
Abstract
The application of patient-derived three-dimensional culture systems as disease-specific drug sensitivity models has enormous potential to connect compound screening and clinical trials. However, the implementation of complex cell-based assay systems in drug discovery requires reliable and robust screening platforms. Here we describe the establishment of an automated platform in 384-well format for three-dimensional organoid cultures derived from colon cancer patients. Single cells were embedded in an extracellular matrix by an automated workflow and subsequently self-organized into organoid structures within 4 days of culture before being exposed to compound treatment. We performed validation of assay robustness and reproducibility via plate uniformity and replicate-experiment studies. After assay optimization, the patient-derived organoid platform passed all relevant validation criteria. In addition, we introduced a streamlined plate uniformity study to evaluate patient-derived colon cancer samples from different donors. Our results demonstrate the feasibility of using patient-derived tumor samples for high-throughput assays and their integration as disease-specific models in drug discovery.
Introduction
High-throughput cancer cell line screening assays are fundamental tools to evaluate drug sensitivity patterns and pharmacogenomic profiling that may guide early-phase clinical trials of novel agents and rational cancer therapeutic strategies. 1 Traditional two-dimensional (2D) cell-based assays are commonly employed to determine the potency of active lead molecules; however, their value in predicting clinical response to novel agents is limited. In recent years, the importance of the tumor microenvironment and the three-dimensional (3D) aspects of solid tumors have promoted strong interest in more precisely mimicking tumor cell growth in vitro. 2 Consequently, novel complex 3D culture systems and more sophisticated xenograft models are currently reinvigorating translational research efforts and predictive biomarker development studies.3–8
Recently, Sato and colleagues 9 succeeded in discovering the growth conditions for mouse intestinal cells that self-organize into so-called organoid cultures. Organoids are developed by seeding dissociated tissue-derived cells into a 3D semisolid extracellular matrix and expanding these cells in defined medium.9,10 The methodology was subsequently refined to establish human organoid cultures for different tumor entities.10–13 Furthermore, the possibility of expansion and storage of an organoid biobank enables the long-term use of patient-derived organoid cultures as experimental tools in basic and clinical research. 14 The application of these physiologically relevant cultures as a new generation of test platforms for future drug discovery efforts could bridge the gap between primary 2D cell-based screening and animal and human trials.15,16 However, the implementation of novel cell-based assay systems to promote a successful development and selection of active compound leads requires reliable and robust test platforms.
Here we apply patient-derived 3D (PD3D) colon cancer organoid samples as 384-well–based cultures to examine whether in vitro assays that comprise primary patient-derived tumor material would be amenable for an automated liquid-handling platform. We focused our study on the validation of assay robustness and reproducibility by running plate uniformity and replicate-experiment studies. In addition, we used various patient-derived colon cancer culture strains to validate the assay conditions and performance on a different organoid sample set.
Materials and Methods
Human Primary and Metastatic Colon Cancer Tissues
All tumors were obtained from patients with high-grade colon carcinoma at clinical centers in Germany (Charité Universitätsmedizin Berlin–Campus Benjamin Franklin, Berlin) and Austria (Medical University of Graz, MUG, Graz) under the regulations of the respective ethical boards. Resident pathologists inspected and classified all resected colon carcinoma tissues. The following patient-derived samples (based on the OncoTrack ID system) were used for this study: 159-MB-P-TF-01-03, 250-MW-P-TF-01-03, 327-MB-P-TF-01-03, and 364-CB-M-MF-01-04.
Generation and Propagation of Patient-Derived Organoid Cell Cultures
Upon receipt of resected colon carcinomas, fatty and necrotic tissue was removed macroscopically. Tumor tissue was rinsed with Hank’s Balanced Salt Solution (Gibco), manually minced using sterile scalpels, and digested in Advanced DMEM/F12 (Gibco) supplemented with 1x P/S, collagenase IV (Sigma-Aldrich), DNaseI (AppliChem), Dispase (StemCell Technologies), and amphotericin B (Sigma-Aldrich) at 37 °C for 60 min. During incubation, the tissue fragments were repeatedly suspended with a 10 mL pipette. The digestion was stopped after 1 h by pelleting the suspension at 300g for 3 min and resuspension in Advanced DMEM/F12, supplemented with 1x penicillin/streptomycin (P/S). To exclude crude undigested tissue fragments but retain multicellular aggregates, 17 the suspension was filtered through a 100 µm cell strainer (Corning). The flow-through was subjected to consecutive filtration using a 40 µm cell strainer. Retained cell aggregates were recovered by rinsing with 10 mL Advanced DMEM/F12 and transferred into a Petri dish. The 40 to 100 µm aggregates and <40 µm flow-through were centrifuged at 300g for 3 min. After depletion of red blood cells using Red Blood Cell Lysis Solution (Miltenyi) and a final pelleting step, both fractions were separately mixed with phenol-red free, growth factor–reduced Matrigel (Corning) and seeded into 24-well plates in 20 µL aliquots. Solidified droplets were carefully overlaid with 500 µL of culture medium (Advanced DMEM supplemented with 1x GlutaMAX, 1x P/S, 10 mM HEPES buffer, 1x N2 Supplement, 1x B27 Supplement [all Gibco], 1 mM N-Acetyl-L-cysteine [Sigma-Aldrich], 20 ng/mL basic fibroblast growth factor [New England Biolabs], and 50 ng/mL epidermal growth factor [Sigma]) according to published protocols. 10 During the first week, 1.25 µg/mL amphotericin B and 10 µM ROCK-II inhibitor Y27632 (Sigma-Aldrich) were added to cultures. The cultures were passaged when the aggregates reached a diameter of approximately 800 µm. Cellular aggregates were released from Matrigel by adding 5 mL Advanced DMEM/F12 and centrifugation. Pellets were digested with TrypLE (Gibco). Trypsinization was stopped with 5 mL Advanced DMEM/F12, and digested cell clusters were replated on a 12-well plate.
