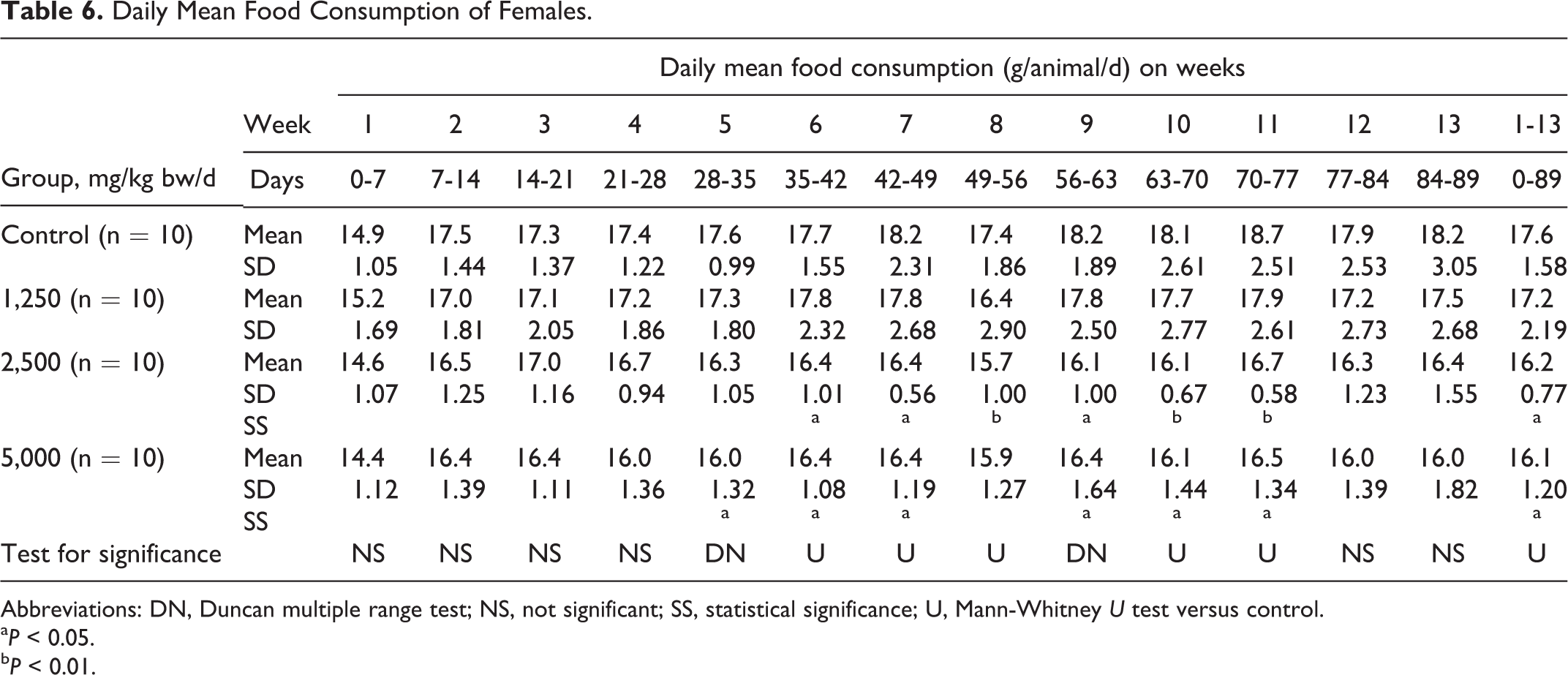
Abbreviations: DN, Duncan multiple range test; NS, not significant; SS, statistical significance; U, Mann-Whitney U test versus control.
a P < 0.05.
b P < 0.01.
Feed Efficiency for Males and Females.
