
Abstract
Many receptor tyrosine kinases (RTKs) represent bona fide drug targets in oncology. Effective compounds are available, but treatment invariably leads to resistance, often due to RTK mutations. The discovery of second-generation inhibitors requires cellular models of resistant RTKs.
An approach using artificial transmembrane domains (TMDs) to activate RTKs was explored for the rapid generation of simple, ligand-independent cellular RTK assays, including resistance mutants.
The RTKs epidermal growth factor receptor (EGFR), MET, and KIT were chosen in a proof-of-concept study. Their intracellular domains were inserted into a series of expression vectors encoding artificial TMDs, and they were tested for autophosphorylation activity in transient transfection assays. Active constructs could be identified for MET and EGFR, but not for KIT.
Rat1 cell pools were generated expressing the MET or EGFR constructs, and their sensitivity to reference tool compounds was compared to that of MKN-45 or A431 cells. A good correlation between natural and recombinant cells led us to build a panel of clinically relevant MET mutant cell pools, based on the wild-type construct, which were then profiled via MET autophosphorylation and soft agar assays.
In summary, a platform was established that allows for the rapid generation of cellular models for RTKs and their resistance mutants.
Keywords
Introduction
Receptor tyrosine kinases (RTKs) are some of the best-validated targets in oncology. Small- and large-molecule inhibitors against epidermal growth factor receptor (EGFR), ERBB2, insulin-like growth factor 1 receptor (IGF1-R), MET, vascular epithelial growth factor receptor (VEGFR), c-kit, and other RTKs have been developed. Despite initial successes, however, treatments often fail due to the emergence of resistance mutations (for a review, see Janne et al. 1 ). Thus, there is a clear medical need for compounds targeting these mutants as a backup strategy.
An essential prerequisite for efficient drug discovery is cell lines that express the target and allow for the measurement of target engagement in a cellular context. Cell lines harboring relevant resistance mutants are rare, however, and even if they are available, their cellular background is often variable, rendering direct side-by-side comparisons challenging. The RTK’s dependency on specific ligands adds another layer of complexity, requiring the adaptation of every assay to the activation dynamics of the given cellular system. Cellular assays that enable rapid screening of compounds targeting resistance mutants, without the need for ligand-mediated activation, might therefore be of great value and help to speed up the drug discovery process. An assay system based on the activation of intracellular RTK domains by artificial transmembrane domains (TMDs) might provide such a system.
The TMD plays a pivotal role in RTK activation, and naturally occurring mutations that lead to activation of the RTKs have been described. In p185c-neu, a Val664→ Glu mutation in the TMD of the rat erbB2 homolog, led to ligand-independent receptor activity and oncogenic transformation.2,3 An activating Gly380 → Arg mutation in the TMD of the human fibroblast growth factor receptor 3 (FGFR3) was shown to cause achondroplasia and thanatophoric dysplasia. 4
In a systematic investigation of the underlying activation mechanism, Chen and colleagues modified the TMD of the p185c-neu RTK using degenerated oligonucleotides to isolate novel transforming derivatives. 5 Whereas several of the novel transforming isolates lacked the originally identified Glu at residue 664, Glu residues were still present in most of the transforming TMDs. Often, they exhibited a spacing of seven residues and thus localized to the helical interface. No clear rules for the prediction of specific sequences for receptor activation and transformation, however, could be derived from an analysis of transforming and nontransforming sequences. To better understand the activation mechanism, entirely novel transmembrane sequences were constructed based on tandem repeats of simple heptad sequences. TMDs that yielded active RTKs required regularly interspersed Glu residues, that is, sequences such as [VVVEVVA]n or [VVVEVVV]n. TMDs consisting only of Val residues were non-activating, as were sequences interspersed with Lys, Ser, or Asp residues. Only either Glu or Gln worked as activating amino acids. The spacing between the Glu residues was systematically varied from two to eight residues, but only the heptad spacing resulted in receptor activation. 5 Bell and colleagues engineered ErbB2 and platelet-derived growth factor receptor (PDGFR) construct [VVVEVVV]n sequences as artificial TMDs, varying the positions of the Glu residues. 6 All constructs gave rise to membrane-bound dimers, but only some TMDs yielded active RTKs. The authors concluded that the activating TMDs dimerized receptor monomers with specific orientations and suggested that the kinase domain is rotationally coupled to the TMD. 6
The role of the TMD in another RTK, VEGFR-2, was investigated by Dell’Era-Dosch and Ballmer-Hofer. 7 When used to replace the native TMD, the majority of the artificial TMDs gave rise to inactive RTKs, although all of the constructs formed dimers. In agreement with Bell and colleagues, Dell’Era-Dosch and Ballmer-Hofer concluded that activating TMDs induced specific conformations of the receptor dimers with the intracellular kinase domains properly oriented in a small set of mutants, leading to kinase transphosphorylation. 7
The ability to promote RTK activation by artificial TMDs led to the idea to develop a robust, miniaturizeable cellular assay system for RTKs without the need for ligand-induced activation.
EGFR, MET, and KIT were chosen for a proof-of-concept study representing clinically established oncology drug targets. Reference compounds with a diverse mode of action are available to validate the cellular systems, and resistance mutants have been described for all of them (e.g., MET M1250T). 8 Thus, we used these RTKs to explore the feasibility of building a cellular assay platform for RTKs based on activation via artificial TMDs.
Materials and Methods
Reagents
The 9E10 myc antibody was purified from hybridoma culture supernatants, whereas the PY-99 antibody was obtained from Santa Cruz Biotechnology (Dallas, TX). Restriction enzymes, polynucleotide kinase, and calf intestine phosphatase were from NEB (Frankfurt, Germany); Phusion polymerase from Finnzymes (Thermo Fisher Scientific, Vantaa, Finland); and rapid ligation kit from Roche Diagnostics (Mannheim, Germany). Met inhibitors (PHA665752, XL184, BMS777607, XL880, MGCD265, JNJ3887605, and PF2341066) were obtained from Biozol (Eching, Germany). All chemicals were, unless stated otherwise, from Roth (Karlsruhe, Germany).
Plasmids
The pcDNA3 V1E–V13E plasmids were generated as published.