Microscopy Analysis
For immunofluorescence imaging, patient-derived organoid cultures were fixed with 4% paraformaldehyde and permeabilized with 0.05% Tween-20 for 30 min. The samples were then washed in phosphate-buffered saline (PBS) with 7.5% bovine serum albumin (BSA). F-actin was stained accordingly with TRITC-labeled phalloidin (Sigma Aldrich). Anti-Ki-67 conjugated to Alexa Fluor 488 (Abcam) was incubated at 4 °C for 24 h and removed by washing with PBS with 7.5% BSA. Nuclei were stained with DAPI (Sigma Aldrich) for 30 min; residual DAPI was washed off with PBS. For image analysis, PBS was removed from wells and mounting medium was added. Microscopy was performed with a Zeiss Axiovert 400 microscope.
For the time-lapse analysis, the growth of the organoid cultures in 384-well plates was monitored using an HC PL APO 10X/0.40 AN (10×) objective, a Hamamatsu ORCA-AG CCD camera, and an inverted motorized microscope (Leica DMI 6000B) coupled with an incubation system to control the temperature and CO2 levels during the course of the experiments. Images were taken every 15 min for 72 h using Leica LAS AF software (version 2.4.1). Confocal microscopy was carried out using a Leica TCS SP5 X confocal microscope equipped with a resonant scanner, a dry 20× Plan Apochromatic, 0.7 AN objective, and Leica LAS AF software (version 2.4.1) for image capturing and the Imaris software (Bitplane) for image analysis.
Semiautomated High-Throughput Drug Response Assays
Organoid cultures were digested with TrypLE (Gibco) until a single-cell suspension was achieved. Disaggregation was stopped with Advanced DMEM/F12, and cells were counted. Suspended in growth factor–reduced Matrigel (Corning), 5000 cells per well were seeded into 384-well plates using a robotic platform (Tecan Freedom EVO MCA 96;
Being,
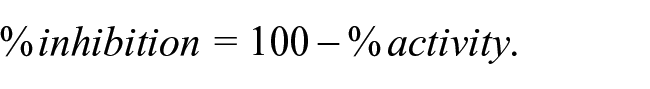
To obtain IC50 values, the four-parameter nonlinear logistic equation (four-parameter logistic CRC) was used:
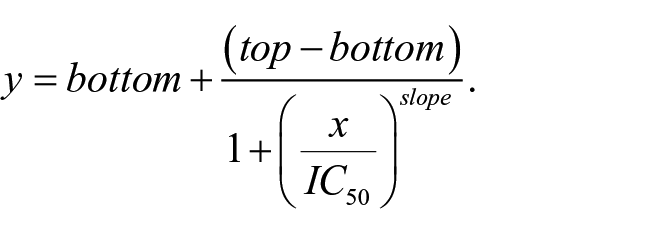
Results
Establishment of Patient-Derived Colon Cancer Organoid Cultures in 384-Well Format
Primary patient-derived colon cancer samples were cultured and expanded as 3D organoid models in Matrigel droplets in a 12-well format as previously described.