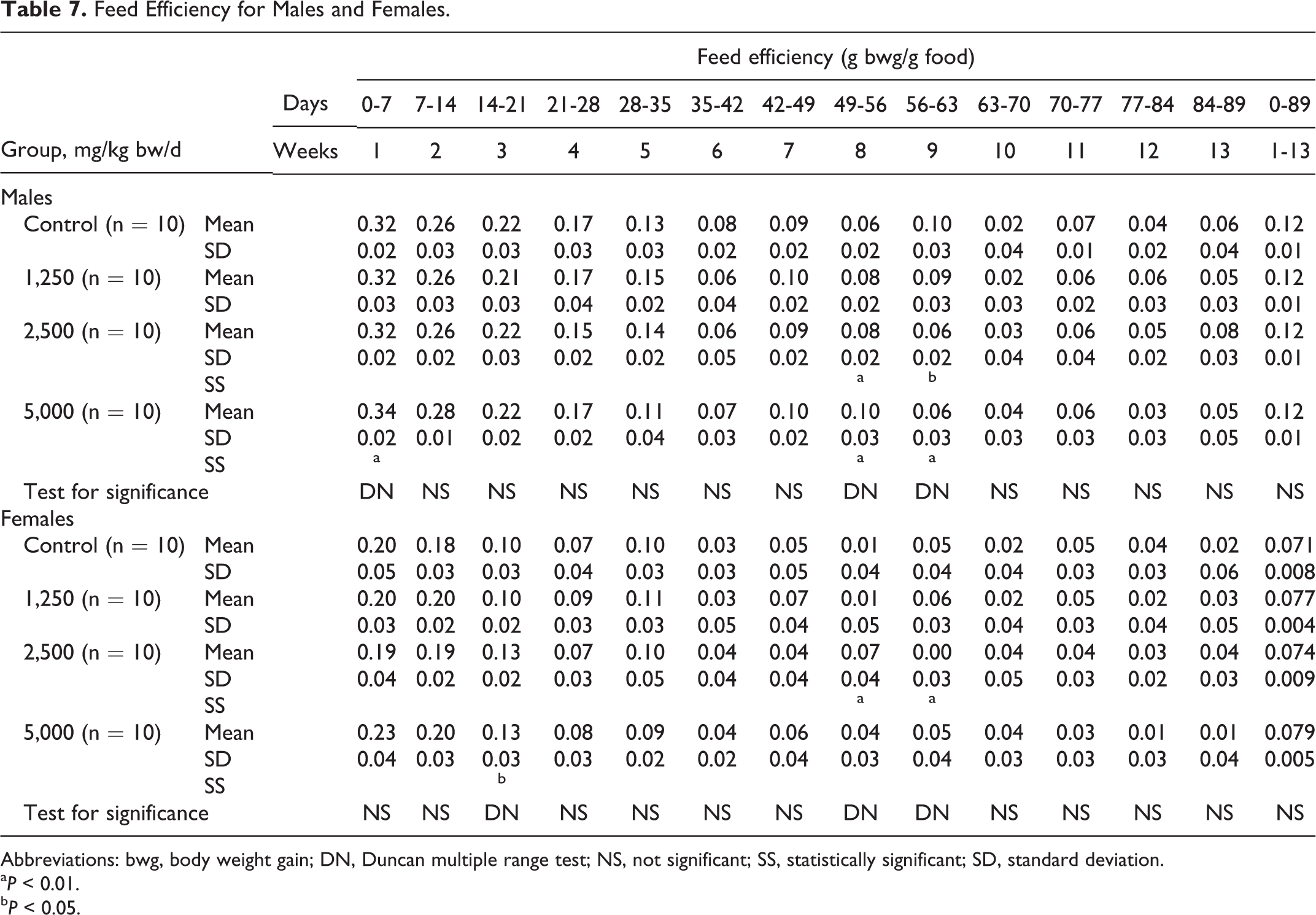
Abbreviations: bwg, body weight gain; DN, Duncan multiple range test; NS, not significant; SS, statistically significant; SD, standard deviation.
a P < 0.01.
b P < 0.05.
A few statistically significant differences compared to controls were observed among the sexes on hematological evaluation as shown in Table 8. Sporadic and/or dose-related statistically significant differences in several clinical chemistry measures compared to controls were also observed in the male and female test groups (see Table 9).
Summary of Hematology.
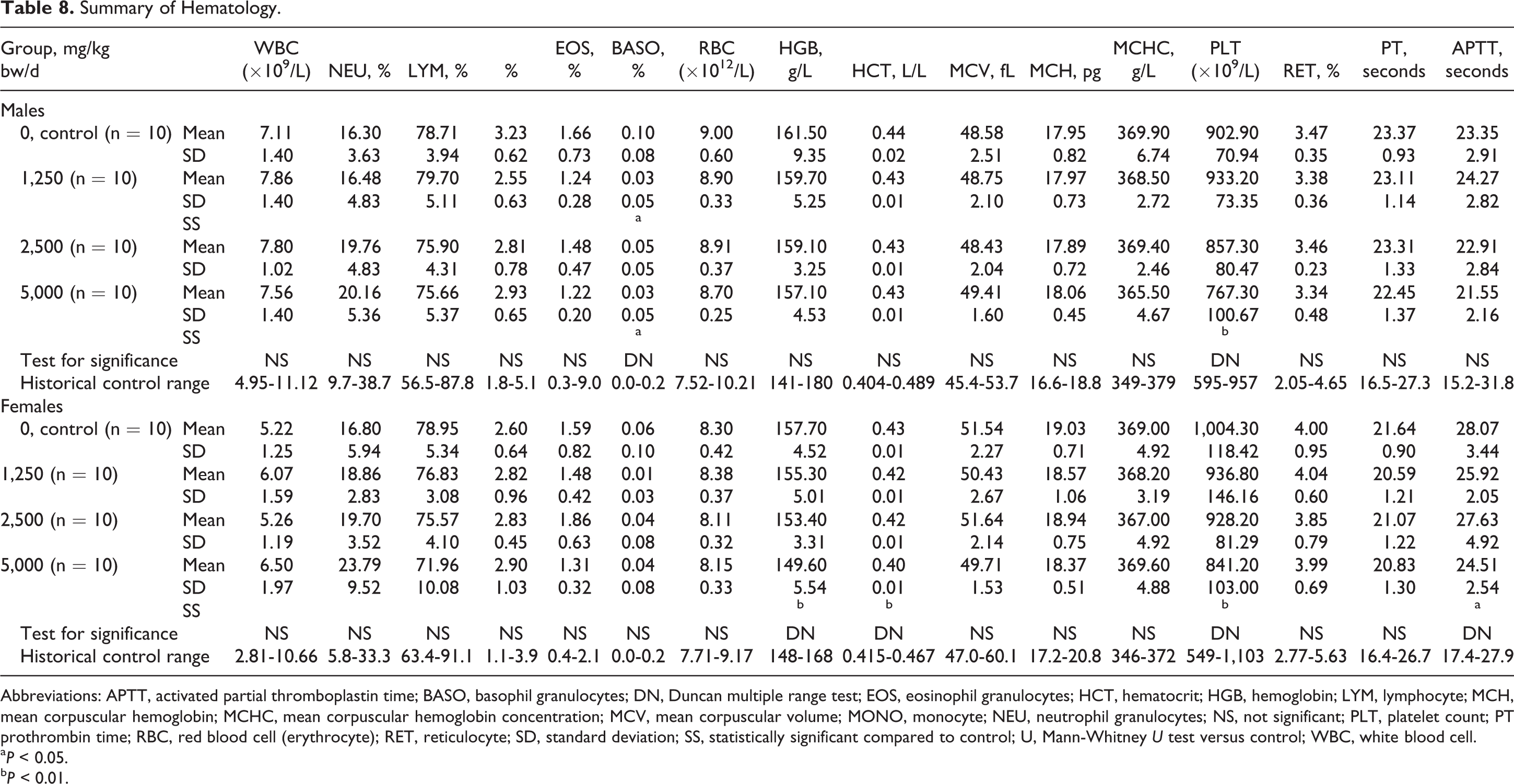