7
The pEF V1E–V13E 2myc6His plasmids were constructed in two steps from the pEF 2myc6His plasmid
8
and the pcDNA3 V1E–V13E plasmids. First, the PCR product from the reaction of 5′SP and 3′SP primers on the pcDNA3 hVEGFR-2-YFP-DeltaEC-V6E plasmid was digested NcoI–EcoRI and ligated into the correspondingly digested and dephosphorylated vector pEF 2myc6His, yielding the plasmid pEF SPTM 2myc6His. In a second step, the artificial TMDs were isolated from the respective plasmids (pcDNA3 V1E–V13E) via PCR, digested BamHI–EcoRI, and inserted into the correspondingly digested and dephosphorylated vector pEF SPTM 2myc6His, yielding the final set of plasmids pEF V1E–V13E 2myc6His (see
Fig. 1B
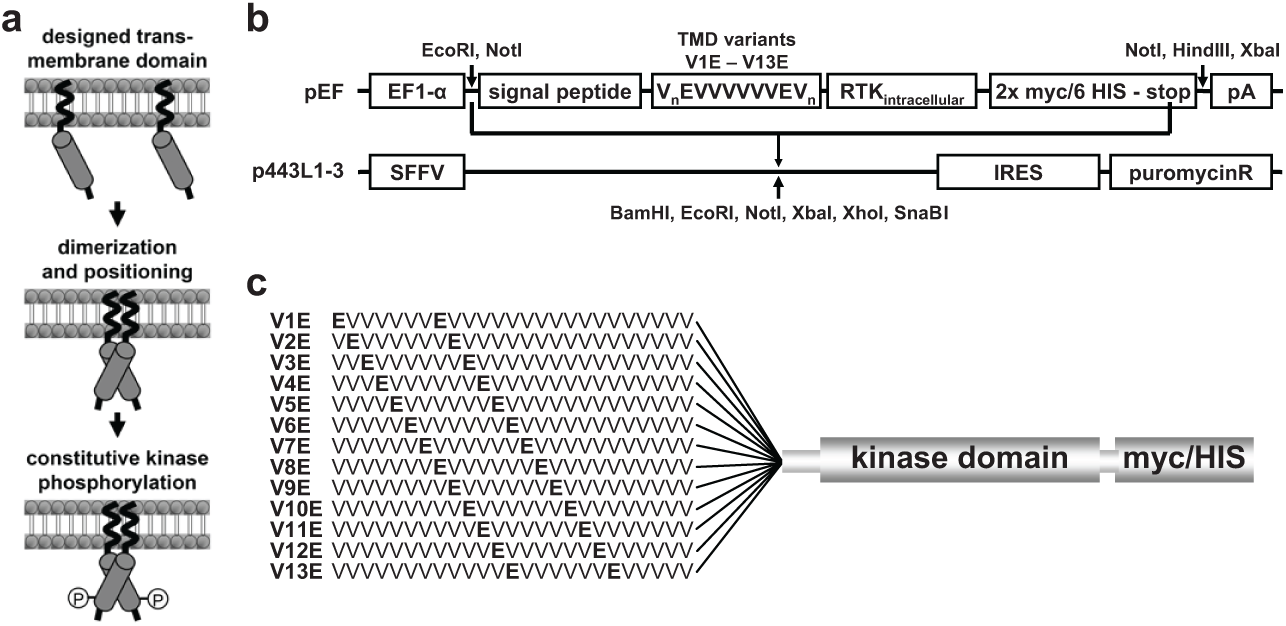
Strategy for the identification of activating transmembrane domains (TMDs) for receptor tyrosine kinases (RTKs): (
To generate TMD RTKs, the cytoplasmatic domains of the RTKs EGFR (amino acids H672-A1210, NCBI/Protein NP_005219.2), KIT (amino acids T544-V976, NCBI/Protein NP_000213.1), and MET (amino acids K956-S1390, NCBI/Protein NP_000236.2) were inserted in frame into BamHI and NcoI sites in the pEF V1E–V13E 2myc6His multiple cloning site using PCR to introduce the respective restriction sites (
Cell Culture
Rat-1, 11 MKN-45, 12 A431, 13 and 293T cells (293tsA1609neo) were kept in Dulbecco’s modified Eagle’s medium (DMEM) Glutamax I (Invitrogen, Darmstadt, Germany) with 10% fetal calf serum (FCS; PAN, Aidenbach, Germany) and 1× penicillin–streptomycin (PAA; Coelbe, Germany) at 37 °C and 10% CO2.
For transient transfections of the pEF V1E–V13E constructs, 50,000 293T cells/well were plated in 100 µl DMEM with 10% FCS (without antibiotics) in flat-bottom 96-well cell culture plates. The next day, the plasmids were transfected using the Lipofectamine 2000 Transfection Reagent (Invitrogen). A total amount of 0.2 µg plasmid was transfected, consisting of 0.02 µg pEF YFP (as a transfection control), and serially diluted pEF V1E–V13E constructs (starting at 0.1 µg, six steps of 1:3 dilutions), and the empty pEF vector to keep the DNA concentration constant. After the transfection, the cells were incubated at 37 °C and 10% CO2 for 48 h. Cell lysates were then tested in an enzyme-linked immunosorbent assay (ELISA) for the presence of phosphorylated recombinant RTKs.
To produce lentiviral supernatants to generate stable cell lines, 293T cells were transfected with the plasmids p443L1-MCS3 IP containing the TMD MET of interest, pMD2G [G glycoprotein of the vesicular stomatitis virus (VSV-G)], and pCMV8-74, which provides the env and gag genes, using the calcium–phosphate precipitation method. After 3–5 h, the supernatant was replaced with 20 ml of fresh medium, and the cells were left for 48 h in the incubator. The supernatant was then harvested and replaced by 20 ml of fresh medium, which was harvested another 24 h later.
Rat-1 cells were transduced twice for 8–12 h in 6-well plates using 2 ml of the viral supernatant containing 4 µg/ml polybrene. One day after the last transduction, the cells were transferred to a 10 cm dish, and cell pools were generated from successfully transduced cells via selection with 3 µg/ml of puromycin, starting the next day.
Western Blot
Cells were lysed in 1× sodium dodecyl sulfate (SDS) sample buffer (Invitrogen) and applied on a 10% polyacrylamide gel electrophoresis (PAGE) gel (Invitrogen). The gel was blotted on a nitrocellulose filter, the transfer and equal protein loading were confirmed using Ponceau S protein stain, and the filters were blocked with 5% skim milk powder in TTBS (20 mM TRIS, pH 7.5, 150 mM NaCl, 0.05% Tween 20). The filter was then incubated with the 9E10 antibody at a 1:1000 dilution in blocking buffer for 90 min, or with the PY99 anti-phospho-tyrosine antibody at a 1:200 dilution in 5% bovine serum albumin overnight; washed three times in TTBS; and then incubated with the secondary peroxidase-labeled antimouse antibody (1:10,000) in blocking buffer for 90 min. The filters were washed again using three changes of TTBS, and the antibody complexes were detected using ECL Plus (Amersham, GE Healthcare Lifesciences, Freiburg, Germany), according to the manufacturer’s instructions.