14
During the culture period, small organoids developed into more complex and larger organoids characterized by numerous budding structures (
Fig. 1A
,
B
). Notably, even complex organoid structures showed structural integrity and Ki-67–positive cells at the outer surface indicating sustained growth and regular morphogenesis during long-term culture (
Fig. 1C
,
D
). To determine whether patient-derived cells could be seeded and subsequently cultured as organoid structures in 384-well format, we aimed at establishing a workflow that entails controlled assay starting conditions with automated liquid-handling platforms (
Fig. 1E
;
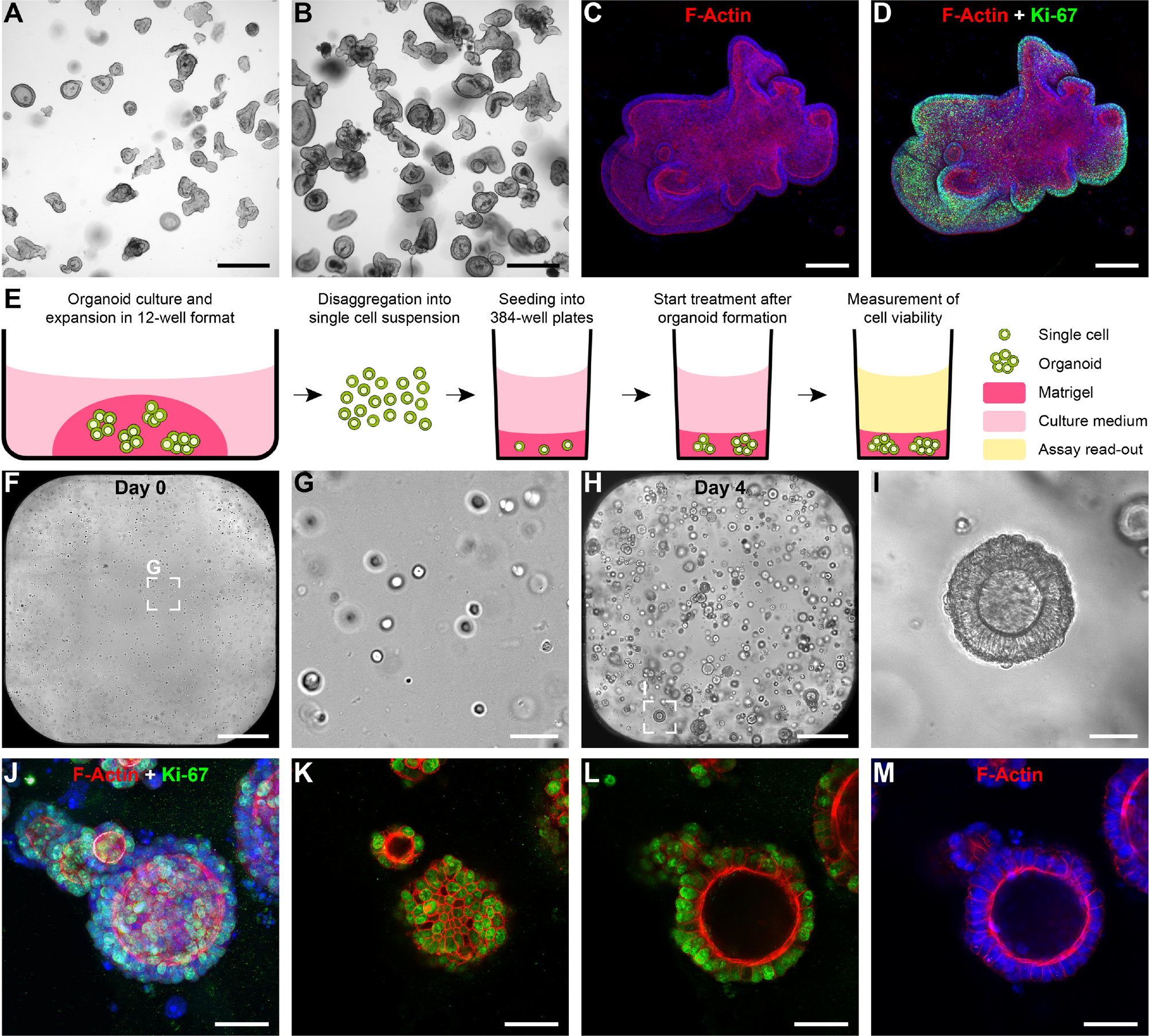
Establishment of patient-derived organoid cultures in 384-well format. (
Spatial Uniformity Assessment of 384-Well Based Organoid Cultures
Given the well-known probability of plate edge or side effects in high-throughput cell-based assay formats due to evaporation during long incubation periods, there has been much interest in developing statistical tools and optimizing the experimental conditions to correct these types of systemic errors.18,19 To evaluate the edge and drift effects in our patient-derived organoid assay, we precultured 384-well plates and added medium containing 0.25% DMSO (vehicle control) at day 4 into all wells. Upon additional 4 d in culture, the plates were analyzed by luminescence measurement of ATP consumption, and the values for the relative luminescence units (RLU) were visualized with the well-level location ( Fig. 2 ). We observed irregular spatial distribution of luminescence/RLU(s) throughout the plate with high values located at the outer wells in the columns 1 and 24 and in the rows A and P ( Fig. 2A , B ). Sealing membranes for multiwell plates are designed to allow effective gas exchange while reducing evaporation. To reduce these well-to-well variations, we tested sealing membranes covering the 384-well plates for defined culture periods. First, we employed the sealing membrane after the preculture period when the vehicle control was added to the plates. We observed a slight reduction of the irregular spatial distribution; however, higher values could still be detected at the corners of the plates ( Fig. 2C , D ). In contrast, when the sealing membranes were used throughout the whole incubation time, including both the preculture period and the treatment period, the RLU values were evenly distributed with no signs of drift or edge effects ( Fig. 2E , F ). These changes reduced the coefficient of variation (CV) across all plates from 19% to 9% ( Fig. 2G ). Per the Assay Guidance Manual, 20 we assessed row and column drift for each of these plates. The largest difference between row means, as a percentage of the overall plate mean, was 59%, 29%, or 17%, in Figures 2A , 2C , and 2E , respectively. Similarly, the column drift results were 37%, 29%, and 15%. Employing sealing membranes throughout the complete assay period might affect the throughput; however, the optimized experimental conditions significantly reduced irregular spatial distribution and well-to-well variations.
