
Abstract
We have established a current good manufacturing practice (GMP) manufacturing process to produce a nanoparticle suspension of 1,1′-methylenebis-4-[(hydroxyimino)methyl]pyridinium dimethanesulfonate (MMB4 DMS) in cottonseed oil (CSO) as a nerve agent antidote for a Phase 1 clinical trial. Bis-pyridinium oximes such as MMB4 were previously developed for emergency treatment of organophosphate nerve agent intoxication. Many of these compounds offer efficacy superior to monopyridinium oximes, but they have poor thermal stability due to hydrolytic cleavage in aqueous solution. We previously developed a nonaqueous nanoparticle suspension to improve the hydrothermal stability, termed Enhanced Formulation (EF). An example of this formulation technology is a suspension of MMB4 DMS nanoparticles in CSO. Due to the profound effect of particle size distribution on product quality and performance, particle size must be controlled during the manufacturing process. Therefore, a particle size analysis method for MMB4 DMS in CSO was developed and validated to use in support of good laboratory practice/GMP development and production activities. Manufacturing of EF was accomplished by milling MMB4 DMS with CSO and zirconia beads in an agitator bead mill. The resulting bulk material was filled into 5-mL glass vials at a sterile fill facility and terminally sterilized by gamma irradiation. The clinical lot was tested and released, a Certificate of Analysis was issued, and a 3-year International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) stability study started. The drug product was placed in storage for Phase 1 clinical trial distribution. A dose delivery uniformity study was undertaken to ensure that the correct doses were delivered to the patients in the clinic.
Introduction
Organophosphate (OP) nerve agents constitute an important class of chemical weapons that function by inhibition of the enzyme acetylcholinesterase (AChE), which leads to accumulation of acetylcholine at muscarinic receptors, causing rapid incapacitation and possible death. 1,2 Various chemical countermeasures have been developed for emergency treatment of OP intoxication, and they are typically delivered by intramuscular (IM) injection. 3 Oxime compounds are primary components of OP emergency countermeasures, because they can reactivate inhibited AChE. The oxime currently fielded by the United States is 2-pralidoxime chloride (2-PAM), a pyridinium oxime. This oxime is only effective against certain nerve agents, so alternative oximes with broad-spectrum efficacy are under development. 4 Bis-pyridinium bis-oximes in general are effective against a broader spectrum of agents, but they have poor thermal stability due to hydrolytic cleavage in aqueous solution. 5 –8 Because stability is greatly enhanced in the solid phase in the absence of water, a formulation that retains the stability of the solid while remaining injectable is desirable.
We previously developed a nonaqueous formulation consisting of a suspension of 1,1′-methylenebis-4-[(hydroxyimino)methyl]pyridinium dimethanesulfonate (MMB4 DMS) nanoparticles in cottonseed oil (CSO). 9 This product, known as Enhanced Formulation (EF, as opposed to the aqueous Original Formulation, or OF), is a non-Newtonian fluid at sufficiently high concentrations and low particle size distributions, exhibiting shear thinning behavior. The EF has a high zero-shear viscosity that allows it to resist rapid particle sedimentation but remains injectable due to the drop in viscosity during injection caused by shear thinning. Because the MMB4 DMS nanoparticles are insoluble in hydrophobic CSO, the chemical stability of MMB4 in EF is vastly superior to that of MMB4 in aqueous solution. The EF exhibit almost no degradation even after 2 years at temperatures up to 80°C. 9
As shown in Figures 1 and 2, various techniques were used during the development to reduce the particle size of MMB4 DMS to accommodate different concentrations in the EF. 9 Range-finding preclinical studies indicated that a solid concentration of approximately 100 mg MMB4 DMS per gram of EF was optimal. 10,11 For maximum physical stability at this concentration, a wet mill process was developed using an agitator bead mill to reduce the MMB4 DMS particle size distribution to the range of approximately 200 to 300 nm. A particle size analysis method based on dynamic light scattering (DLS) was also developed to measure the particle size range of MMB4 DMS nanoparticles in CSO suspension.
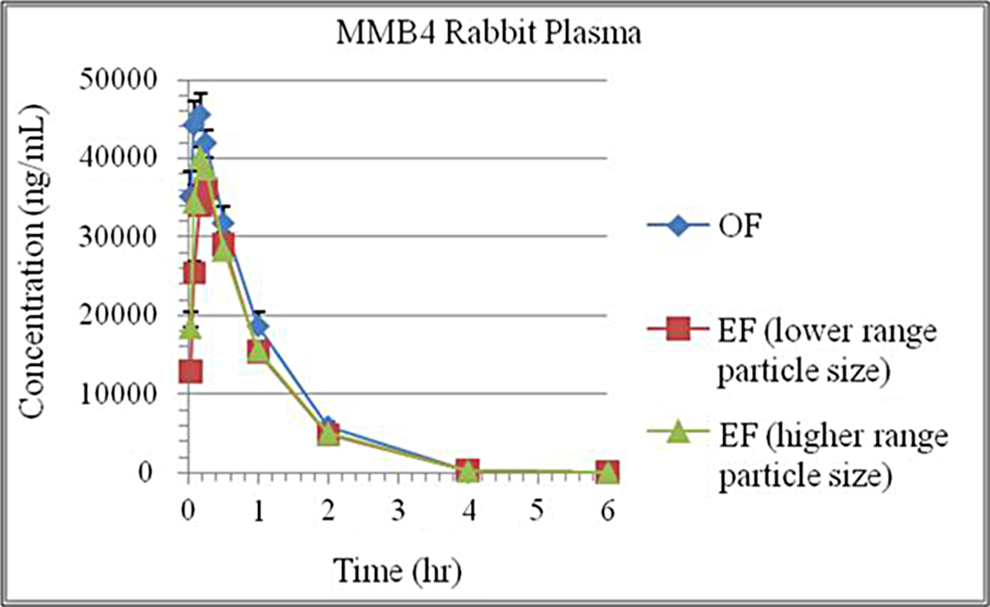
Rabbit plasma concentration of MMB4 following intramuscular injection. The MMB4 DMS OF and MMB4 DMS EF show a similar T max and C max for a 25 mg/kg dose delivered at a concentration of 100 mg/g. MMB4 DMS indicates 1,1′-methylenebis-4-[(hydroxyimino)methyl]pyridinium dimethanesulfonate; EF, enhanced formulation; OF, original formulation.
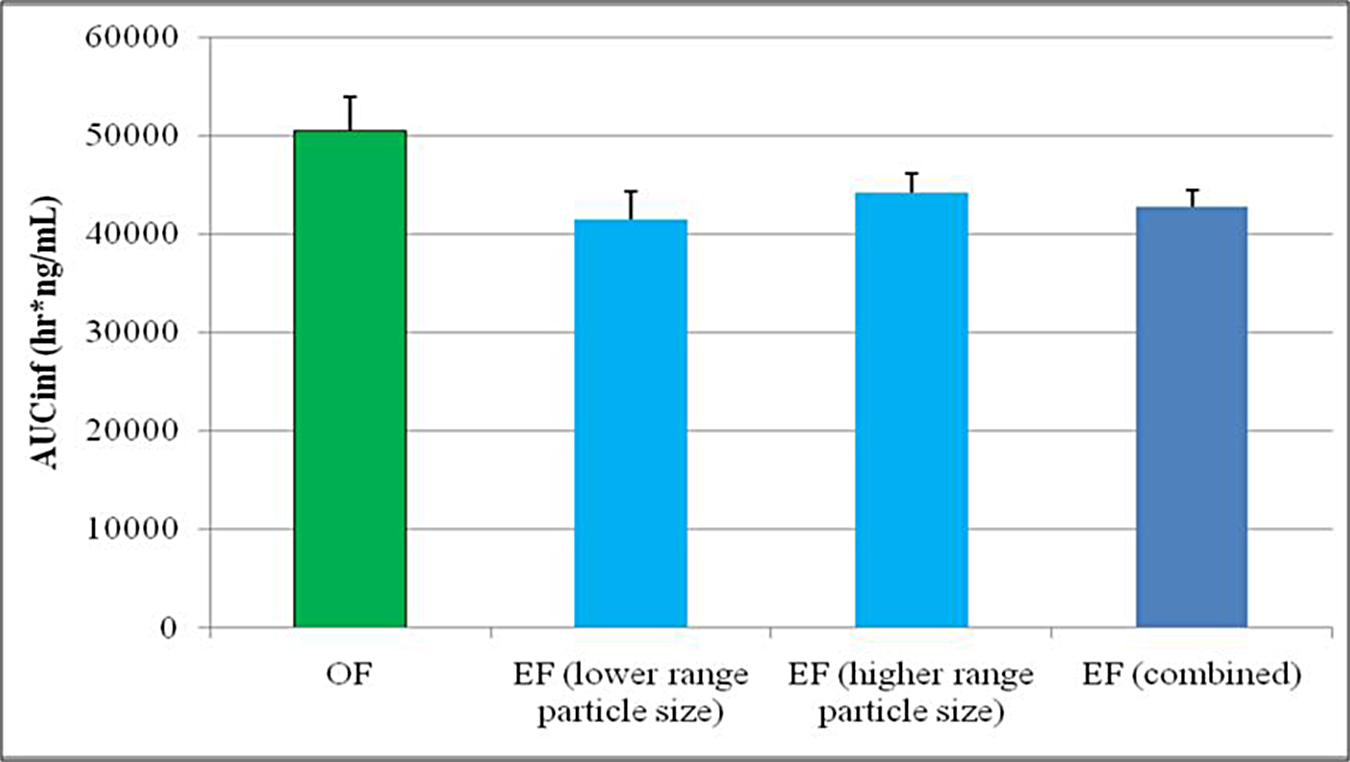
Comparison of MMB4 DMS AUCinf for MMB4 DMS OF versus MMB4 DMS EF. AUC, area under the curve; MMB4 DMS, 1,1′-methylenebis-4-[(hydroxyimino)methyl]pyridinium dimethanesulfonate; EF, enhanced formulation; OF, original formulation.
The goal of the work reported herein was to produce and characterize batches of MMB4 DMS EF to supply preclinical and clinical studies. We report the development and validation of the aforementioned particle size analysis method by DLS. We also report manufacturing of EFs by a process compliant with good laboratory practice (GLP) for preclinical studies and current good manufacturing practice (cGMP) for a Phase 1 clinical trial. 12 Preclinical studies in rabbits found no statistically significant difference in pharmacokinetic (PK) properties between EF with different particle sizes, and the absorption/elimination profile of EF was very similar to OF. 13 The clinical trial batch was manufactured using cGMP, including milling and compounding of the MMB4 DMS API and CSO, filling of the product into vials, terminal sterilization by gamma irradiation, and clinical dose delivery studies. The batch met release specifications, stability studies have been initiated in accordance with the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) guidance, and a Phase 1 clinical trial is underway.