Fluorescence-Activated Cell Sorting (FACS)
Cells were harvested from the plates, washed with PBS, and fixed in methanol. After an overnight incubation at 4 °C, the cells were rehydrated in PBS for 30 min, blocked using PBS containing 1% FCS and 0.2% Tween 20, and then stained using blocking solution containing the primary antibody at the appropriate dilution (9E10 : 1:1000) for 30–45 min. The cells were then washed three times for 5 min, and incubated with the secondary fluorescein isothiocyanate (FITC)-labeled antibody at the appropriate dilution for 30–45 min. Cells were then washed again and analyzed using a FACSCalibur (BD Heidelberg, Germany).
Biochemical Kinase Activity Assay
All kinase activity assays were done using 96-well, scintillator-coated plates (FlashPlate® Basic Microplate, #SMP200, PerkinElmer, Waltham, MA). The assay for all enzymes contained 70 mM hydroxyethyl piperazineethanesulfonic acid (HEPES)–NaOH, pH 7.5, 3 mM MgCl2, 3 mM MnCl2, 3 µM sodium orthovanadate, 1.2 mM DTT, 0.3 µM adenosine triphosphate (ATP)/[γ-33P]-ATP (corresponding to the apparent ATP-Km of the respective kinase, approx. 5×105 cpm per well), 2.5 to 6.3 nM protein kinase (ProQinase GmbH, Freiburg, Germany), and 0.125 or 0.25 µg/50 µl substrate Poly(Ala,Glu,Lys,Tyr) 6:2:5:1 (Sigma, Munich, Germany). The reaction cocktails were incubated at 30 °C for 60 min. The reaction was stopped with 50 µl of 2% (v/v) H3PO4, and plates were aspirated and washed two times with 200 µl 0.9 % (w/v) NaCl. Incorporation of 33Pi was determined with a microplate scintillation counter (Microbeta, PerkinElmer). All assays were performed with a Beckman Coulter SAGIAN Core System.
Cellular Phosphorylation Assay
Cells were seeded in 96-well cell culture plates (25,000 cells/well), and on the following day medium was exchanged for FCS-free medium. Compounds solved in DMSO were added to eight semilogarithmic diluted concentrations in duplicates for 1.5 h (final DMSO concentration, 1%). Cells treated with Lapatinib (EGFR, 1E-5M) or BMS777607 (MET, 1E-5M) were defined as low control (n = 8). Cells treated with DMSO (1%) alone were defined as high control (n = 8). Thereafter, cells were lysed in lysis buffer (20 mM Tris-HCl, 135 mM NaCl, 1% NP-40, 10% glycerol, 100 mM NaOVanadate, and complete PhosphoSTOP), and the phosphorylation status of respective substrates was determined by sandwich ELISA, using antibodies directed against the pan-protein (e.g., anti-myc, anti-MET, or anti-EGFR) as capturing, and the biotinylated phospho-tyrosine antibody PY99 as detecting antibody. Detection was done with the strepavidin-based horseradish peroxidase labeled ABC reagent (Thermo, Schwerte, Germany).
Proliferation Assay
Cells (5000 cells/well) were seeded in the inner wells of 96-well-plates in 150 µL complete medium. On the next day, serially diluted compounds were added to the medium to reach the final concentration and incubated at 37 °C at 5% CO2 for 72 h. Subsequently, 15 µL Alamar blue reagent was added and fluorescence at 590 nm was measured after 3–5 h incubation at 37 °C, 5% CO2 using a fluorometer. As low control, cells were treated with 1E-05M staurosporine (sixfold values). As high control, cells were treated with 0.1% DMSO (solvent control, sixfold values).
Soft Agar Assay
For each cell line, 96-well suspension cell culture plates were prepared. One hundred microliter of the soft agar bottom layer (0.6% final concentration in complete medium) was poured and left to set. Fifty microliters of the soft agar top layer (0.4% final concentration) containing the corresponding cells (7500 cells/well) were added on top and allowed to set, and the plates were incubated at 37 °C, 10% CO2. Next day, serially diluted compounds were added into the inner wells of the plate. Subsequently, assays were incubated in cell culture incubators for 8 days. Finally, assays were automatically photographed and subsequently developed using Alamar blue. After 3–5 h of incubation at 37 °C, fluorescence intensity was determined (excitation: 560 nm; emission: 590 nm). As low control, cells were treated with 1E-05M staurosporine (sixfold values). As high control, cells were treated with 0.1% DMSO (solvent control, sixfold values).
Results
Transient Transfection Assay to Identify Activating TMDs for MET, EGFR, and KIT
The aim of this study was to develop a robust, miniaturizeable cellular phosphorylation assay platform for RTKs based on ligand-independent activation of the RTKs using artificial TMDs 5 ( Fig. 1a ). From a set of artificial TMDs consisting of a valine helix interspersed with glutamic acids in a heptad spacing, Dell’Era-Dosch and Balmer-Hofer had successfully identified activating (Glu residues at positions 6 and 13; V6E) as well as nonactivating TMDs (Glu residues at 10 and 17; V10E) for the RTK VEGFR-2. 7 In a small proof-of-concept study, this VEGFR-2 result was confirmed (data not shown) and we thus went on to try to identify activating TMDs for the RTKs EGFR, MET, and KIT.
To identify the optimal position of the Glu residues within the artificial TMD for each of the RTKs, an easy, fast, and reliable test system was established. Expression plasmids using an EF1alpha promoter (pEF) were prepared encoding all potential variants of the artificial VnEVVVVVVEVm TMDs (V1E to V13E), an N-terminal signal peptide for efficient membrane integration, a multiple cloning site to insert the cytoplasmic domain of the RTK of choice, and a C-terminal combined 2xmyc/6His tag to identify and isolate the recombinant protein ( Fig. 1b ).
The cytoplasmic domains of wild-type MET, EGFR, and KIT were inserted into this set of plasmids, yielding vectors that encode myc-tagged membrane-anchored intracellular MET, EGFR, KIT-proteins differing only in the position of the Glu residues in the artificial TMD ( Fig. 1c ).
The plasmids were transiently transfected into HEK 293T cells, the cells were lysed after 48 h, and the autophosphorylation of the recombinant intracellular RTK fragments measured via a sandwich ELISA detecting tyrosine phosphorylation of myc-tagged proteins. Serial dilutions of the expression plasmid (1:3, starting at 0.1 µg/96-well plate) were applied to avoid massive overexpression of the constructs, which was found to overcome their dependence on the TMD configuration (data not shown). A flowchart of the generation of the TMD-activated RTKs is shown in Figure 2 .