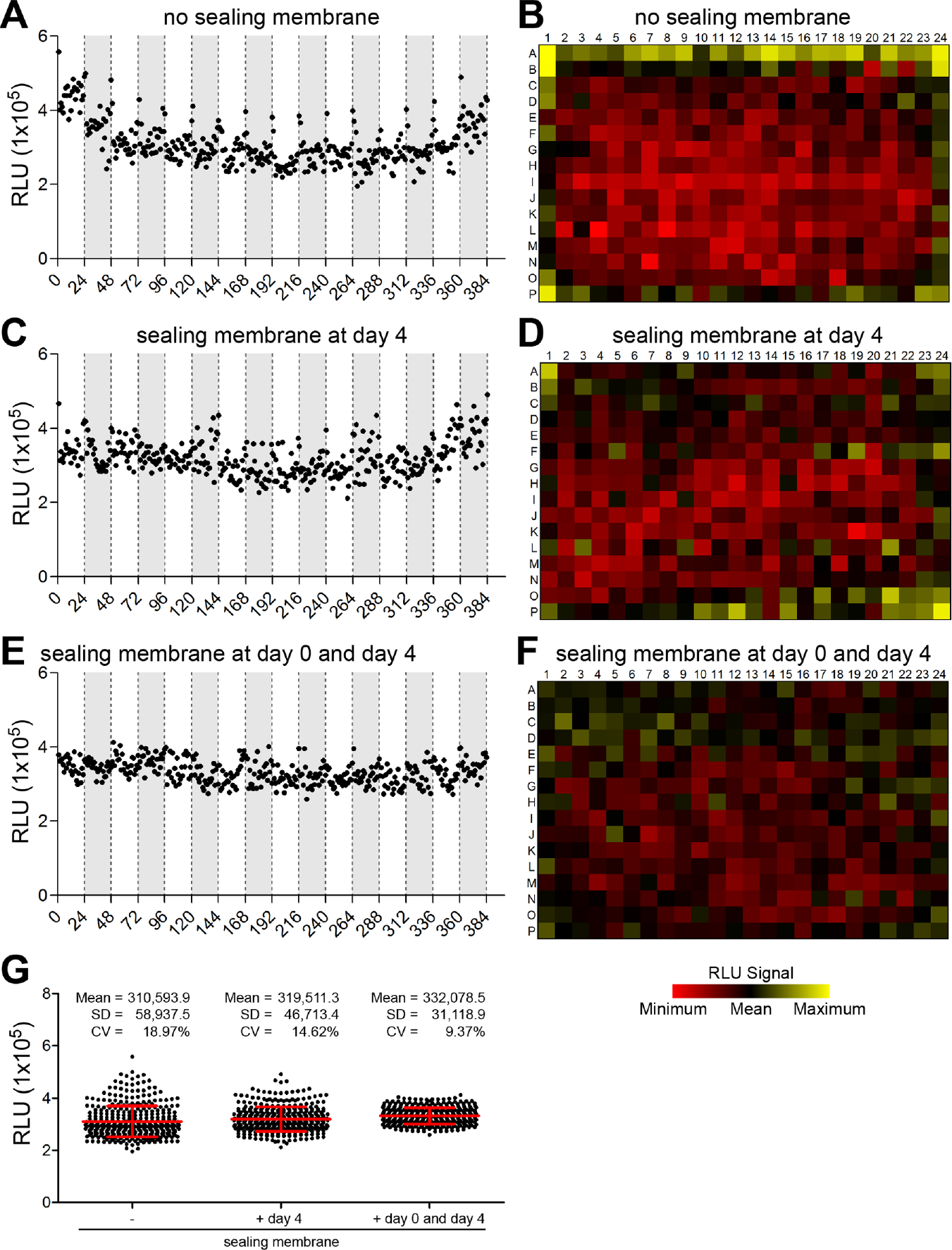
Spatial uniformity assessment with optimized assay conditions. Patient-derived organoid samples in 384-well format were precultured for 4 d and subsequently treated with the vehicle control. (
Assay Validation of Organoid Cultures in 384-Well Format by Plate Uniformity Study
New high-throughput screening assays should be comprehensively validated for robustness of assay performance and pharmacologic relevance.