Abbreviations: APTT, activated partial thromboplastin time; BASO, basophil granulocytes; DN, Duncan multiple range test; EOS, eosinophil granulocytes; HCT, hematocrit; HGB, hemoglobin; LYM, lymphocyte; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; MONO, monocyte; NEU, neutrophil granulocytes; NS, not significant; PLT, platelet count; PT prothrombin time; RBC, red blood cell (erythrocyte); RET, reticulocyte; SD, standard deviation; SS, statistically significant compared to control; U, Mann-Whitney U test versus control; WBC, white blood cell.
a P < 0.05.
b P < 0.01.
Summary of Clinical Chemistry.a
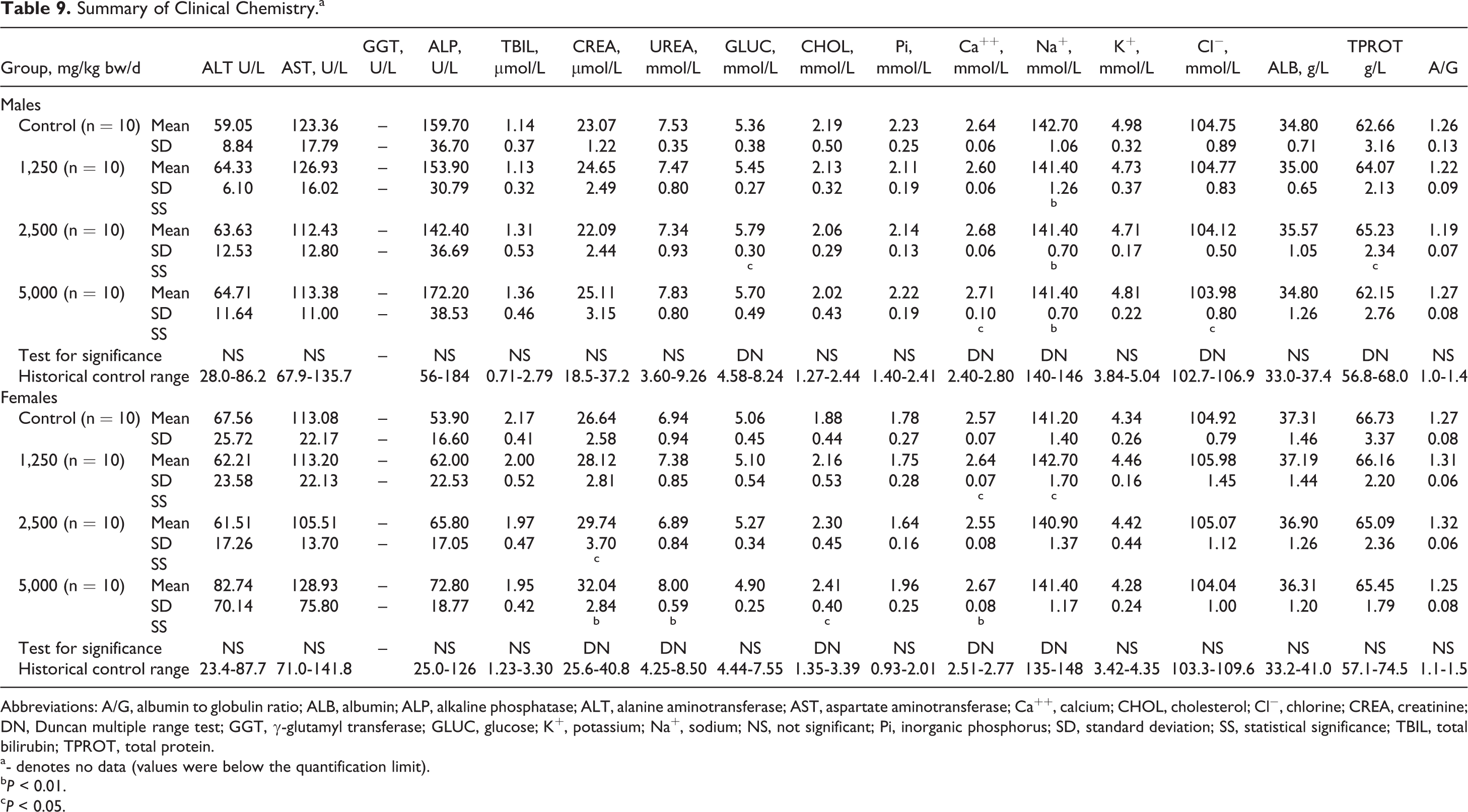
Abbreviations: A/G, albumin to globulin ratio; ALB, albumin; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; Ca++, calcium; CHOL, cholesterol; Cl−, chlorine; CREA, creatinine; DN, Duncan