Materials and Methods
Materials
High-performance liquid chromatography (HPLC)-grade acetonitrile, methanol, and phosphate-buffered saline were purchased from Fisher Scientific (Pittsburgh, Pennsylvania) and used as received. Reagent-grade methanesufonic acid and 4-pyridine aldoxime (4-PA) were purchased from Sigma-Aldrich and used as received. Cottonseed oil (CSO, United States Pharmacopeia [USP]/National Formulary grade) was purchased from Welch, Holme, & Clark Co, Inc (Newark, New Jersey). The MMB4 DMS was manufactured by SwRI and Cambrex (Charles City, Iowa). Syringes (1, 3, and 5 mL; Monoject [Fisher Scientific ]) and needles (16 ga 1.5in and 21 ga 1 in; Becton-Dickinson, Franklin Lakes, New Jersey) were purchased from Fisher Scientific.
High-performance liquid chromatography
The quantification of MMB4 and its degradation products were analyzed by HPLC (Waters HPLC system, Waters, Milford, Massachusetts). Separations were performed on reversed phase columns with ultraviolet (UV) or diode-array detectors.
For MMB4, HPLC was performed on a Waters 2695 or Agilent 1100 system (Agilent Technologies, Santa Clara, California) on a reversed-phase C18 column (Waters Atlantis HILIC, 4.6 × 150 mm, 5 μm) at a flow rate of 1.3 mL/min, equipped with a UV detector set at 297 nm. The mobile phase consisted of 80/20 MeCN/0.05 mol/L ammonium formate buffer (pH 4). The major–minor peaks of MMB4 DMS and the impurity have retention times of 18, 21, and 25 minutes, respectively.
For quantification of the degradation product from MMB4 DMS, HPLC was performed on a reversed-phase C18 column (Discovery C-18, 3.5 × 150mm; Supelco, Bellefonte, Pennsylvania) at a flow rate of 1.5 mL/min with photodiode array detector set at 215 nm. The mobile phase consisted of a gradient of 90/10 (v/v) 25 mmol/L K2HPO4, pH 7.0/MeOH (solvent A2) and methanol (solvent B2) with the following conditions: 100% solvent A2 (0-20 minutes), 50% solvent A2/50% solvent B2 (20-25 minutes), and 100% solvent A2 (25.5-30 minutes). The 4-PA was eluted after 7 minutes.
Particle Size Characterization
The particle size distributions of MMB4 DMS nanoparticles were determined by DLS, also known as photon correlation spectroscopy (PCS), using a Brookhaven ZetaPALS 90Plus BI-MAS (Brookhaven Instruments Corp, Holtsville, New York). This method is discussed in more detail in the Results and Discussion section. Samples were prepared by diluting EF with CSO to approximately 50 µg/mL and sonicating in a 100-W bath sonicator for approximately 60 minutes. The DLS experiments were conducted at a controlled temperature of 74°C, and results are reported as unimodal intensity-weighted median particle sizes.
Milling
To reduce the particle size to submicron levels, an agitator bead mill (Glen Mills Dyno-Mill Multi-Lab, Glen Mills, Clifton, New Jersey) was used in a wet milling process. Milling of MMB4 DMS in the presence of CSO was conducted in an enclosed vessel filled with zirconia grinding media. For the relatively small-scale milling for the preparation of preclinical study samples, the milling set up consisted of the Dyno-Mill (Glen Mills) with a jacketed grinding chamber cooled by circulating coolant mixture of water and glycerin, a peristaltic pump to introduce the suspension into the grinding chamber, a single reservoir equipped with an overhead mechanical stirrer for the collection of the mixture exiting the grinding chamber, and a nitrogen line to blanket the product with dry nitrogen. The suspension sample was continuously cycled through the mill by pumping from the reservoir into the grinding chamber and back to the reservoir until either the predetermined grinding time was reached or the desired particle size in the suspension was obtained. The temperature of the milled product inside the grinding chamber was monitored through an inline thermometer, a maximum allowable temperature was set at 70°C, and the milling process was conducted under a blanket of dry nitrogen to minimize thermal degradation of the product (70°C was never reached during any of the milling processes reported herein). For the larger scale cGMP production of the Phase 1 clinical trial batch, the set up was modified to include 2 reservoirs—the suspension was pumped into the grinding chamber from one reservoir while the milled material was collected in a second reservoir. The material in the second reservoir was then pumped back through the mill and into the first reservoir. This process was repeated until the predetermined grinding time was reached or the desired particle size in the suspension was obtained. This change was made to ensure that the entire product went through the milling chamber, with the same number of passes to promote product uniformity. A schematic of the milling process can be found in the Results and Discussion section.
Ultraviolet–Visible
The MMB4 absorbs strongly in the UV region at 292 nm. The response was measured using a spectrophotometer and quantitated against a reference standard. The absorbance at this wavelength was affected by the pH of the solution. The standards and samples were diluted in a buffer to maintain constant pH.
Sterilization Validation
The sterilization validation for the MMB4 DMS EF drug product was performed at North American Science Associates, Inc (NAMSA; Northwood, Ohio). Bacteriostasis and fungistasis, bioburden recovery validation, and dose verification determination were performed by NAMSA. All the materials for this validation were procured by NAMSA. Sterilization of the drug product for dose verification and terminal sterilization were performed by Steris Isomedix (Libertyville, Illinois).
Sterile Fill
The MMB4 DMS EF was sterile filled into 5-mL glass vials under cGMP conditions at Albany Molecular Research, Inc (AMRI; Burlington, Massachusetts). Two thousand glass vials (20-mm diameter, 5-mL capacity) composed of borosilicate glass were sterile filled under a nitrogen atmosphere by means of a peristaltic metering pump, each with 3.43 g of MMB4 DMS EF. The vial closures consisted of 20-mm bromobutyl stoppers, Fluorotec Flip-Off caps, and crimped aluminum seals. Vials, stoppers, caps, and crimp seals were sourced from West Pharmaceutical (Exton, Pennsylvania).
Preclinical Study
This study (Comparative Pharmacokinetic Study of Original and Enhanced MMB4 DMS Formulations in the Male New Zealand White Rabbit [Battelle Study Number CG920771-C2]) included 3 dose groups containing 6 animals each. All doses for this study were prepared from formulated MMB4 DMS EF and MMB4 DMS OF at a target dose of 0.25 mg/kg based on individual body weights. The doses were based on actual animal body weight that was provided on the day of the fill.
Results and Discussion
Particle Size Analysis
Accurate particle size analysis of MMB4 DMS EF is essential, because the physical properties of the product are defined by the critical parameters of concentration and particle size distribution. At a given concentration, a formulation with smaller particles has a higher viscosity, because the greater overall surface area of suspended particles creates stronger aggregate interparticle interactions. 14 For EF to function as an injectable product, the viscosity must be sufficiently high as to prevent rapid particle sedimentation, which could lead to inhomogeneous dose delivery and/or needle blockage. Viscosity must likewise be sufficiently low as to enable injection through a standard needle (typically 22-23 gauge). Due to the profound effect of particle size distribution on product quality and performance, it must be controlled during the manufacturing process. Therefore, particle size analysis is used as an in-process test to define the end point of the milling process (see following sections on GLP and cGMP milling).
A particle size analysis (PSA) method for MMB4 DMS EF was developed and validated for use in support of GLP/GMP development and production activities. Since the particle size reduction step (milling) and formulation compounding step (dispersion in CSO) are combined in the bead mill, a PSA method capable of measuring the particles in CSO was preferable. Attempts to remove the particles from suspension and measure them as a dry powder could result in aggregation, making it difficult to determine the size of the suspended particles.
Several techniques were evaluated, including optical, electron, and force microscopy, analytical centrifugation, and PCS. Ultimately, PCS (also known as DLS) gave the most reproducible results. The DLS measures the time-correlated intensity of scattered light from a laser incident on a liquid sample, as the dispersed particles undergo thermally induced Brownian motion. As such, the sample must be nearly optically transparent (concentration is typically optimized for a certain range of detected intensity, chosen such that the assumption of single scattering events is reasonably accurate). The EF samples typically have concentrations in excess of 10% by mass (100 mg of MMB4 DMS per gram of suspension), making them nearly optically opaque. Therefore, dilutions of several 1000 times are necessary to reach a target concentration of approximately 50 µg/g. Even at this low concentration, the samples still have relatively high viscosity of CSO (approximately 60 cP at 20°C). Although the viscosity of the suspension is an input parameter in the DLS experiment, the instrument is generally designed to work with lower viscosity samples. Therefore, the method for EF characterization uses the maximum temperature of the instrument, which is 74°C. At this temperature, the viscosity of CSO drops to approximately 13 cP.
Preliminary experiments indicated low replicate precision, but this problem was alleviated by bath ultrasonication of the samples immediately prior to measurement. This process presumably dissociates loosely aggregated particles, making the measured particle size distribution more homogeneous. Replicate precision is demonstrated in the method by triplicate sample preparation and analysis. The data analysis can be optimized for single (unimodal) or multiple (multimodal) populations of particles and can be weighted by scattered intensity or by particle volume, surface area, or number of particles. In this case, the reported data are obtained from a unimodal fit to produce a lognormal particle size distribution and are reported with intensity weighting. To indicate the width of the particle size distribution, the data are reported as D10, D50, and D90, which are the points in the distribution where 10%, 50%, and 90% of the population, respectively, have a size smaller than the reported value.
The DLS method was validated to demonstrate suitability for characterization of MMB4 DMS EF in accordance with cGMP for pharmaceutical products. The validation focused on the aspects of precision, accuracy, and robustness in accordance with USP General Chapter <429>, Light Diffraction Measurement of Particle Size, 15 and USP General Chapter <729>, Globule Size Distribution in Lipid Injectable Emulsions. 16 System suitability was established by measurement of a standard consisting of an aqueous dispersion of monodisperse 500 nm diameter polystyrene latex nanoparticles on each day that samples were measured and demonstrating a deviation of D50 from the particle size specified in the Certificate of Analysis supplied with the standards of not more than 10% (approximately 50 nm).