20
The plate uniformity study is required to assess uniformity and separation of the assay readout signals. We performed a plate uniformity study over three independent runs with six uniform signal plates for Max (maximum signal) and Min (minimum signal) signals and with six CRC plates (
Fig. 3
;
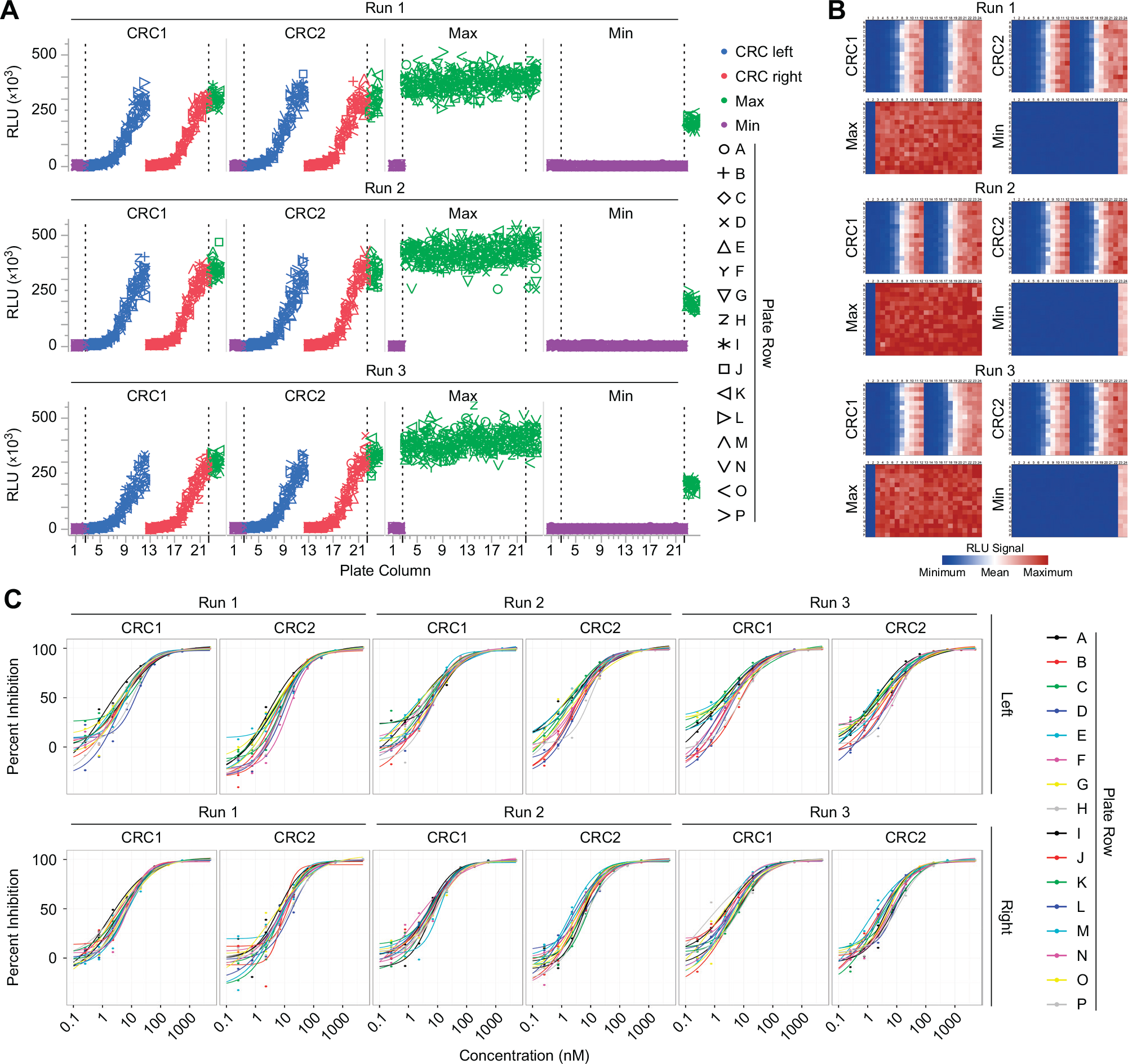
Assay validation by plate uniformity study. The plate uniformity study was carried out over three independent runs consisting of two concentration-response curve (CRC) plates (CRC1 and CRC2), one plate with the maximum signal (Max) and one plate with the minimum signal (Min) for each run for the patient-derived organoid sample 250-MW-P-TF-01-03. (
Results of the plate uniformity study. a
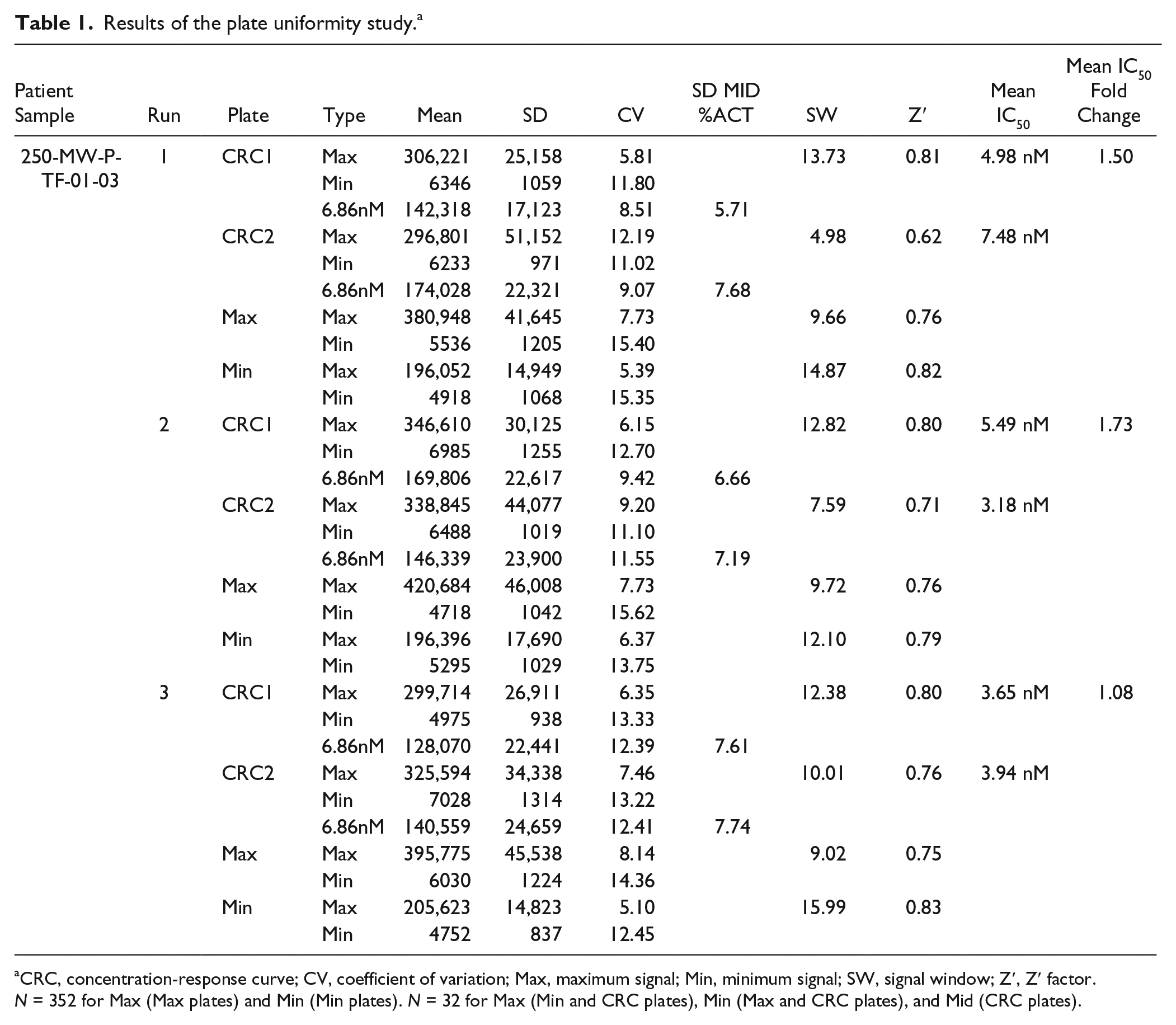
CRC, concentration-response curve; CV, coefficient of variation; Max, maximum signal; Min, minimum signal; SW, signal window; Z′, Z′ factor. N = 352 for Max (Max plates) and Min (Min plates). N = 32 for Max (Min and CRC plates), Min (Max and CRC plates), and Mid (CRC plates).