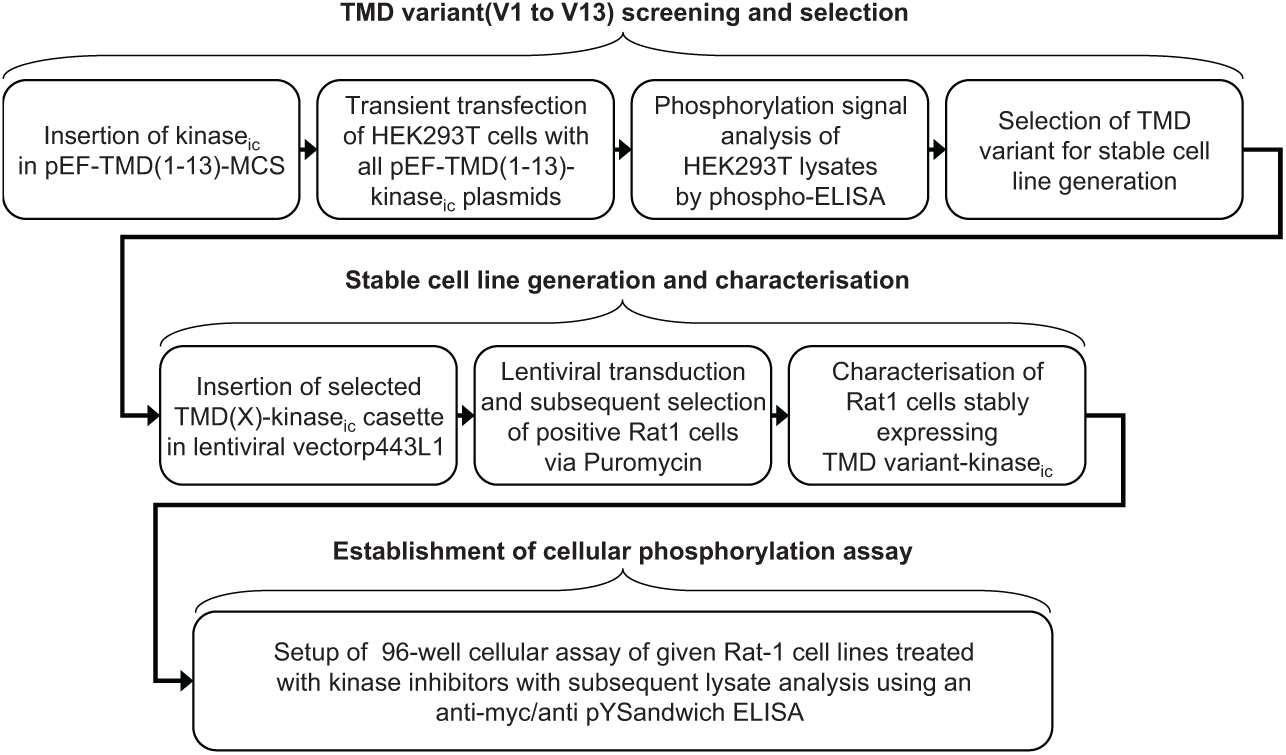
The flowchart developed here to identify activating transmembrane domains for receptor tyrosine kinases.
As described previously, explained by the structure of α-helices, a repetitive pattern of activating Glu residues was observed for all RTKs tested here. 6 Whereas the EGFR and MET intracellular kinase domains were activated efficiently, KIT could be activated to only a rather modest level ( Fig. 3 ).
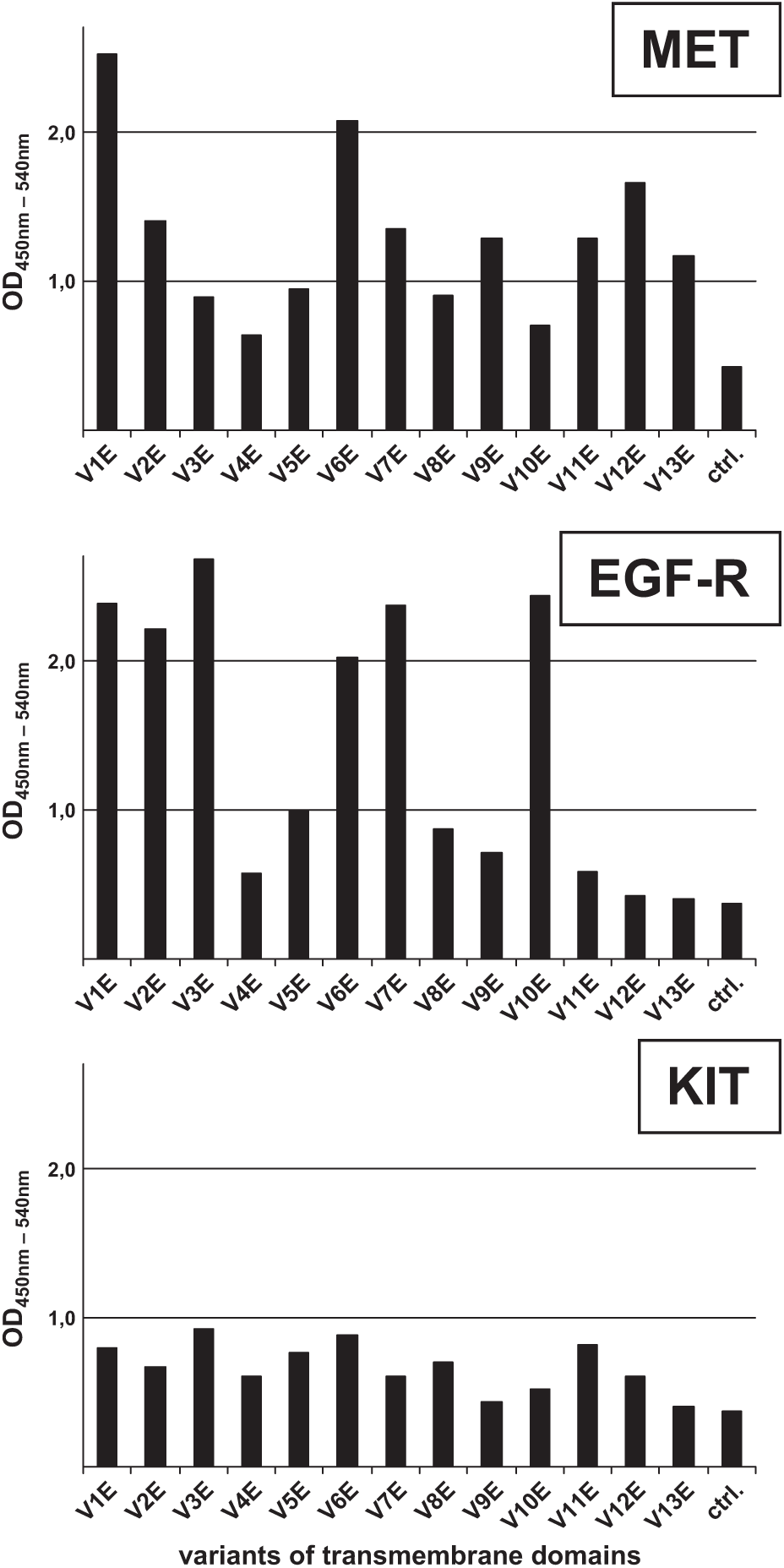
Screening for the appropriate activating transmembrane domain (TMD) for epidermal growth factor receptor (EGFR), MET, and KIT. 293T cells were transiently transfected with the pEF V1E–V13E receptor tyrosine kinase (RTK) constructs. Subsequently, autophosphorylation of expressed TMD–RTK constructs was quantified by a sandwich ELISA (enzyme-linked immunosorbent assay) using the myc-antibody 9E10 as the capturing antibody and a phospho-tyrosine specific antibody (PY99) as the detecting antibody. ctrl., lysates of mock transfected cells.