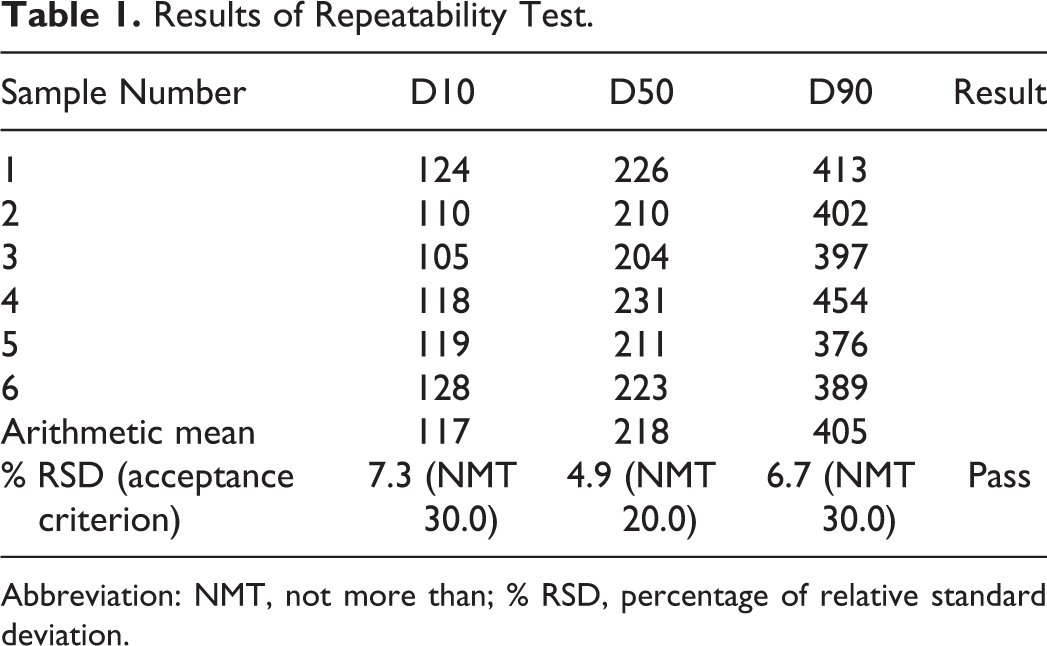
Precision was established by demonstration of repeatability and of intermediate precision. Repeatability was demonstrated by preparation of 6 samples by separate dilutions from a single lot of MMB4-DMS EF stock. Results and acceptance criteria are presented in Table 1.
Results of Repeatability Test.
Abbreviation: NMT, not more than; % RSD, percentage of relative standard deviation.
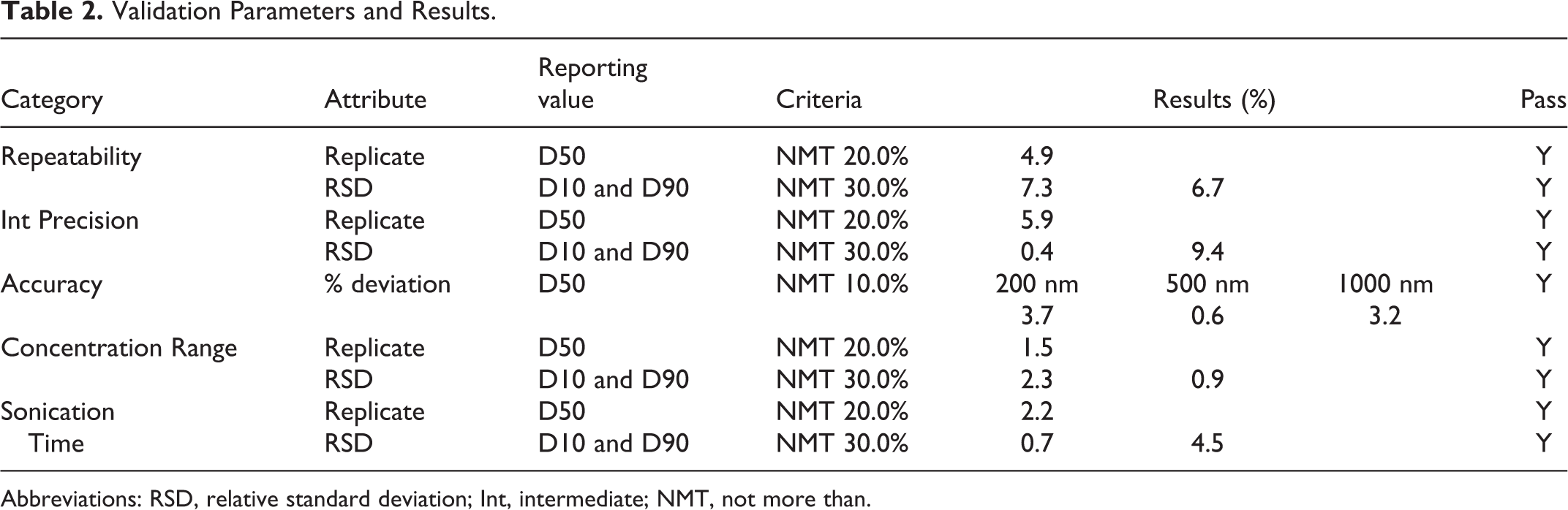
Intermediate Precision was demonstrated by a second analyst testing 6 preparations of a single lot of MMB4-DMS EF stock on a different day from the Repeatability experiment above. The same EF lot was used for both experiments. Results and acceptance criteria are presented in Supplementary Tables S1 to S3 and summarized in Table 2.
Validation Parameters and Results.
Abbreviations: RSD, relative standard deviation; Int, intermediate; NMT, not more than.
Due to the nature of the EF product, it is not feasible to analyze the particle size distribution by another method without potentially altering the size of the particles. Therefore, accuracy was determined by DLS analysis of particle size standards that have the National Institute of Standards and Technology-traceable accuracy. These standards consisted of monodisperse polystyrene latex spheres in aqueous suspension. The 3 particle size standards chosen had nominal diameters of 200, 500, and 1000 nm, respectively. The mean value of D50 for each standard was calculated and compared to the specified mean diameter in the Certificate of Analysis for the same standard. Results are presented in Table 3.
Results of Accuracy Test.
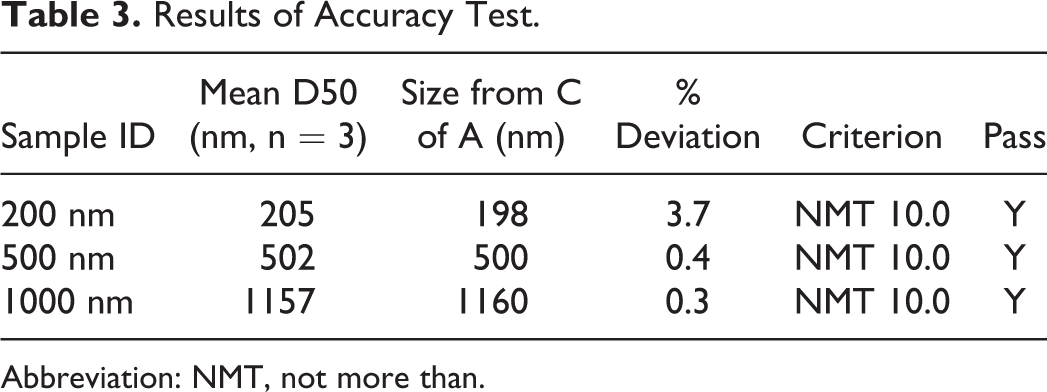
Abbreviation: NMT, not more than.
Robustness was assessed by varying the concentration range and ultrasonication time. The dilution factor of the DLS samples was varied around the nominal concentration of 0.05 mg/mL. Three samples from the same batch were tested with concentrations of approximately 0.04, 0.05, and 0.06 mg/mL. From 3 replicates at each concentration, the arithmetic means were calculated separately for D10, D50, and D90. The results and acceptance criteria are shown in Supplementary Tables S4 and S5 and summarized in Table 2.
The previously determined optimum sonication time to achieve a dispersion of primary particles is typically 60 minutes. To assess robustness, this time was varied by ±10 minutes. Sonication times of 50, 60, and 70 minutes were each tested with triplicate preparations from the same EF batch. From the 3 replicates at each sonication time, the arithmetic means were calculated separately for D10, D50, and D90. Results and acceptance criteria are shown in Supplementary Tables S6 and S7 and summarized in Table 2. Table 2 shows a summary of all of the PSA validation results.
The degrees of precision, accuracy, and robustness demonstrated by these studies validate this method for particle size analysis of EF samples under GLP conditions. This method was used to characterize all MMB4 DMS EF samples used in the GLP definitive preclinical studies and in the cGMP production of the batch used for the Phase 1 clinical supply and ICH stability studies.
GLP Milling
The MMB4 DMS was milled with CSO to reduce the MMB4 DMS particle size into the nanometer range to maintain the physical stability and injectability of the suspensions. Formulations with solid concentrations of MMB4 DMS in CSO at or below 120 mg/g concentration were prepared using an agitator bead mill (Dyno-Mill Multi Lab). Process parameters included the size of milling beads, the milling speed (agitator shaft rotation speed), milling time, and the MMB4 DMS solid concentration in CSO. In our experiments, the zirconia milling media diameter ranged from 0.1 to 1.5 mm, the drug concentration in the suspension ranged from 1 to 12% w/w, and the mill rotor speed ranged from 2400 to 6000 rpm. The jacketed milling chamber was cooled with circulated coolant that kept the temperature of the suspension sample below 70°C inside the milling chamber to mitigate possible drug degradation.
Two MMB4 DMS EF were milled and compounded to produce target particle sizes of 250 and 300 nm. To achieve the desired particle sizes, 2 separate milling operations were conducted at target solid concentrations around 100 mg/g, with different size grinding beads and agitator shaft speeds for 2 hours each. Table 4 summarizes the formulations obtained from the processes, and Table 5 shows the particle size distributions. These formulations were selected for the GLP preclinical studies discussed below.
MMB4 DMS EF Batches Used for Definitive Preclinical Study.
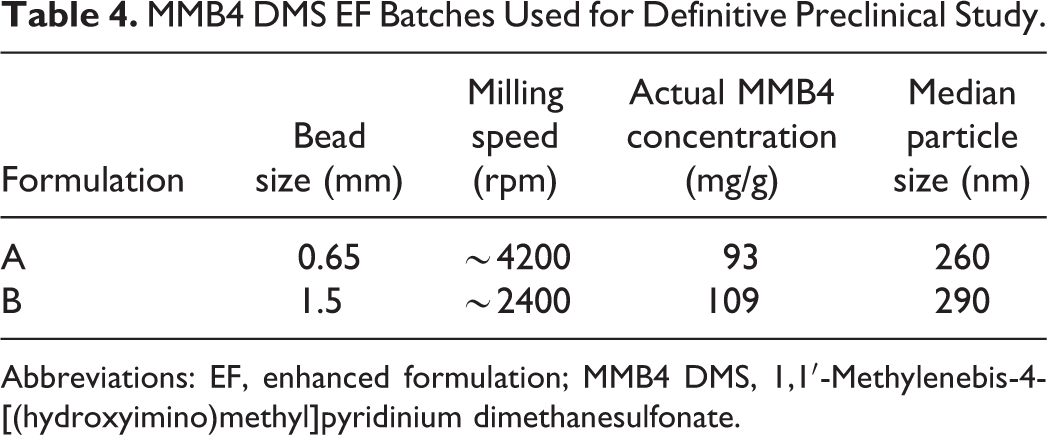
Abbreviations: EF, enhanced formulation; MMB4 DMS, 1,1′-Methylenebis-4-[(hydroxyimino)methyl]pyridinium dimethanesulfonate.