Assay Transition to Organoid Culture Strains Derived from Different Patients
Our results suggest that a novel assay platform for PD3D colon cancer organoid cultures could be established and validated in 384-well format. However, it is important to note that assay variations of different patient-derived culture strains could occur. For that reason, we aimed at evaluating additional organoid cultures from three different colon cancer patients (sample IDs 159-MB-P-TF-01-03, 327-MB-P-TF-01-03, and 364-CB-M-MF-01-04) by plate uniformity assessment (
Fig. 4A–D
). Given the complex and time-consuming expansion period to obtain sufficient material to perform a full plate uniformity study over three runs (i.e., 12× 384-well plates;
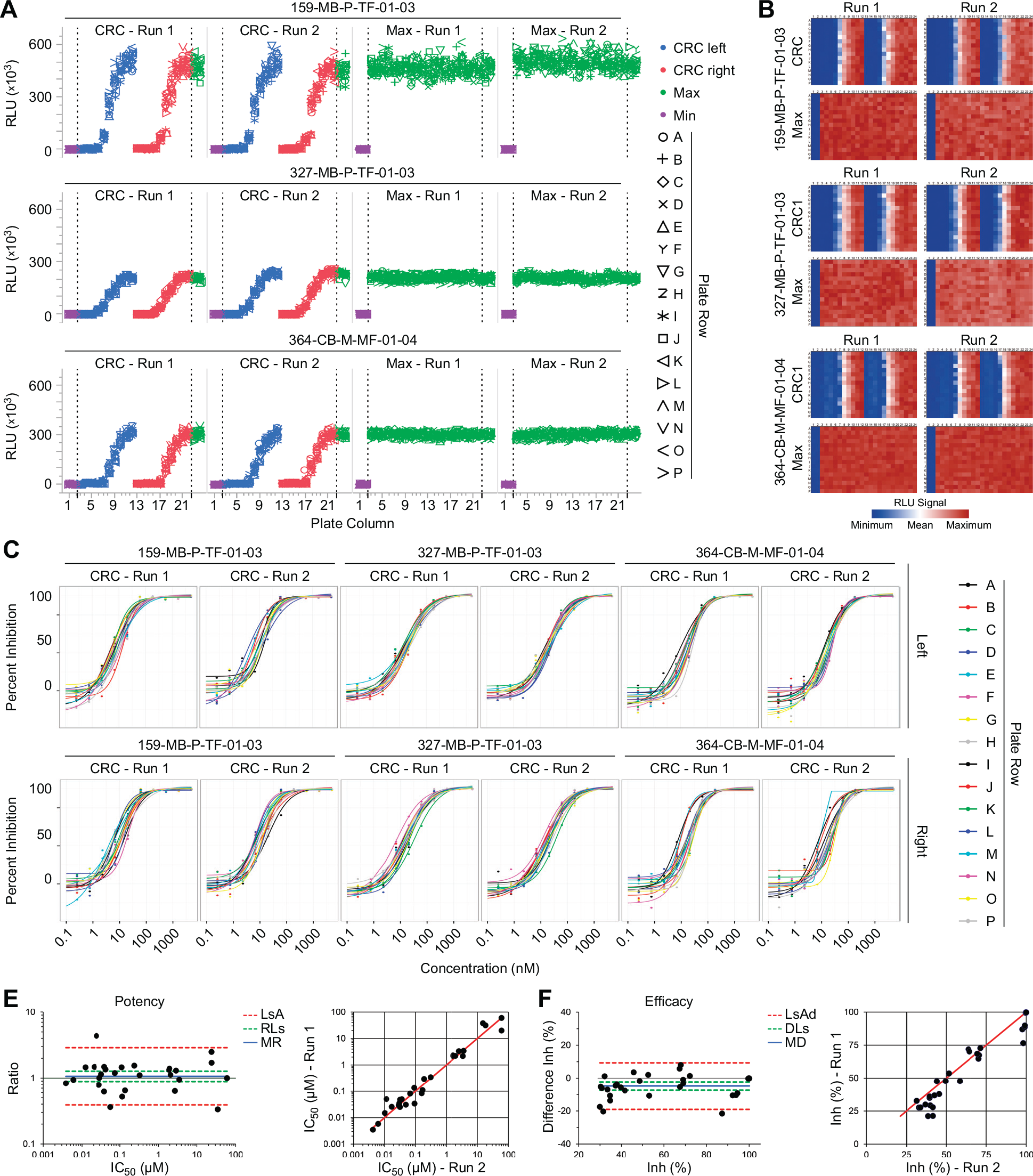
Streamlined plate uniformity study for different patient-derived samples and replicate-experiment study. (
Results of the streamlined plate uniformity studies for different patient-derived samples.