multiple range test; GGT, γ-glutamyl transferase; GLUC, glucose; K+, potassium; Na+, sodium; NS, not significant; Pi, inorganic phosphorus; SD, standard deviation; SS, statistical significance; TBIL, total bilirubin; TPROT, total protein.
a- denotes no data (values were below the quantification limit).
b P < 0.01.
c P < 0.05.
Some sporadic macroscopic changes were observed in the male and female control and test article groups with similar frequencies and/or without dose relation and are presented in Table 10. Organ weight examination revealed some apparent dose-related and statistically significant changes in organ weights, organ weight to body weight ratios, and organ weight to brain ratio in females (see Table 11). For males, the only statistically significant difference for organ weight measures and ratios compared to control was a possibly dose-related slightly lower mean epididymides weight in the 5,000 mg/kg bw/d group (data not shown).
Summary of Macroscopic Findings.a
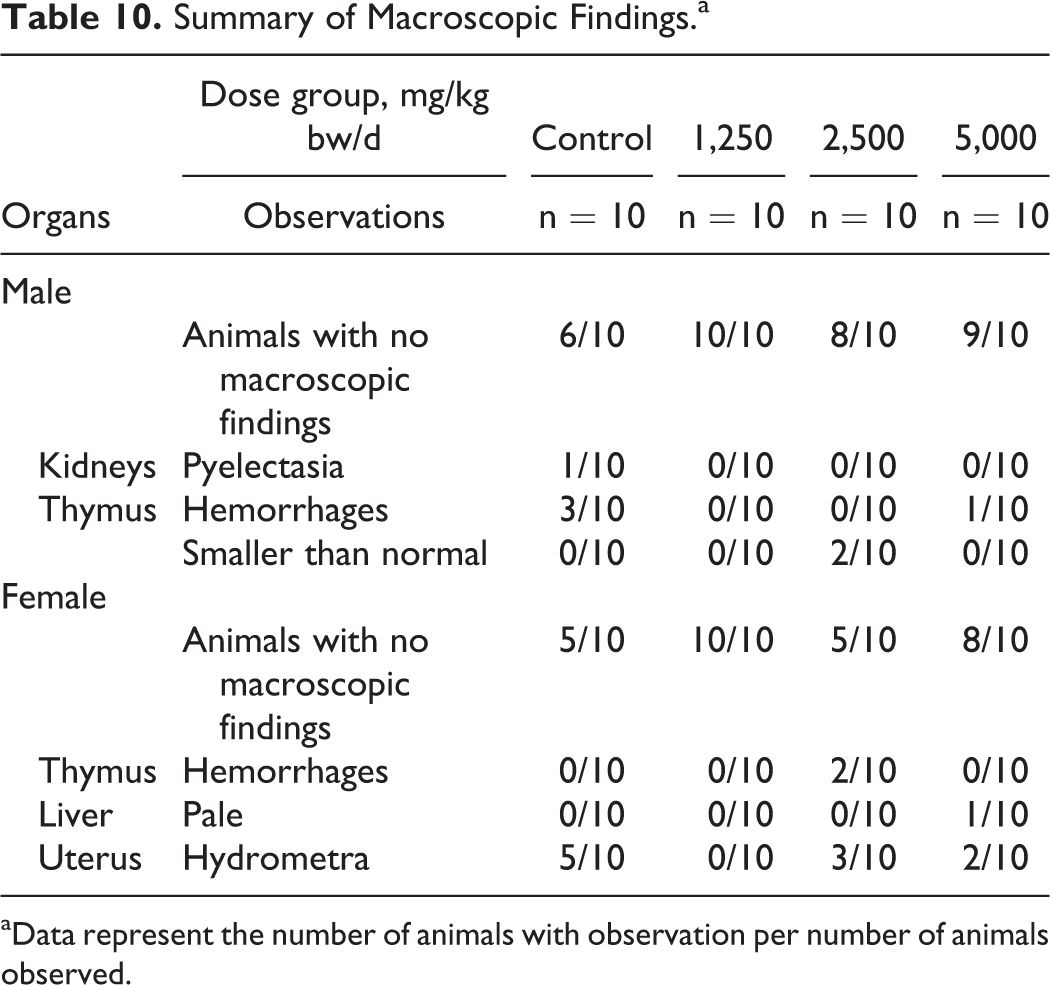
aData represent the number of animals with observation per number of animals observed.
Summary of Selecteda Absolute and Relative Organ Weights in Female Animals in the 90-Day Repeated-Dose Oral Toxicity Study.
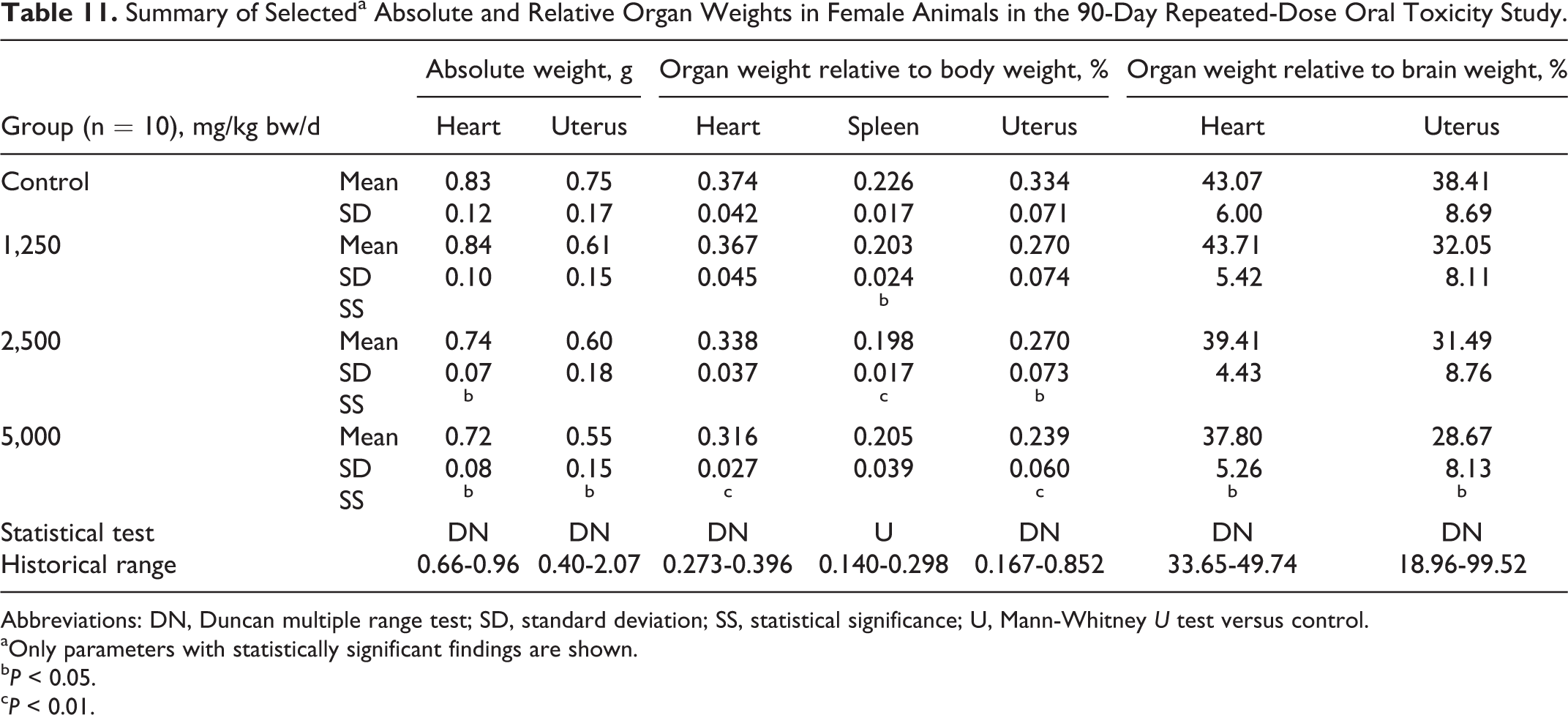
Abbreviations: DN, Duncan multiple range test; SD, standard deviation; SS, statistical significance; U, Mann-Whitney U test versus control.