Particle Size Distributions of MMB4 DMS EF Batches Used for Definitive Preclinical Study.
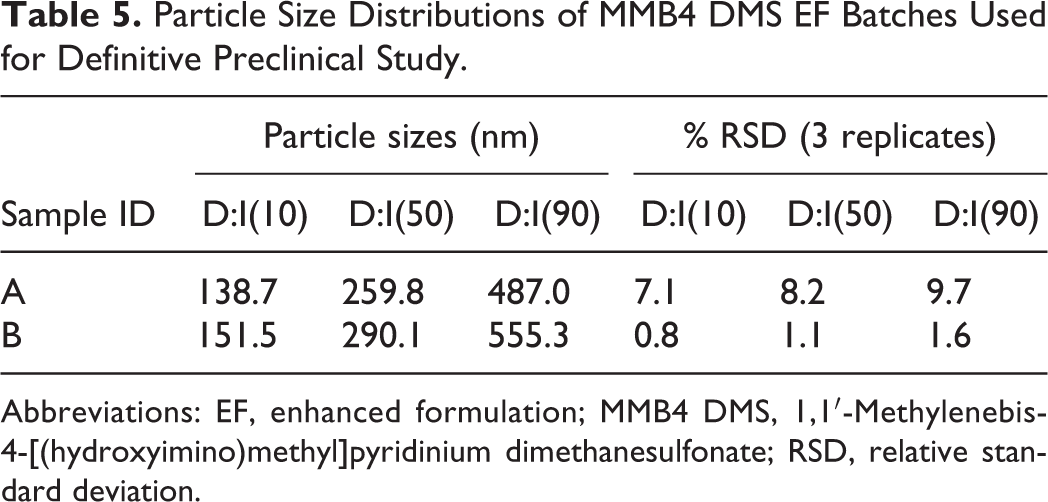
Abbreviations: EF, enhanced formulation; MMB4 DMS, 1,1′-Methylenebis-4-[(hydroxyimino)methyl]pyridinium dimethanesulfonate; RSD, relative standard deviation.
GLP Definitive PK Animal Study
Early preclinical non-GLP MMB4 DMS EF range-finding animal studies demonstrated that the systemic exposure of MMB4 DMS EF in New Zealand White Rabbits following an IM injection was independent of particle size (200-6000 nm) and dose proportional through 120 mg/g. 10 The objective of this GLP bridging confirmatory study was to compare the systemic exposure of MMB4 DMS OF, an aqueous MMB4 DMS formulation that possesses acceptable C max and T max values for the treatment of nerve agent intoxication versus MMB4 DMS EF, a nanoparticle suspension formulation, in male New Zealand White Rabbits following an IM injection. 13 The primary goals of this study were to confirm that the exposure was independent of particle size and to complete the bridge, comparing the systemic exposure of MMB4 DMS OF versus MMB4 DMS EF following an IM injection.
Three formulations were tested in this study, a MMB4 DMS OF aqueous formulation and 2 MMB4 DMS EF nanoparticle formulations (one at 250 nm and one at 300 nm). All formulations were prepared at MMB4 DMS concentration of 100 mg/g and dosed at a level of 25 mg/kg for the rabbit.
These GLP bridging confirmatory studies show no apparent formulation effects on the MMB4 DMS PK parameters related to formulation type, OF versus EF, and confirm exposure was independent of particle size, with in differences attributed to animal-to-animal variability. Observed and predicted C max and T max, area under the curve values, absorption half-life and elimination half-life values, clearance, and apparent volume of distribution are similar across all the 3 groups.
An important manufacturing outcome of these studies was the confirmation that there was no effect on the MMB4 DMS PK due to particle size. This information will be of great value when setting acceptable particle size specification for the manufacture of the MMB4 DMS EF drug product.
Preclinical Dose Delivery Uniformity Study
The purpose of this study was to confirm dose delivery uniformity of 3 animal dose groups for GLP animal study, Comparative Pharmacokinetic Study of Original and Enhanced MMB4 DMS Formulations in the Male New Zealand White Rabbit. 13 Additional syringes were prepared for dose groups 2 and 3 to evaluate the uniformity of the dose containing MMB4 DMS in the nanoparticle suspension formulation. Uniformity was assessed by evaluating the MMB4 concentration of the filled syringe (actual concentration) and the MMB4 concentration of the expelled formulation (effective concentration) from the syringe. The percentage difference was then calculated. Dose uniformity was evaluated at the time of syringe fill and within 2 weeks after dose delivery in animals. The syringes were identified uniquely with a sample ID (which may be different from the parent sample), as they were prepared and are reference to the animal group and syringe number. The results are summarized in Tables 6 and 7.
Dose Uniformity Results for Group #2 (260 nm Particle Size).
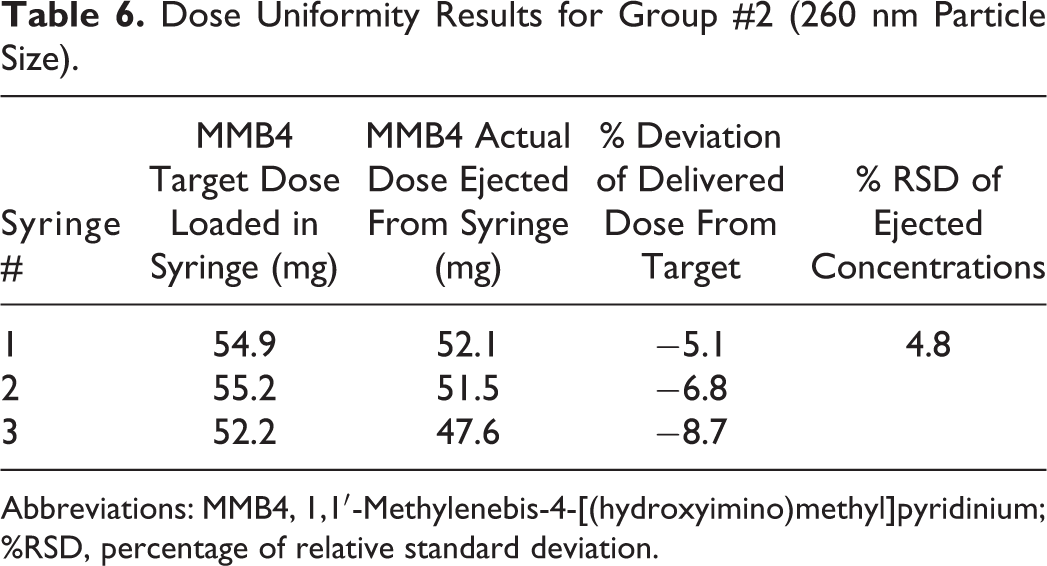
Abbreviations: MMB4, 1,1′-Methylenebis-4-[(hydroxyimino)methyl]pyridinium; %RSD, percentage of relative standard deviation.
Dose Uniformity Results for Group #3 (290 nm Particle Size).
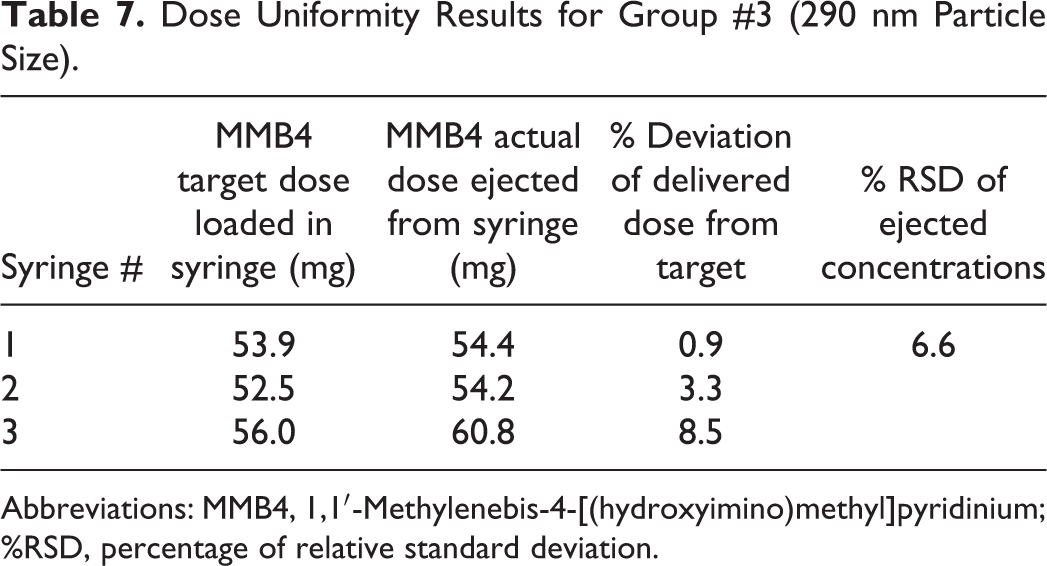
Abbreviations: MMB4, 1,1′-Methylenebis-4-[(hydroxyimino)methyl]pyridinium; %RSD, percentage of relative standard deviation.
Syringes were filled from 2 MMB4 DMS EFs at concentrations of approximately 93 and 89 mg/g and an approximate particle size range of 260 and 290 (D50), respectively. The syringes were filled within 10% of the target dose. Vials were filled from 1 lot of aqueous MMB4 DMS OF at approximately 94 mg/mL.
Comparison of the MMB4 assay values between the target dose and the expelled dose were within 10% of the target dose. Uniformity of dose was achieved within each dose group as demonstrated by the precision of the concentration results of the expelled formulations (Tables 6 and 7).
Current Good Manufacturing Practice Manufacturing
Milling
As in the GLP milling described previously, MMB4 DMS was milled with CSO to reduce the particle size to maintain physical stability and injectability of the suspension. To produce a batch of drug product suitable for use in a human clinical trial, it was necessary to modify the process to comply with cGMP for pharmaceutical manufacturing. In the cGMP milling process, CSO was first charged into a stainless steel vessel equipped with an overhead agitator and blanketed with nitrogen, followed by the addition of MMB4 DMS. The mixture was pumped via a peristaltic pump into the grinding chamber, and the milled sample exiting the chamber was collected in another stainless steel vessel (also equipped with an overhead agitator and blanked with nitrogen). The mixture in the second vessel was then pumped into the grinding chamber. This process was repeated until the desired particle size range was achieved as measured by in-process PSA testing. The temperature inside the grinding chamber was controlled by circulating coolant through the outer jacket. A schematic of the wet milling set up is shown in Figure 3.