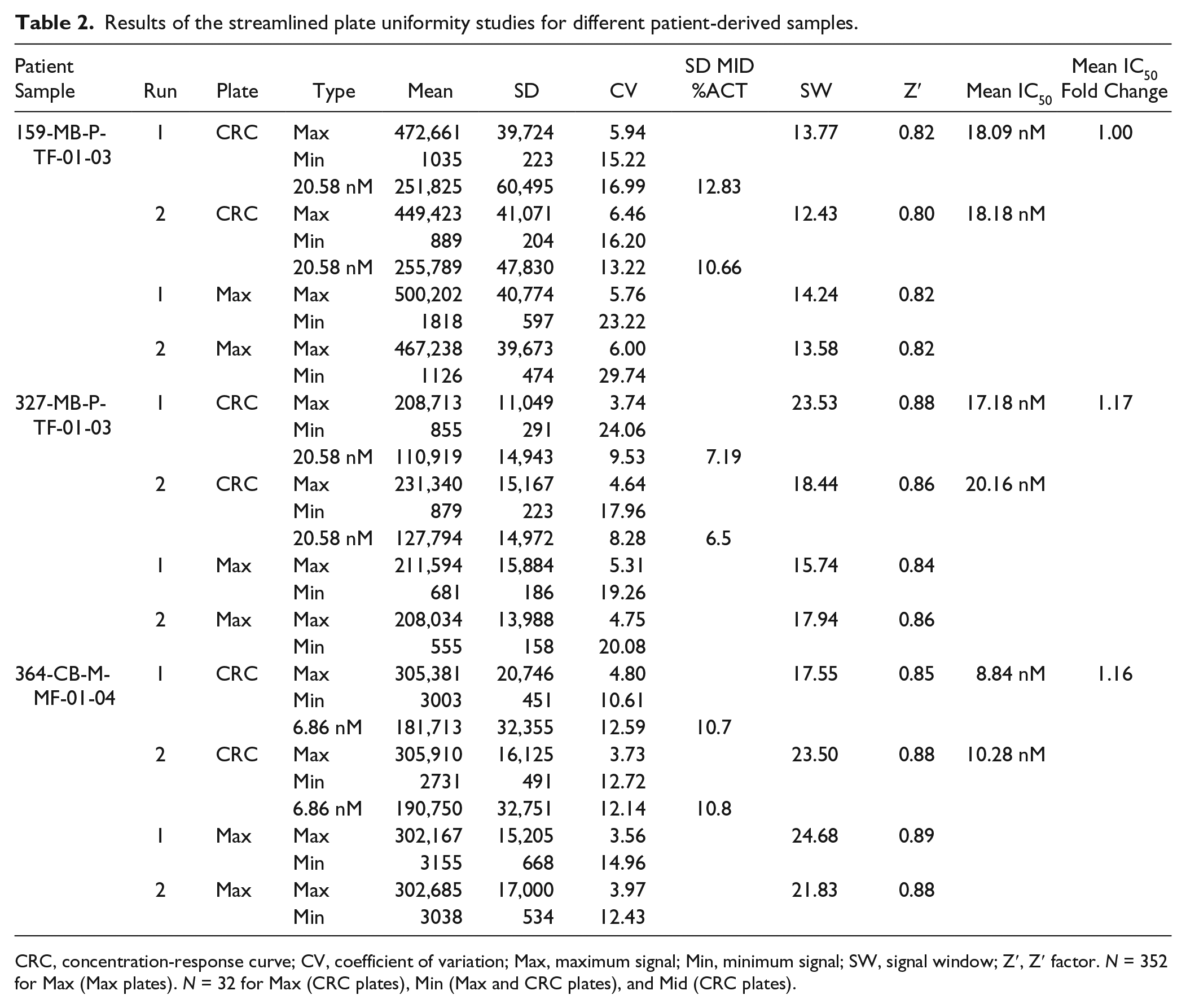
CRC, concentration-response curve; CV, coefficient of variation; Max, maximum signal; Min, minimum signal; SW, signal window; Z′, Z′ factor. N = 352 for Max (Max plates). N = 32 for Max (CRC plates), Min (Max and CRC plates), and Mid (CRC plates).