TMD variant V1E, with Glu residues at positions 1 and 8, drove expression and activation of the recombinant MET construct most effectively, followed by V6E and V12E. For EGFR, this was V3E and V10E, followed by V7E.
For the intended proof-of-concept study, the V1E MET and V10E EGFR constructs were chosen for the generation of stably expressing Rat-1 cells (referred to from now on as TMD MET WT and TMD EGFR WT, respectively).
Generation and Characterization of Rat-1 Cells Expressing TMD MET WT and TMD EGFR WT
To generate stable cell pools, the TMD MET WT and TMD EGFR WT 2myc/6His cassettes were inserted into lentiviral vector p443L-1-3 modified to harbor an IRES-puromycin element downstream of the insertion site for the expression cassette ( Fig. 1b ), which allows for selection of successfully transduced Rat-1 cells.
FACS analysis of the puromycin selected cell pools using the myc-specific mAb 9E10 suggested expression of the recombinant constructs (
As a biological validation, the sensitivity of the recombinant MET and EGFR constructs to reference compounds was tested in the generated recombinant Rat-1 TMD MET WT and TMD EGFR WT cells, and compared to MKN-45, a human gastric carcinoma cell line dependent on MET overexpression, 14 and A431, a vulvar squamous cell line dependent on EGFR, 15 respectively. PHA665752, XL184, BMS777607, XL880, MGCD265, JNJ3887605, and PF234106616–20 served as reference compounds for MET, and erlotinib, gefitinib, lapatinib, and AG1478 for EGFR.21,22 Cells were treated with serially diluted inhibitors for 90 min, ligand-stimulated if required (only A431), and lysed, and the lysates analyzed for the autophosphorylated RTK by ELISA using a pan-MET (for MKN-45), a pan-EGFR (for A431), or a myc-antibody (for the recombinant Rat-1 cells) for capturing, and a generic phospho-tyrosine antibody for detection. Detection limits and Z’-factors were determined as a metric for the quality and robustness of the cellular phosphorylation assay. 23 The dynamic range of the cellular phosphorylation assays was between fourfold and sixfold (TMD MET) and from 10 to 14 fold (TMD EGFR), with Z’-factors from 0.4 to 0.8 (data not shown), indicating excellent assay systems.
The six MET reference compounds inhibited MET autophosphorylation in the Rat-1 TMD MET WT cells with IC50 values comparable to those obtained for MKN-45 ( Fig. 4a ). Also, the IC50 values of the four EGFR reference compounds tested correlated well between the TMD EGFR WT cells and the A431 cells, the only exception being erlotinib ( Fig. 4b ).
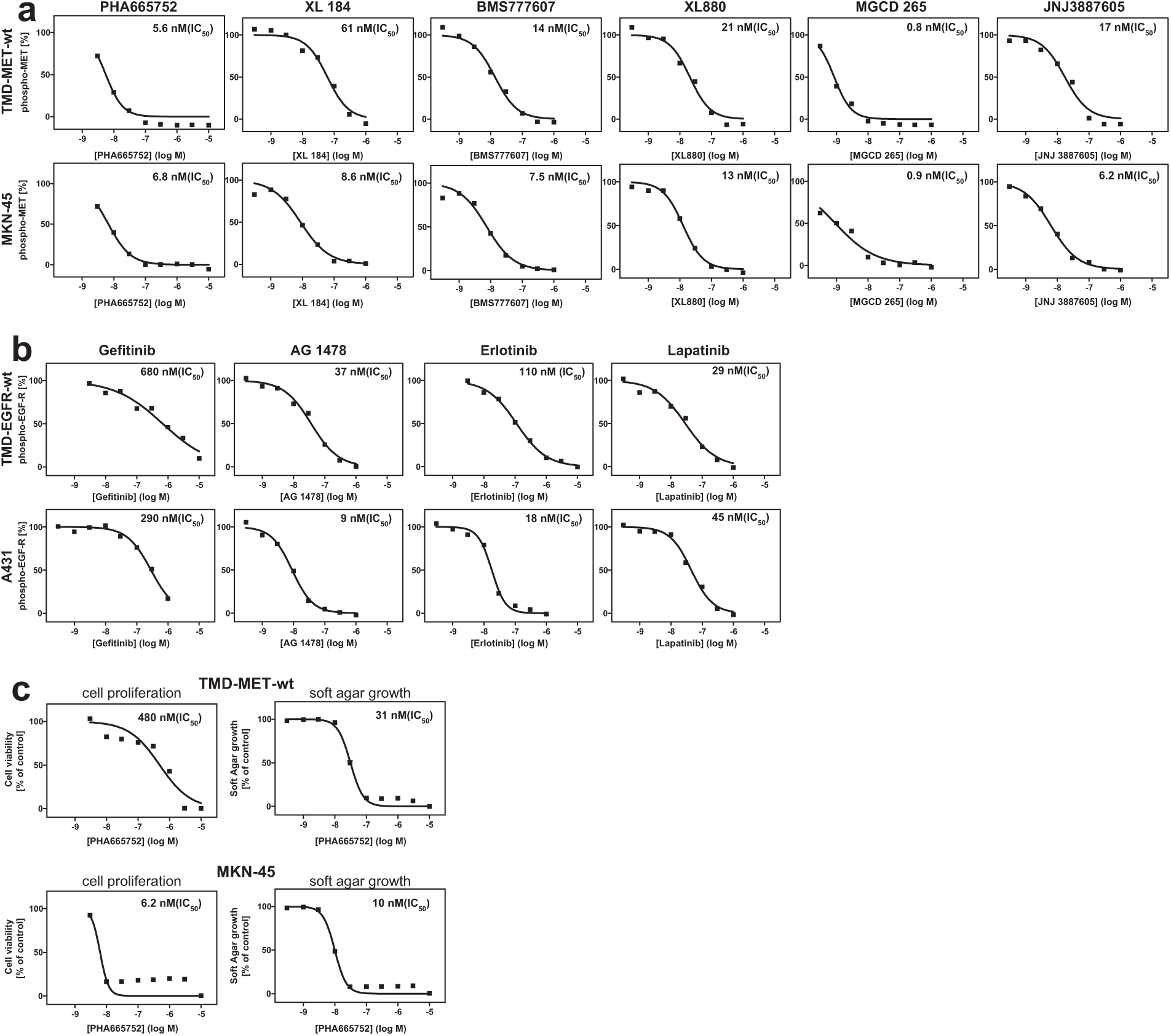
Characterization of transmembrane domain (TMD) epidermal growth factor receptor (EGFR) and MET wild-type expressing Rat-1 cells. Rat-1 cells were transduced with a lentivirus encoding TMD EGFR WT or TMD MET WT, and subsequently selected using puromycin. The resulting cell pools were analyzed for TMD EGFR WT or TMD MET WT inhibition in cellular phosphorylation assays using EGFR and MET reference inhibitors. (
Thus, the recombinant EGFR and MET cell lines appeared to represent valid cellular assay systems for the testing of compounds targeting these RTKs. The homogeneous cellular background enables straightforward cross-comparisons, and the lack of a requirement for ligand activation allows for an easy side-by-side plate layout.