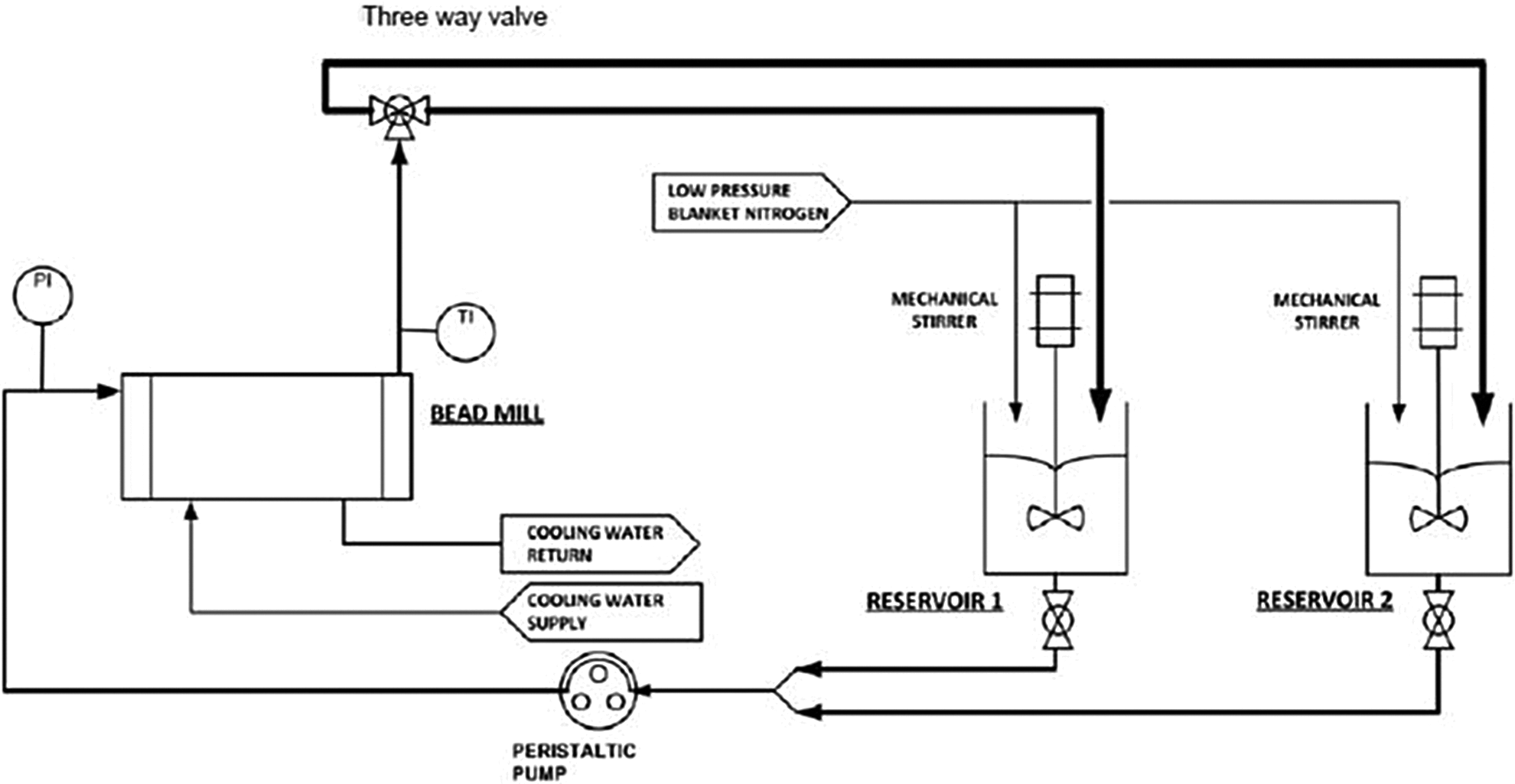
Schematic of wet milling set up.
The MMB4 DMS EF clinical supply batch was wet milled in CSO at 100 mg/g as shown in Figure 3. The mill was charged with 1.5 kg MMB4 DMS and 13.5 kg CSO with 4.4 kg 0.65-mm zirconia grinding beads. The agitator speed was set at approximately 3300 rpm. Samples were collected for particle size analysis starting at 2 hours and every hour thereafter until the measured particle size reached the specified targets (D10 ≤ 160 nm; D50 ≤ 300 nm; and D90 ≤ 550 nm). The intensity-weighted DLS PSA data, using lognormal unimodal fits, are shown in Figure 4. The D10, D50, and D90 represent the particle sizes for which 10%, 50%, and 90% of the particles are smaller than the given size, showing the approximate width of the particle size distribution. The particle size of the MMB4 DMS starting material was approximately 400 microns. 9 It is therefore evident that the majority of particle size reduction occurred within the first 2 hours of milling. Particle size reduction continued thereafter, albeit more slowly, to an equilibrium median (D50) size below 300 nm after 10 hours. The size distribution also narrowed slightly at longer milling times. The final particle size distribution of the clinical trial batch is shown in Table 8.
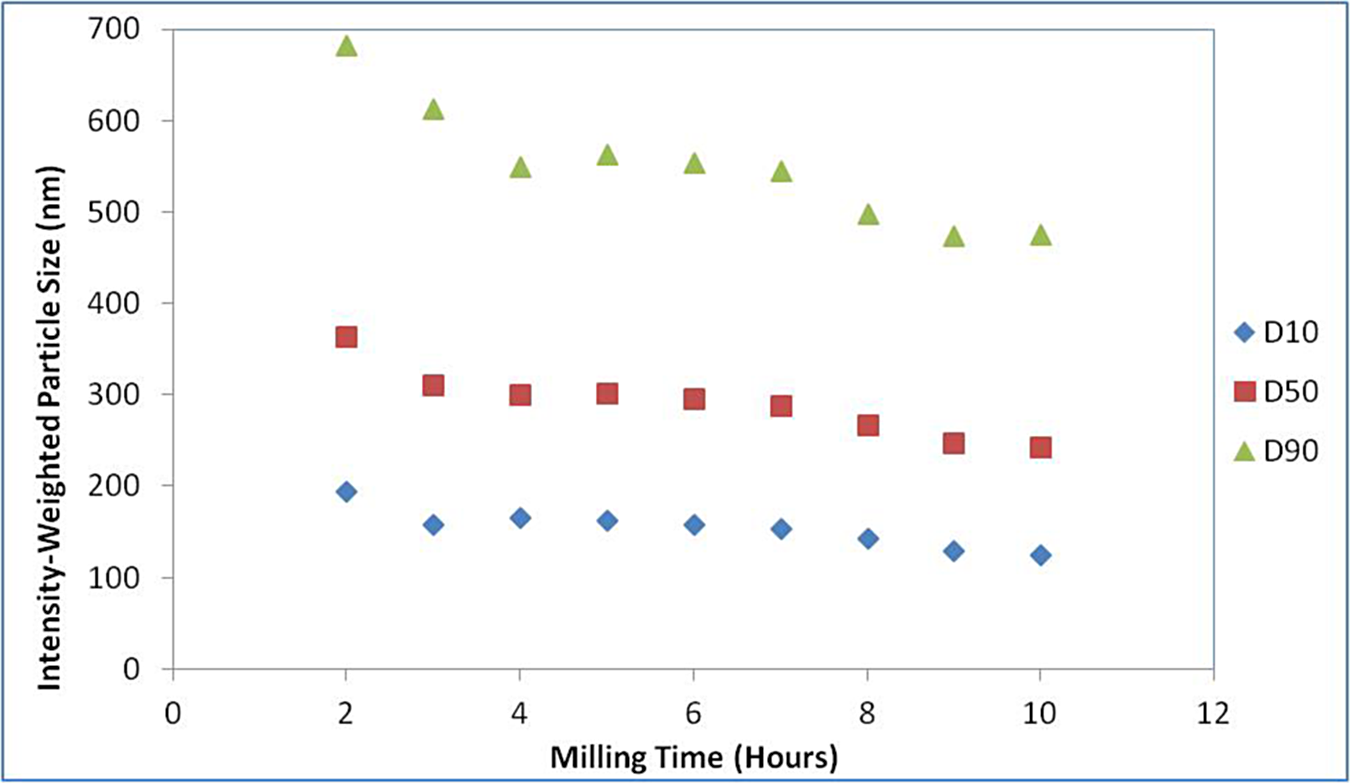
Particle size reduction as a function of bead milling time. D10, D50, and D90 represent the particle sizes for which 10%, 50%, and 90% of the particles are smaller than the given size, showing the approximate width of the particle size distribution.
Particle Size Analysis of EF Clinical Trial Batch.
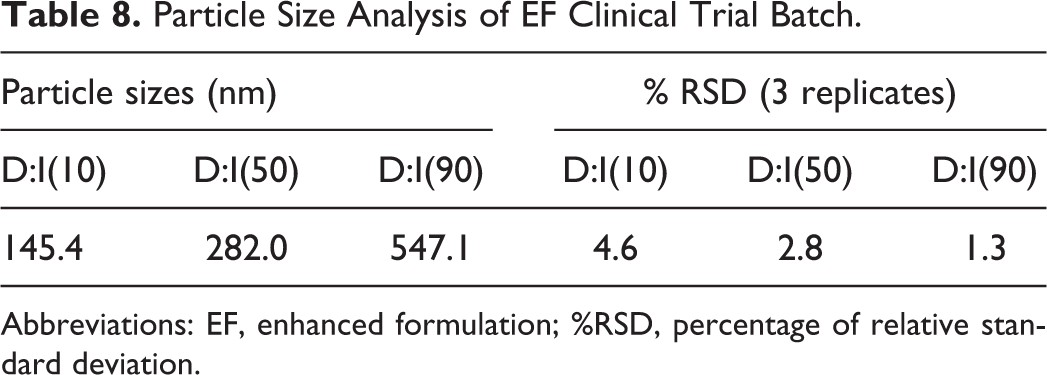
Abbreviations: EF, enhanced formulation; %RSD, percentage of relative standard deviation.
The milled product was tested against the specifications set for the “compounded MMB4 DMS EF” before releasing for sterile filling.