aOnly parameters with statistically significant findings are shown.
b P < 0.05.
c P < 0.01.
Histopathological examination revealed several microscopic lesions in the kidneys, lungs, thymus, or uterus in individual animals or with similar frequency in control and high-dose animals among the sexes. Several lesions were also observed upon examination of mid-dose animals in which macroscopic lesions were observed (see Table 12). There was no morphological evidence of acute or subacute injury (degeneration, inflammation, necrosis, etc) in the alimentary system, liver, pancreas, cardiovascular system, hematopoietic system, musculoskeletal system, male or female reproductive system, or the central or peripheral nervous system. The structure and cytomorphology of the endocrine glands were the same in the control and treated animals.
Summary of Histopathology Findings.a,b
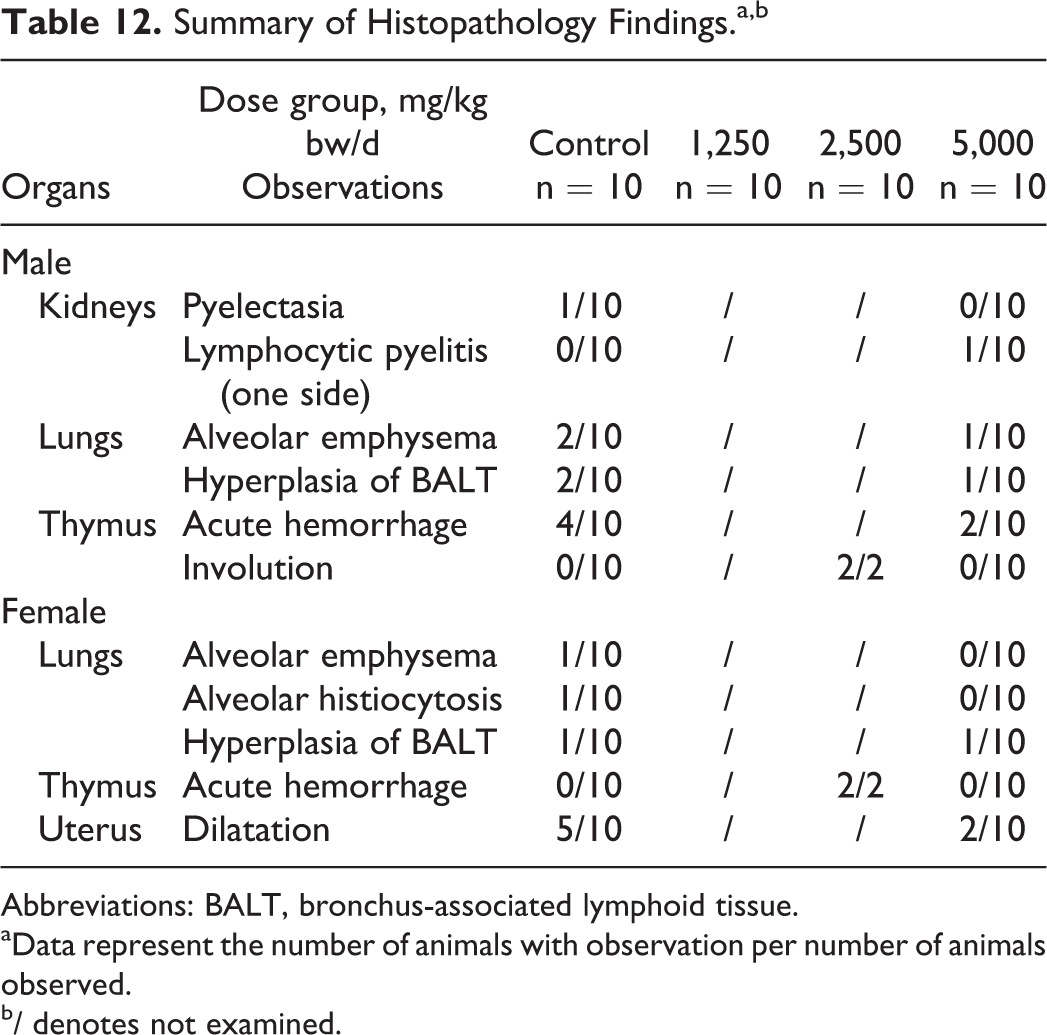
Abbreviations: BALT, bronchus-associated lymphoid tissue.
aData represent the number of animals with observation per number of animals observed.
b/ denotes not examined.