The biology of the recombinant MET cell line was then investigated in some more detail.
MKN-45 proliferation in 2D tissue culture was potently inhibited by PHA665752; however, Rat-1 TMD MET WT cells were significantly less sensitive ( Fig. 4c ), suggesting that Rat-1 TMD MET cells in contrast to MKN-45 cells were not dependent on MET for proliferation in 2D culture.
Because MET plays an important role in 3D biology (motility, invasion, adhesion, and growth 24 ), Rat-1 TMD MET WT cells were next tested for anchorage-independent soft agar growth. Whereas parental Rat-1 cells did not grow in soft agar, Rat-1 TMD MET WT cells readily formed colonies (data not shown), indicating that the recombinant TMD MET WT construct had transforming activity in Rat-1 cells. Supporting an essential role for the MET kinase in the 3D growth of the recombinant Rat-1 cells, the MET inhibitor PHA665752 inhibited soft agar growth of both cell lines, MKN-45 and the recombinant MET-expressing rat-1 cells, with similar potencies ( Fig. 4c ).
Thus, in both our simple MET autophosphorylation assay platform and the biologically more relevant 3D soft agar growth assay, the Rat-1 TMD MET WT cells showed sensitivities to reference compounds comparable to the MET-addicted tumor cell line MKN-45.
Generation and Characterization of Rat-1 Cells Expressing TMD MET Mutants
Resistance mutations that prevent kinase inhibitors from binding to the catalytic cleft are a major cause for tumor progression after initial positive response in patients treated with tyrosine kinase inhibitors. A number of such mutants have been described for MET. 1 There is a medical need for compounds targeting these mutants, but their discovery relies on the availability of cellular assays for mutant MET isoforms.
Because the resistance is often conferred by alteration of just a single residue in the T-loop of the kinase domain, we speculated that the same recombinant TMD as for the WT MET kinase, V1E, might induce the proper confirmation for an active kinase. If so, this would significantly accelerate the generation of a panel of cell lines harboring resistance mutants of MET. Thus, constructs encoding resistance mutants of MET with the V1E TMD were prepared (compare
FACS analysis using a myc-specific monoclonal antibody detected by a fluorescently labeled anti-mouse antibody suggested that the TMD MET mutant Rat-1 cell pools were completely shifted to higher fluorescence, demonstrating homogeneous expression of the various TMD MET mutants (
To functionally test the recombinant cell lines, they were plated in 96-well plates and treated for 90 min with the MET inhibitor BMS777607 as a low control, the vehicle DMSO as a high control, and PHA665752 as a Type I and BMS777607 as a Type II kinase inhibitor.16,17 The autophosphorylation status of the TMD MET mutants was then determined using the previously described sandwich ELISA.
As expected for a Type I inhibitor, PHA665752 was active on wild-type TMD MET and the M1250T mutant, showing clearly reduced potency on MET F1200I, and no activity at all on the Y1230 and the D1228 mutants ( Fig. 5 ). In stark contrast, the Type II inhibitor BMS777607 potently inhibited wild type as well as most of the TMD MET mutants, including Y1230 and D1228. The only mutant that showed some resistance (approx. 20-fold) was F1200I ( Fig. 5 ). Thus, despite the constitutive activation of the MET RTK, the assay system successfully identified and distinguished both Type I and Type II inhibitors. The dynamic range of the cellular phosphorylation assays was between three- and sixfold, with Z’-factors from 0.4 to 0.8 (data not shown), indicating an excellent assay system.