Sterile Fill
A sterile fill was necessary to repackage the MMB4 DMS cGMP bulk product into smaller containers to be used for measuring and administering individual doses during the Phase 1 clinical trial. The cGMP batch was filled into 5-mL borosilicate glass vials under a nitrogen atmosphere, each with 3.43 g of MMB4 DMS EF. The vials were sealed with bromobutyl stoppers and caps with crimped aluminum closures. Due to antimicrobial properties of the MMB4 DMS EF and single-use intentions of the container closure, no preservative was required. Manufacturing practices were used to produce a bulk product with a low bioburden (microbial load). Cottonseed oil, the compounding vehicle for the particle size reduction step and the carrier for the active pharmaceutical ingredient, was filtered through a 0.2-µm sterilizing filter to remove foreign particles and lower the bioburden. Bioburden testing of the bulk product showed zero colony forming units (CFUs). Nonparticle formulations are usually delivered to a filling facility nonsterile. The material is then sterile filtrated through a 0.2-µm filter in a controlled environment before entering the filling cleanroom environment. Due to the nature of the MMB4 DMS EF, a nanoparticle suspension, the formulation was not sterile filtered. However, aseptic filling in a cleanroom into a sterilized container closure did help keep the bioburden low. After inspection for foreign particles, the MMB4 DMS lot was conditionally released from the fill facility for terminal sterilization.
Terminal Sterilization
The clinical supply of MMB4 DMS EF was terminally sterilized by gamma irradiation. This method of sterilization was selected due to higher assurance of product sterility compared to aseptic processing. Because of the size of the suspended particles and the physical properties and nonaqueous suspension nature of the drug product, filtration via a 0.2-µm sterilizing filter was impossible. The use of ethylene oxide or e-beam sterilization was undesirable because of the potential for residual gas and high temperature exposure created by these methods.
Development work leading to the sterilization of the drug product began with high gamma exposures to evaluate the effects on the drug product. Vials containing 3.5 mL of MMB4 DMS EF were irradiated at 25 and 40 kGy, respectively. These doses were verified via dosimeters. The contents of the vials were analyzed by HPLC, and the results were compared to unsterilized drug product. Degradation was noted in the high gamma doses (40 kGy); however, the 25 kGy irradiated vials showed no effect when compared to the controls. The lower dose, 25 kGy, was selected as the maximum tolerated dose for the drug product.
The bacteriostatic and fungistatic potentials of the MMB4 DMS EF drug product were challenged with Bacillus subtilis, Candida albicans, and Aspergillus brasiliensis. 17 Sterile samples of the drug product were inoculated with each organism and placed in separate culture vessels. Positive controls were set up simultaneously with each organism seeded into the culture medium. All cultures were incubated at 28°C to 32°C for 5 days. The standard plate count method was used to determine the microbial count (CFUs) challenging each test or control system. Bacteriostatic and fungistatic characteristics were not shown to be associated with the drug product.
Bioburden determination of a drug product was the first step in determining the sterilization dose. It was important to evaluate the bacterial load of a drug product to ensure the sterilization dose is effective. Bioburden recovery testing was performed on the drug product to validate the processing methods used to determine bioburden. Insufficient recovery would result in underestimation of the drug product’s true bioburden and could lead to an inadequate sterilization dose. The recovery data from validation testing provided a recovery factor that is applied to results obtained by routine bioburden testing. This simply normalizes the results based on the bioburden recovery study. In the current study, bioburden testing was performed on 3 sterile vials of MMB4 DMS EF. Each vial was inoculated with Bacillus atrophaeus, incubated inverted at 30°C to 35°C for 48 hours, and then counted using the total plate count method. The estimated efficiency of the method was established as a percentage of the microorganisms recovered based on the inoculum population. The validated recovery for the bioburden procedure was 97%.
Bioburden testing of MMB4 DMS EF followed to evaluate the aerobic bacterial load of the drug product. This quantitative test was performed to determine the number of viable microorganisms present in the drug product. 18 Ten vials were tested per specifications. The average CFUs detected was 5 per vial. Based on the bioburden validation recovery of 97%, this bacterial load was adjusted to 5.2 CFU/vial.
Due to the maximum tolerated dose (25 kGy) determined during development, the option to pursue a sterilization dose that followed the VDmax protocol 19 was selected. Using table 4 of Association for the Advancement of Medical Instrumentation Technical Information Report (AAMI TIR) 33: 2005, the maximum tolerated dose of 25 kGy, and the adjusted average bioburden of 5.2 CFU/vial, the appropriate sterilization doses to provide a 10−6 sterility assurance level (SAL) were VDmax17.5, VDmax20.0, VDmax22.5, and VDmax25.0 (superscripts denote radiation doses in kGy). Because a dose range must be set in order to irradiate the drug product, the lowest VDmax dose was selected. Using this minimum sterilization dose of 17.5 kGy and the adjusted average bioburden of 5.2 CFUs, a verification dose was determined using table A.2 of AAMI TIR 33: 2005. The verification dose for the MMB4 DMS EF was 3.3 kGy.
In accordance with the AAMI TIR 33:2005 for Method VDmax17.5, 10 vials of drug product were randomly selected from the single batch of MMB4 DMS EF drug product (SwRI Lot CEF-08032011) and irradiated at the verification dose of 3.3 kGy + 10%. Sterility testing of the 10 vials incubated over a 14-day period indicated no positive sterility vials. Since the 10 units of product irradiated at the 3.3 kGy met the acceptance criteria (no more than one positive test) set forth in the AAMI guidelines for test of sterility, then the minimum routine sterilization dose to assure a 10−6 SAL would be 17.5 kGy for this single production batch.
The MMB4 DMS EF drug product was gamma irradiated at a range of 17.5 to 25 kGy. Confirmation of doses was verified via dosimeters placed throughout the contents of the boxes containing the drug product. All irradiation doses were within the specified range. Sterility of the drug product was confirmed during release testing of the clinical product at the analytical facility. Following issuance of a Certificate of Analysis, ICH stability studies were initiated.
ICH Stability Study
ICH stability studies have been initiated to evaluate stability, shelf life, and potential degradents. The MMB4 DMS EF degrades slowly and primarily to 4-PA that is tracked as a precise marker for stability. Analysis results from samples stored at accelerated conditions indicate that the drug product is stable at 50°C and 60°C. Storage conditions for the drug product have been set to 20°C to 25°C.
Samples of the drug product have been placed on stability according to the plan shown in Table 9. Stability testing will be conducted for 36 months.
Stability Test and Time Points for MMB4 DMS EF 100 mg/g.
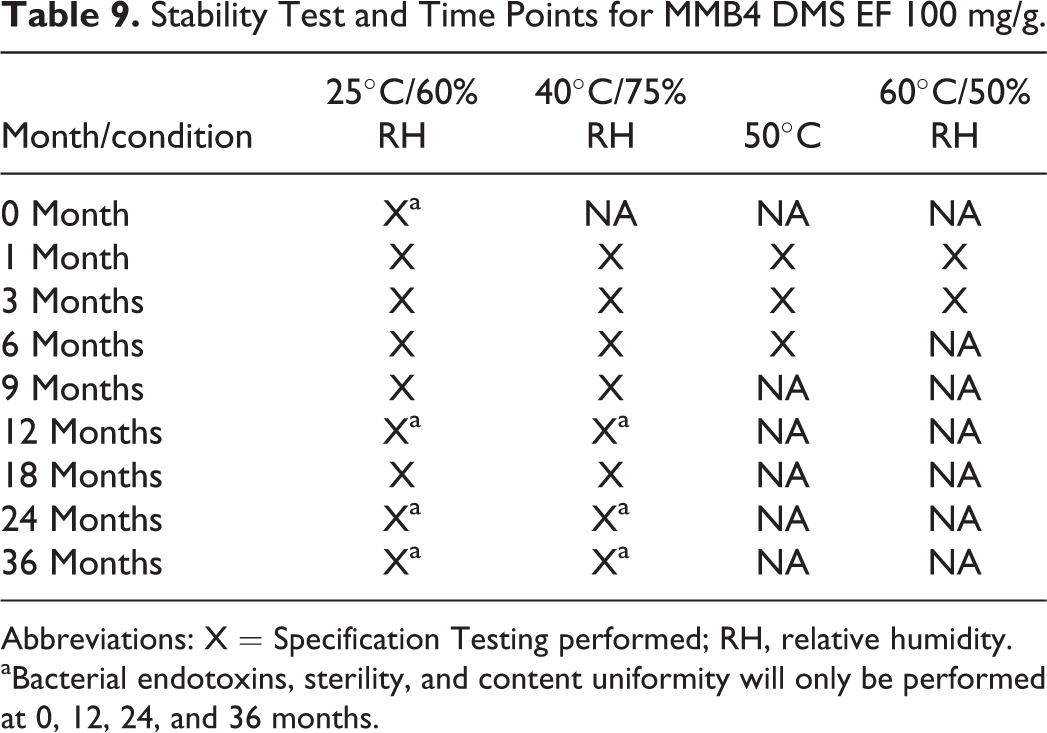
Abbreviations: X = Specification Testing performed; RH, relative humidity.
aBacterial endotoxins, sterility, and content uniformity will only be performed at 0, 12, 24, and 36 months.
Specification testing for MMB4 DMS EF will include all of the following: appearance, identity (IR), identification (HPLC), MMB4 concentration, related substances, 4-PA, particle size, foreign matter, Water (Karl Fischer), bacterial endotoxins (at 0, 12, 24, and 36 months only), sterility (at 0, 12, 24, and 36 months only), and content uniformity (at 0, 12, 24, and 36 months only).
Data have been obtained up to the 9-month time point. Stability-indicating parameters, which include MMB4 assay and known impurity (4-PA), are provided in Figure 5 and Figure 6 for the respective time points.