Discussion and Conclusions
In the performed bacterial reverse mutation test, all of the validity criteria regarding the investigated strains, negative and positive controls, S9 activity, and number of investigated analyzable concentration levels were fulfilled. Apart from the lower values obtained in the initial mutation test in S typhimurium TA98 and E coli WP2 uvrA, the test item did not show cytotoxic effects throughout the study. Because the validation criteria were met, the results were considered unequivocally negative based on the lack of dose-related or reproducible biologically relevant increases in revertant colonies in any strain under the tested conditions. The chromosomal aberration test and mammalian micronucleus test were unequivocally negative and unremarkable.
Although the digestive and metabolic fate of CLL is unknown, there is a hypothetical potential that some level of ingested CLL may be digested or catabolized into
Creatine use in dietary supplements usually includes a loading dose of 20 to 30 g divided into 4 equal doses for 5 to 7 days followed by a 1 to 10 g/d maintenance dose, although some daily dose recommendations go as high as 20 to 30 g. 4,36 –38 In a recent review, Barcelos et al discussed the conflicting reports on creatine use concluding that creatine supplementation induces no real modification in liver metabolism in humans, while side effects of liver toxicity or renal toxicity from creatine supplementation are regarded as inconclusive. 36 Additionally, studies have reported mixed results of creatine’s effect on creatinine (CREA) excretion in humans with some studies showing an increase 39 –42 and others showing no increase. 37,43 –46 In the current 90-day study, we provide the beginnings of evidence supporting the safety of CLL.
The slightly soft stool observed in the intermediate- and high-dose groups was likely caused by the relatively large concentration of the administered test article and was not considered toxicologically relevant due to the absence of related changes in body weight, food consumption, or biochemical parameters in the intermediate- and high-dose groups. Week-by-week body weight gain, food consumption, and feed efficiency changes were occasionally statistically significant, although the changes were minor and transient and did not cause relevant changes in mean body weight or cumulative body weight of these groups. Cumulative food consumption in the 2,500 and 5,000 mg/kg bw/d female groups was statistically significantly lower than control; however, the effect of the changes was low in magnitude, lacked correlating histopathological findings, and there were no significant effects on cumulative feed efficiency, cumulative body weights, or cumulative body weight gain (see Tables 5–7). Therefore, the variability observed was not considered to be toxicologically significant.
Slight but statistically significant dose-related decreases compared to control were observed in hematological parameters, mean hemoglobin (HGB), and platelet count in the female 5,000 mg/kg bw/d group (see Table 8). The decrease in HGB was accompanied by a statistically significant decrease in hematocrit (HCT) in the same group, but in the case of HCT, a possible dose relationship was not as clearly evident. Hemoglobin was within the historical control range of the laboratory, while HCT was marginal to the range. Platelet counts were well within the historical control range, and although a statistically significant decrease in related parameter, activated partial thromboplastin time (APTT), was also observed, this occurred without a dose relationship, and the control value for APTT was marginally elevated above the historical control range, suggesting the statistically significant finding was an artifact. Because of the above reasons and the absence of correlating histopathological findings, these dose-related and statistically significant hematological changes in the female high-dose group were not considered toxicologically relevant.
In the clinical chemistry examination, statistically significant dose-related changes in cholesterol and CREA were also observed in the female animals (see Table 9). The cholesterol value was statistically significantly increased compared to control at the high dose, but remained within the historical control range and lacked correlating histopathological findings. Creatinine was statistically significantly increased compared to controls in the mid- and high-dose groups, and in the female high-dose group, the related parameter, urea, was statistically significantly increased without a clear dose relationship. Both CREA and urea lack sensitivity for renal pathology; when toxicological alterations do occur in these parameters, they present following histological damage, 47 which was not observed in this study. Thus, because both remained well within the historical ranges and there were no associated histopathological findings, these results were not considered to be toxicologically relevant. As noted above, studies of creatine intake by humans have shown mixed results for effects on plasma CREA levels; however, it is unknown whether CLL is digested to creatine in the digestive tract or absorbed and metabolized to creatine in vivo, and if so, to what extent. As such, no conjecture as to whether the effects observed on CREA levels might be attributed to a normal biological response to increased creatine levels can be made.
The remaining statistically significant alterations in clinical pathological parameters in the female groups and all statistically significant clinical pathological findings in the male groups occurred sporadically without clear dose relationships and remained within historical control ranges, and correlating histopathological findings were absent. Therefore, they were considered indicative of normal biological variation and unrelated to administration of the test article.