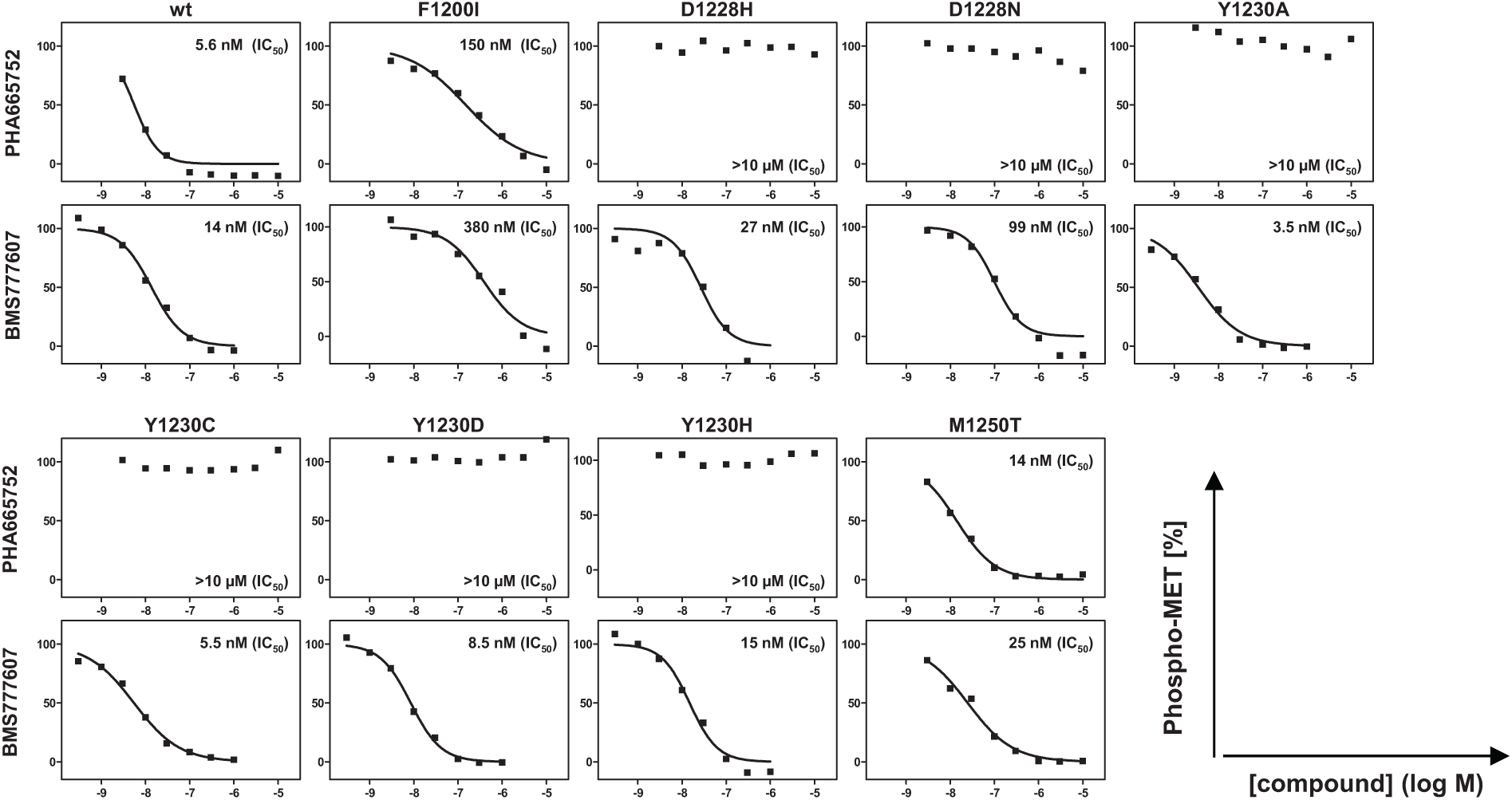
Generation of Rat-1 cells expressing transmembrane domain (TMD) MET mutants. Rat-1 cells were selected for stable expression of TMD MET mutants after lentiviral transduction of the respective TMD MET mutant. The derived cell pools were characterized using two reference inhibitors in cellular phosphorylation assays. Cellular phosphorylation assay: The inhibition profiles of two MET inhibitors (PHA665752 and BMS777607) were determined with all established TMD MET mutant and TMD MET WT Rat-1 cell lines. The autophosphorylation status of the respective MET variants was analyzed by sandwich ELISA using the myc-antibody 9E10 (for Rat-1 TMD MET WT) as capturing antibody and a phospho-tyrosine specific antibody (PY99) as detecting antibody.
The observation that a TMD that was able to activate the wild-type RTK was indeed transferrable to its kinase mutants held true not only for MET but also for EGFR (data not shown), suggesting that this system would allow the ready generation of mutant kinase panels for other RTKs as well.
Comparative Analysis of Clinical MET Inhibitors in Biochemical and Cellular Assays
In vitro kinase assays are high throughput and relatively easy to set up, but highly reductionist, whereas cellular assays are lower throughput but provide a more relevant context for the target. The availability of the same panel of wild-type and mutant MET kinase constructs for cellular as well as in vitro kinase assays enabled us to perform direct side-by-side comparisons. Because the resistance mutants also induced anchorage-independent growth of Rat-1 cells similar to the wild-type construct, comparisons were possible not only on the level of cellular autophosphorylation assays but also on the level of 3D soft agar growth assays.
Thus, a set of seven current or past clinical candidate MET inhibitors was subjected to parallel analyses using biochemical kinase assays, cellular autophosphorylation assays (plated in 2D), and soft agar growth assays (plated in 3D).
Generally, similar effects were observed in biochemical and cellular autophosphorylation assays. Three of the seven MET inhibitors—PHA665752, JNJ-38877605, and PF231066—showed potent activity toward wild-type V1E-MET as well as the M1250T mutant, but were completely blocked by mutations in D1228 or Y1230. In contrast, XL184, BMS777607, XL880, and MGCD-265 were not affected by these mutations. All tested compounds, however, lost potency by circa 10-fold when assayed against the F1200I mutant. Interestingly, MGCD-265 proved up to 60-fold more active in the cellular phosphorylation assay than in the biochemical assay ( Table 1 ).
Profiling of MET inhibitors on the established transmembrane domain (TMD) MET mutant and TMD MET WT Rat-1 cells comparing results obtained from analyses of biochemical kinase activity, cellular phosphorylation, and soft agar growth assays. Shown are results as IC50 values in nM, ratios between biochemical and cellular phosphorylation activity, as well as ratios between soft agar and cellular phosphorylation activity (italic).
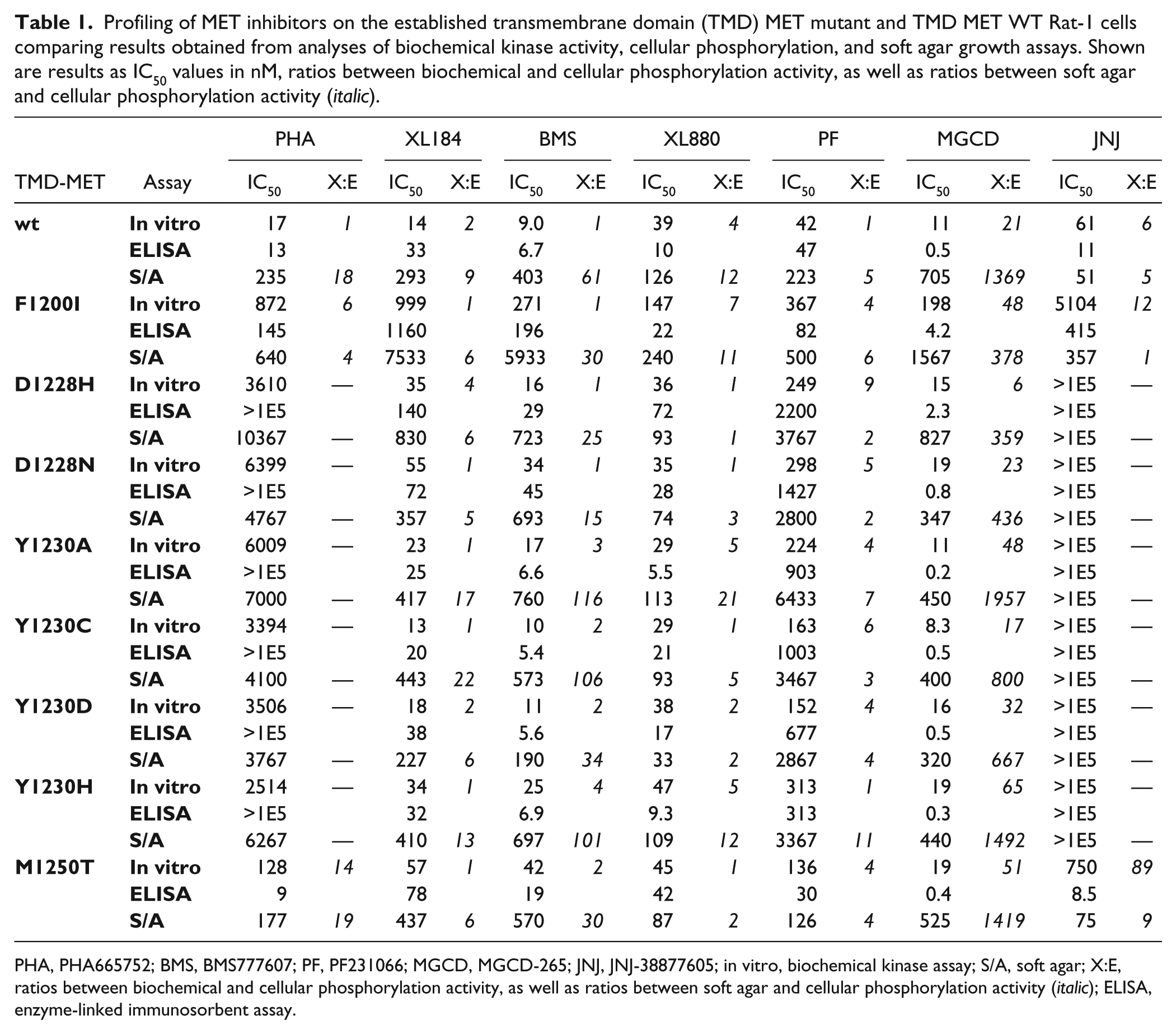
PHA, PHA665752; BMS, BMS777607; PF, PF231066; MGCD, MGCD-265; JNJ, JNJ-38877605; in vitro, biochemical kinase assay; S/A, soft agar; X:E, ratios between biochemical and cellular phosphorylation activity, as well as ratios between soft agar and cellular phosphorylation activity (italic); ELISA, enzyme-linked immunosorbent assay.
In the 3D growth assay, all inhibitors were generally less active than in the biochemical or cellular phosphorylation assays ( Table 1 ). XL880 was the only exception, showing IC50 values of around 50 nM in all biochemical and cellular test systems, on wild-type as well as mutant MET. MGCD-265 ranged on the other side of the spectrum, with its soft agar growth inhibitory activity reduced by about three magnitudes compared to the other assays. Besides differences in biological complexity, stability of the compounds during the extended incubation applied in the soft agar growth assays (8 days) as compared to the other assays (90 min), and drug penetration through the soft agar matrix also might affect compound efficacies.