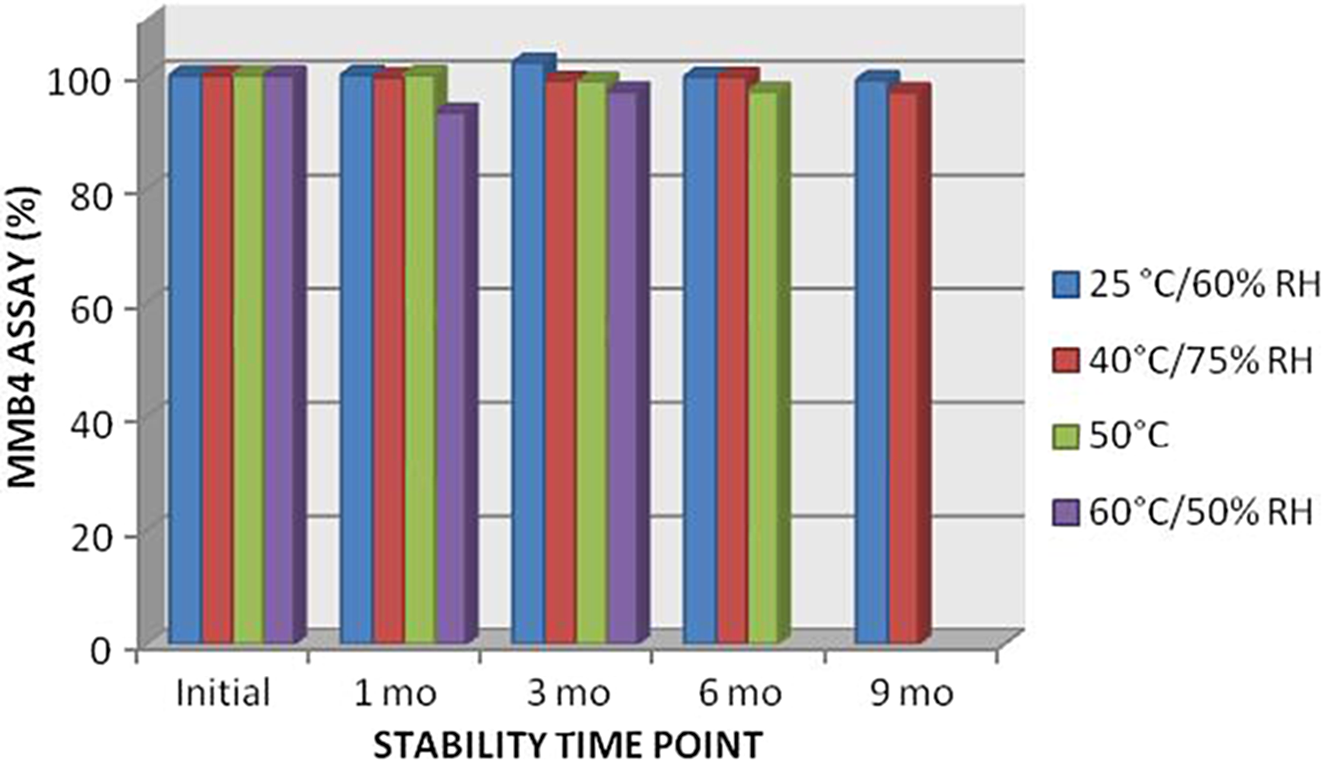
MMB4 assay values from Phase 1 clinical lot through 9 months. 60 °C samples were held for a maximum of 3 months, and 50 °C samples for 6 months. MMB4 indicates 1,1'-methylenebis-4-[(hydroxyimino)methyl]pyridinium.
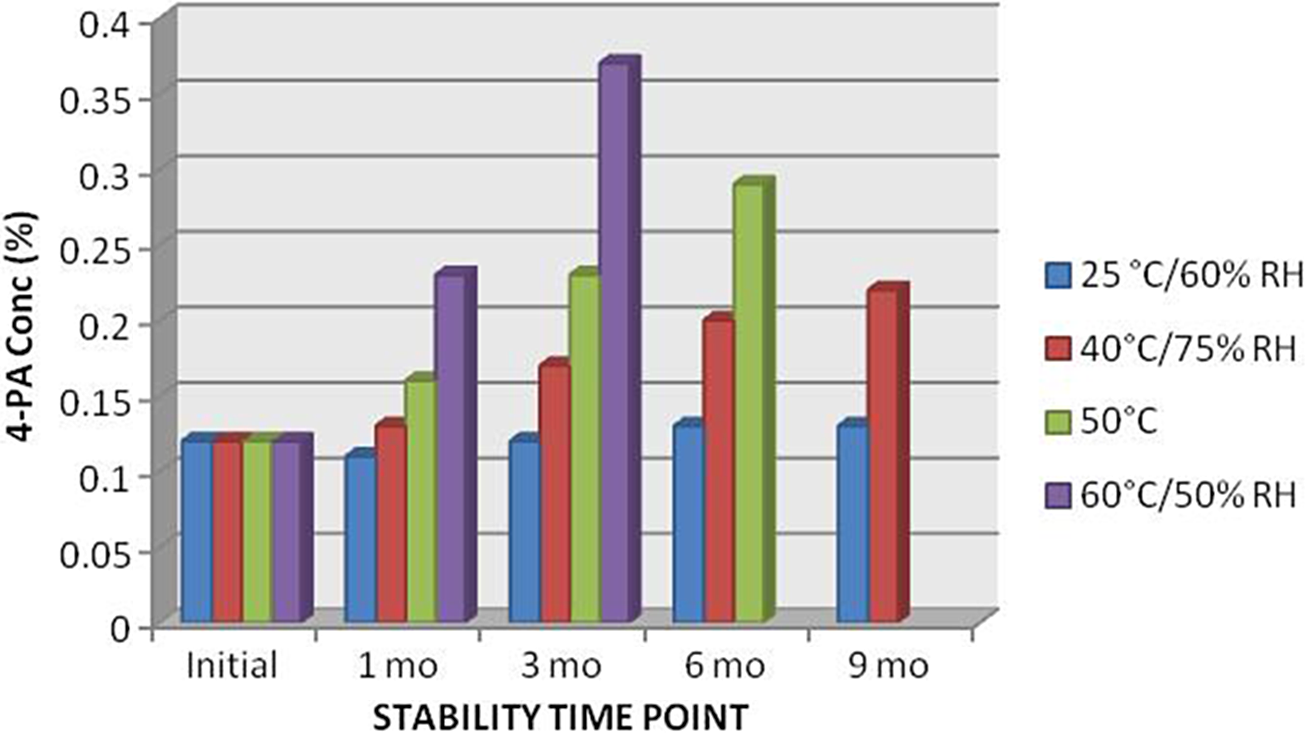
4-pyridine aldoxime concentration from Phase 1 clinical lot through 9 months. 60 °C samples were held for a maximum of 3 months, and 50 °C samples for 6 months.
The stability of the drug product is being maintained at the standard storage conditions of 25°C/60% relative humidity. At this condition, the drug product maintains its stability as indicated by the assay value that remains approximately 100%. Additionally, degradation captured by monitoring 4-PA concentration is less than 0.15%. Conclusions regarding shelf life and additional degradents will be made after stability testing is complete under this plan.
The clinical lot of drug product is currently being stored and stability monitored. The clinical supply material has been distributed to the clinical site for the Phase 1 clinical study.
Clinical Dose Delivery Uniformity Study
A study to evaluate dose delivery uniformity of MMB4 DMS EF was conducted. The MMB4 DMS EF is being used in a Phase 1 clinical study (A Phase 1 Double-Blind, Placebo-Controlled, Single-Center Dose Escalation Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of Intramuscular Administration of a New Oxime, MMB4 Dimethanesulfonate (DMS) Enhanced Formulation (EF)) in human volunteers. The dose delivery method was developed to ensure uniformity of the dose being delivered to human participants. This method was transferred to the clinic to ensure the accurate preparation and administration of the drug product to human participants by the clinical staff. Two pharmacists were trained on this procedure, and their were efforts verified based on the assay and the delivered weight of the doses prepared were evaluated.
The clinical lot of MMB4 DMS EF is supplied in single-use 5-mL borosilicate vials. Eighty-five vials were sent to the clinic for training, familiarization, and dose preparation. The pharmacists were trained to handle and prepare doses, including an overview of the handling, preparation, and calculations for dose determination.
To ensure homogeneity, the contents of the vials were thoroughly shaken by hand for no less than 30 seconds. Subsequent mixing by vortex for at least 1 minute ensured visible homogeneity of the drug product. The pharmacists were instructed to fill syringes (1, 3, and 6 mL with attached needles) according to the dose-escalation protocol in the Phase 1 clinical study for patients ranging from 70 to 85 kg.
Weight tolerances were calculated for the drug product to ensure the delivery of the product to be within target specification. Six syringes for each dose group were loaded to the specified weights according to the dose-escalation protocol. The actual dose weights as percentages of target weights are documented in Figure 7.
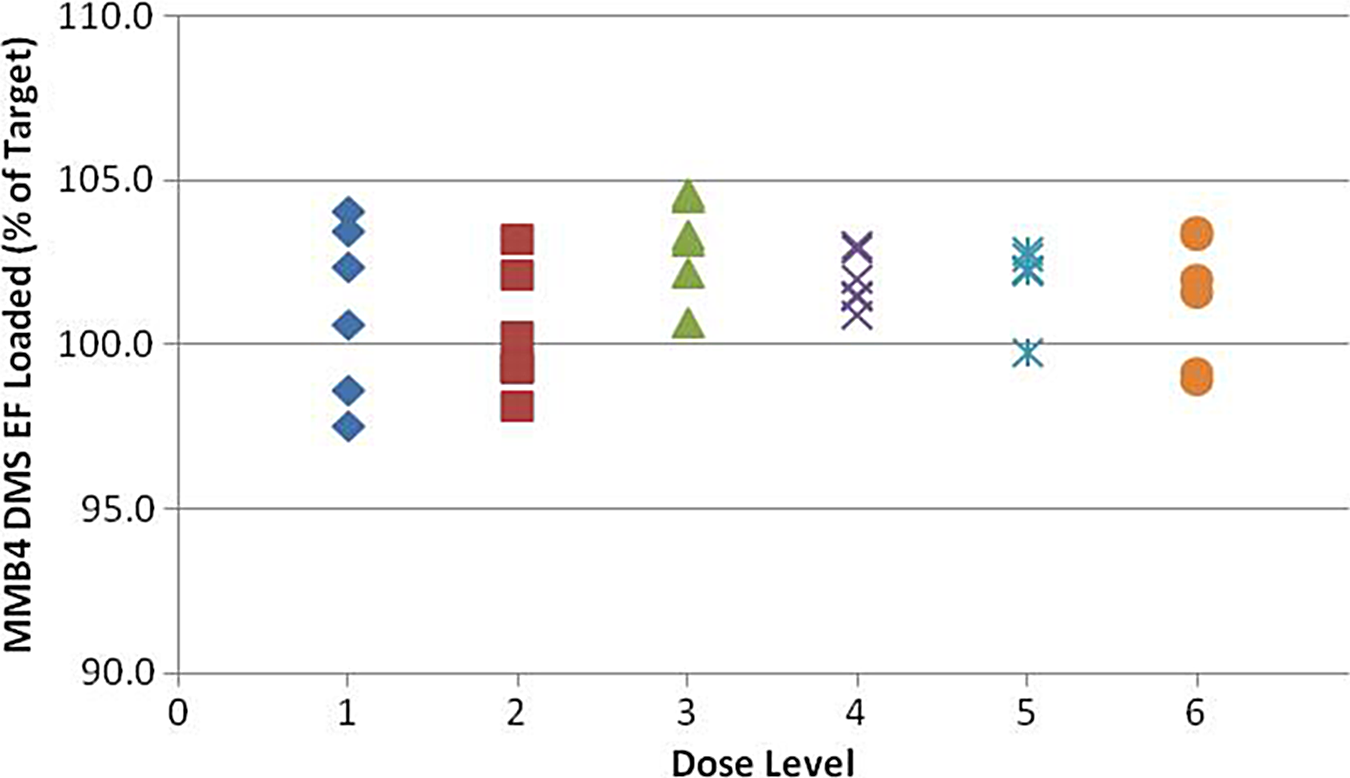
MMB4 DMS EF loading per dose group (by weight). MMB4 DMS indicates 1,1′-methylenebis-4-[(hydroxyimino)methyl]pyridinium dimethanesulfonate; EF, enhanced formulation.