Although several of the statistically significant organ weight changes compared to their corresponding control groups appeared dose-related, and in female animals were carried across absolute and relative evaluations, they were all of small degree, remaining well within historical control ranges, and had no correlated histopathological findings (see Table 11); therefore, they were not considered to have biological significance.
Thymic hemorrhages were observed both macroscopically and microscopically with similar frequencies among the sexes in control and treated animals (see Tables 9 and 11). These together with alveolar emphysema, which also occurred with similar frequency in controls and high-dose animals, were considered to be associated with the hypoxia, dyspnea, and circulatory disturbance that occurs during exsanguination, as this is common in experimental rats. 48,49 On histological examination of the smaller than normal thymuses observed macroscopically in 2 mid-dose males, the lack of histopathological changes such as degeneration, inflammation, or fibrosis leads to the conclusions that these changes were consistent with accelerated involution of the thymus, an individual phenomenon common in this age of experimental rats, and not test article-related. 50,51 Pyelectasia was observed macroscopically and microscopically in a single control male only, and one-sided lymphocytic pyelitis was observed on histopathological examination of a single high-dose female; these were considered individual findings unrelated to the administration of the test article. 52 Dilatation of uterine horns was observed with greatest frequency in controls and was considered due to a slight neurohormonal phenomenon connected with the proestrus phase of the inner reproductive organs. 49,53,54
In conclusion, CLL was not mutagenic under the applied conditions of the bacterial reverse mutation test at concentrations up to 5,000 µg/plate or up to the maximum recommended concentration of 2,000 μg/mL, both with and without metabolic activation, in V79 Chinese hamster lung cells in the in vitro mammalian chromosomal aberrations test. Under the in vivo conditions of the mammalian micronucleus test, CLL did not exhibit genotoxic potential at doses up to 2,000 mg/kg bw in male Crl:NMRI BR mice. Finally, CLL did not cause adverse effects and no target organs were identified in male or female Hsd.Han Wistar rats after the consecutive 90-day repeated-dose gavage administration of 1,250, 2,500, or 5,000 mg/kg bw/d. Based on the observations made in this study, an NOAEL was estimated as 5,000 mg/kg bw/d in male and female Hsd.Han:Wistar rats. Future investigations of CLL in pharmacokinetics and nonrodent species would be valuable additions to the currently available research.
Footnotes
Authors’ Note
AIBMR Life Sciences, Inc was contracted by the study sponsor, as an independent third party, to determine appropriate study protocols and dose selections, place the studies, approve the study plans, and monitor the toxicological studies herein described and to analyze and interpret the resulting data and prepare the manuscript. Toxi-Coop Zrt was contracted by AIBMR to develop the study plans and conduct, analyze, interpret, and report the results of the toxicological studies herein described.
Acknowledgments
The authors thank the following individuals for their contributions to the work: Viktória Balogh, Ibolya, Bogdán, Katalin Böröczki, István Buda, Tamás Buda, Timea Csörge, Kata Eszter Diószegi, Biermanné Major Erika, Mónika Fekete, Zsuzsanna Frank, Zsuzsanna Gaál, Irén Somogyi Háriné, Ildikó Hermann, Brigitta Horváth, Istvánné Horváth, Jurácsikné Sereg Kornélia, Klára Fritz Kovácsné, Nóra Pongrácz Kurdiné, Adrienn Laczó, Marcell Mardár, Viktória Matina, Edit Kővári Mesterháziné, Ágota, Jó Schüllerné, János Stáhl, Ákosné Szabó, Éva Szabó, Zsuzsanna Szabó, Mariann Lennert Szabóné, Edit Szám, Beatrix Szilágyi, Márta Tenk, and Ruth Weimer for the performance of experimental tasks and/or collection of data and Jared Brodin for administrative support in preparation of the manuscript.
Author Contributions
Robin A. Reddeman contributed to analysis and interpretation of data, drafted the manuscript, and critically revised the manuscript. Róbert Glávits contributed to analysis and interpretation of data and critically revised the manuscript. John R. Endres contributed to conception and design, contributed to analysis and interpretation of data, and critically revised the manuscript. Timothy S. Murbach contributed to conception, contributed to analysis and interpretation of data, contributed to drafting of manuscript, and critically revised the manuscript. Gábor Hirka contributed to conception and design, contributed to analysis and interpretation of data, and critically revised the manuscript. Adél Vértesi contributed to conception and design, contributed to acquisition, analysis, and interpretation of data, contributed to drafting of manuscript, and critically revised the manuscript. Erzsébet Béres contributed to conception and design, contributed to acquisition, analysis, and interpretation of data, contributed to drafting of manuscript, and critically revised the manuscript. Ilona Pasics Szakonyiné contributed to conception and design, contributed to acquisition, analysis, and interpretation of data, contributed to drafting of manuscript, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors disclose that financial support for the research described herein was provided by Vital Pharmaceuticals, Inc (Weston, Florida).