The next step in assay validation was to assess the reproducibility of the outcome measure, in this case the IC50 (potency) and maximum inhibition (efficacy;
Fig. 4E
,
F
). To illustrate this for one of the organoid models (sample ID 159-MB-P-TF-01-03), we tested 16 compounds in 10-point concentration-response format in two independent runs (
Discussion
The application of 3D culture systems as disease-specific human drug-screening models has enormous potential to connect compound screening and clinical trials. Recent technical advances allow for engineering a cellular microenvironment that more closely mimics the physiological situation compared with conventional 2D cell cultures.23–26 Primary organoid cultures with the property to self-organize and maintain the complexity of the tissue of origin are novel experimental patient-specific 3D model systems that could represent a new generation of drug-screening technology for pharmacogenomic profiling.15,16 A successful establishment of novel and complex technologies in the drug discovery pipeline requires implementation of automatic high-throughput platforms into the workflow as well as comprehensive assay validation studies.
Our results demonstrate that primary patient-derived tumor material can be used to establish a robust in vitro drug sensitivity assay. First, complex 3D structures are formed from single cells in miniaturized 384-well format with regular organoid morphogenesis. Our results are consistent with previous findings that single cells suspended into an extracellular matrix self-organize into luminized organoid structures.9,13 We provide further experimental support for this concept for a 384-well–based assay system that is also applicable to drug-screening methodologies. Second, our approach allows for the use of robotic platforms for cell seeding and compound administration and thus facilitates high-throughput and reproducible screening studies. The high accuracy of the automatic platforms is particularly important in complex long-term 3D assays, as minor well-to-well differences might lead to significant readout variations. Third, we subjected the organoid drug sensitivity assay to systematic plate uniformity assessment and could validate the assay as a robust and reproducible high-throughput platform. After assay optimization, the patient-derived organoid platform passed all relevant validation criteria of assay performance and pharmacologic relevance. 20 Moreover, we introduced a streamlined plate uniformity study and demonstrated its validity for patient-derived colon cancer samples from different donors.
Despite the clear value of complex PD3D assays in high-throughput format, several challenges remain. It is conceivable that patient-derived samples from different tumor entities show diverse requirements for their establishment in 384-well format, and these different conditions must be taken into consideration when validating new assay platforms. Thus, it is recommended to perform a comprehensive plate uniformity study for each tumor tissue type established as a 3D organoid culture assay system and, as presented herein, subsequently apply a streamlined plate uniformity study for patient-specific samples derived from different donors from the respective tumor entity. Also, the requirement for a constant supply of fresh patient material would exclude their use as high-throughput platforms for large compound screens. However, the propagation of patient-derived tumor specimens has been recently reported for colon, pancreatic, and prostate tumors.14,27,28 In addition, human pluripotent stem cells have been demonstrated as a source for colon, gastric, lung, and pancreatic organoid cultures.11,12,29,30 For the development of novel patient tailoring strategies, a comprehensive correlation of compound response data with the genomic analysis is essential. Because of the complexity of multiple alterations in colorectal cancer 31 and consequently in PD3D organoid models, a large set of patient-derived organoid samples is critical to dissect a diverse response data set. Notably, Matano et al. 32 pioneered the use of the CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats–CRISPR-associated 9) system in human intestinal organoids. With this technology, diverse oncogenic mutations could be engineered into organoids derived from normal colon. 32 Despite the experimental progress, to date, patient-derived organoid cultures do not mimic effects of the immune system, vascularization, and other effects of stromal elements. By increasing our understanding of the impact of the microenvironment on tumor progression, we may be able to generate predictive data from more biologically relevant experimental assays that incorporate multicellular constituents and physical properties of a tumor. Further investigation is required to establish sophisticated co-culture tumor models (e.g., with cancer-associated fibroblasts or endothelial cells33–37) as reproducible and standardized tools for translational research and drug discovery.
In summary, the study presented herein shows the establishment and validation of primary organoid cultures as an automated drug sensitivity platform. We anticipate that robust high-throughput technologies for patient-derived samples will be instrumental tools in the drug discovery pipeline as an invaluable link to disease-specific human models.
Footnotes
Acknowledgements
We gratefully acknowledge Jesús Castañon, José Luis Díaz-Puentes, Wenling Zhang, Bruna Calsina, Ana Hermoso, Ester Arroba, Julia Gutiérrez, Eduardo Goicoechea, and Laura Álvaro, from Eli Lilly and Company; Cathrin Davies and Yvonne Welte from Charité Universitätsmedizin Berlin, Germany; Caroline Schweiger from Medical University Graz; and Ulrich Keilholz from Charité Comprehensive Cancer Center for fruitful discussions and technical assistance. We thank Diego Megias and the CNIO Confocal Microscopy Unit for technical support.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The research leading to these results has received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement No. 115234 (OncoTrack), resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies in kind contribution.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.