The syringes were filled to the specified weight range. All weights were verified with the use of a 4 place balance. To confirm the delivery of the weights, the contents of each syringe were expelled into a vial. The weights of the delivered doses were verified according to each dose-escalation group and summarized in Figure 8.
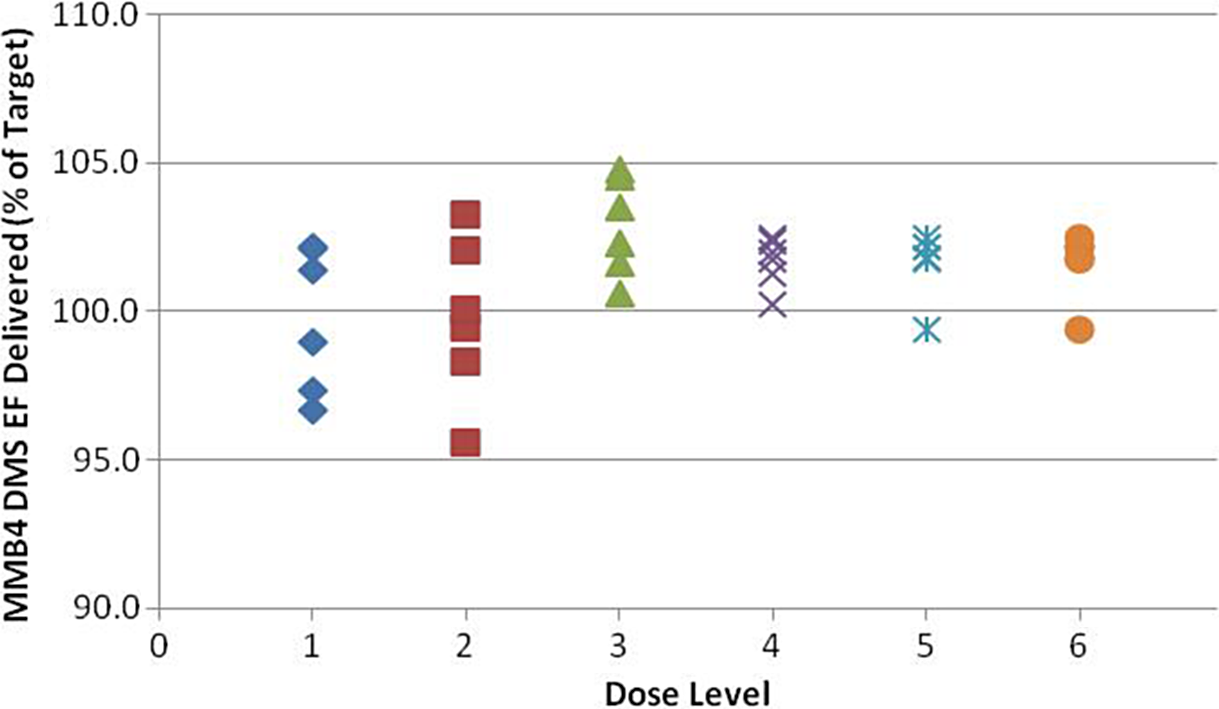
MMB4 DMS EF delivered from syringes (by weight). MMB4 DMS indicates 1,1′-methylenebis-4-[(hydroxyimino)methyl]pyridinium dimethanesulfonate; EF, enhanced formulation.
Confirmation of the dose in the drug product was determined by HPLC assay. Assay values for all syringes filled with MMB4 DMS EF were within 90 to 110% (10% of the target dose) for each respective dose group. Each pharmacist filled 3 syringes per dose group for a total of 18 syringes. The assay results for each pharmacist are presented in Figure 9.
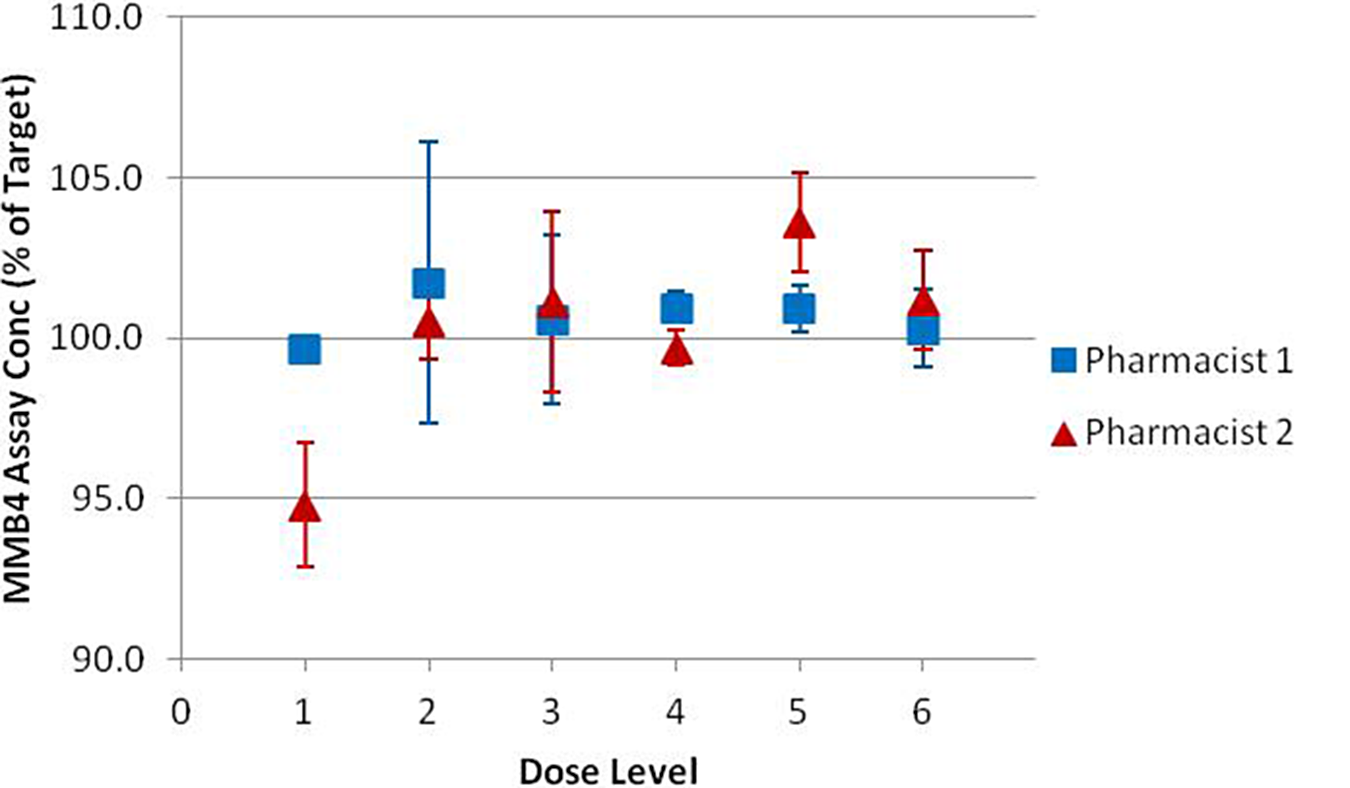
MMB4 assay concentration results from each pharmacist.MMB4 indicates 1,1′-methylenebis-4-[(hydroxyimino)methyl]pyridinium.
Stability of the drug product was determined by evaluating a filled syringe 24 hours after filling. The negligible amount of 4-PA found in MMB4 DMS EF at T = 0 and T = 24 hours indicates that the samples (filled syringes) are stable for at least 24 hours. As such, the syringes could be prepared, if necessary, 24 hours prior to administration without the risk of degradation.
The loading recommendations for the Phase 1 dose-escalation study of MMB4 DMS EF ensure the drug product is delivered within 10% of the target dose.
Conclusion
We have developed and manufactured a novel nerve agent antidote based on a viscous suspension of MMB4 DMS nanoparticles in CSO. This EF has superior thermal stability to aqueous MMB4 and all other aqueous bis-pyridinium oxime formulations and remains injectable due to shear thinning of the non-Newtonian fluid. As demonstrated by the results reported herein, we have successfully developed and implemented GLP and cGMP manufacturing processes to produce batches of MMB4 DMS EF for preclinical and clinical studies, respectively. We have also developed and validated a particle size analysis method using DLS.
Manufacturing of EF was accomplished by milling MMB4 DMS with CSO and zirconia beads in an agitator bead mill at solid concentrations of approximately 100 mg/g to produce particle size distributions with median sizes between 250 and 300 nm. The clinical trial batch production milling was carried out under cGMP conditions. The resulting bulk material was filled into 5-mL glass vials at a sterile fill facility and terminally sterilized by gamma irradiation. The clinical lot was tested and released, a Certificate of Analysis was issued, and a 3-year ICH stability study started. The drug product was placed in storage for Phase 1 clinical trial distribution. A dose delivery uniformity study was undertaken to ensure that the correct doses would be delivered to the patients at the clinic.
The MMB4 DMS EF is a promising candidate for a next-generation nerve agent antidote emergency treatment system. The MMB4 has shown efficacy against a broad spectrum of nerve agents, 7 and the EF delivery system enables a much longer shelf life compared to traditional aqueous bis-pyridinium oxime formulations. The EF platform of a viscous nanoparticle suspension is also being investigated with other APIs and holds great promise as a delivery system capable of significantly increasing the stability shelf life of any drug or other compound that is subject to hydrothermal degradation.
Footnotes
Acknowledgments
The authors acknowledge the editorial assistance of Dr Vincent Brown.
Authors’ Note
The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or reflecting true views of the Department of the Army or the Department of Defense.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the U.S. Army Chemical Biological Medical Systems under SwRI grant number W9113M0810001 and Battelle contract number SP070000D3180DO0599.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.