Taken together, the platform presented here should prove a versatile tool for the discovery of MET inhibitors, and also provide the flexibility and ease to rapidly incorporate newly discovered resistant MET mutants into the panel. Once extended to other RTKs, it will allow for the rapid screening and profiling of tyrosine kinase inhibitors.
Discussion
The identification of naturally occurring mutations in the TMDs of RTKs, which led to constitutive ligand-independent autophosphorylation, provided the basis for the development of a series of artificial TMDs that activate intracellular kinase domains of RTKs in a sequence-specific manner. 5 The present study took advantage of this know-ledge to generate a panel of cell lines expressing wild-type and mutant artificial TMD-activated RTKs and use them as cellular test systems for the profiling of kinase inhibitors.
Using a set of plasmids that encode an N-terminal signal peptide, the full panel of artificial TMDs described by Chen and colleagues, 5 a multiple cloning site to insert the intracellular part of the RTKs, and a C-terminal combined myc/His tag for detection, we identified artificial TMDs that efficiently activated the RTKs MET and EGFR. Major peaks of activity were separated by 5–6 positions (refer to Fig. 3 ), corresponding to two full turns of the α-helix with smaller peaks found in between, corresponding to a single turn. 6 A similar pattern was also found for KIT, but the activation levels were much lower ( Fig. 3 ). The presence of an autoinhibitory juxtamembrane domain in KIT (and other Type III RTKs like c-FMS, PDGFR, and FLT3)25,26 may explain the failure of the TMD domain to fully activate KIT. Omission of this sequence might provide a potential way to activate these RTKs.
In their analysis of the RTK VEGFR-2, Dell’Era-Dosch and Ballmer-Hofer 7 reported V6E as the optimal TMD; however, for MET, TMD V1E was the most efficient activator, whereas V3E and V10E worked best for EGFR. Thus, the optimal TMD will likely have to be determined individually for each RTK—a task that should be greatly facilitated by the set of optimized plasmids and the rapid transfection assay followed by the ELISA-based autophosphorylation readout described in this article.
The ease of the approach is further substantiated by our observation that the same TMD could be used to generate panels of wild-type and clinically relevant resistance mutants, as established here for V1E and MET (and also for V10E and EGFR—data not shown), which may be due to the fact that mutant RTKs often only differ by single amino acids from their wild-type counterpart.
After identifying the proper TMD configuration for EGFR and MET, stable cellular systems were established by transducing Rat-1 cells with the corresponding constructs. The resulting cellular phosphorylation assay systems proved reliable as characterized by Z’-factors (>0.4) and dynamic ranges (>3×). Furthermore, comparison of the recombinant Rat-1 TMD MET- and EGFR-expressing cells with the endogenously MET- or EGFR-overexpressing tumor cell lines MKN-45 and A431 revealed similar sensitivities to a panel of MET and EGFR kinase inhibitors in this cellular phosphorylation assay. The reason why erlotinib was found to be 10× less potent in the rat cell line than in A431 ( Fig. 4b ) is currently unclear. Carey found in another—artificial—system an IC50 of 25 nM for erlotinib [i.e., slightly closer to the A431 (18 nM) than the Rat-1 value (110 nM)]. 27
However, although MET inhibition was equally effective in MKN-45 and Rat-1 TMD MET cells, only the proliferation of MKN-45 cells was affected in both 2D and 3D, suggesting that the proliferation of Rat-1 cells in 2D was independent of MET ( Fig. 4c ). An explanation for this observation might be that whereas introduction of the TMD-MET construct did not markedly alter the in vitro 2D growth behavior of Rat-1 TMD MET cells compared to the parental Rat-1 cell line, their growth in soft agar was mediated by MET, potentially introducing a dependency on MET activity in the 3D environment. MKN-45 has acquired a 15–20-fold MET amplification, 28 leading to ligand-independent pathway activation, possibly while being adapted to grow in vitro, using FCS, or in mice, both of which lack hepatocyte growth factor to stimulate human MET. 29 This massive MET amplification may lead to a profound dependence on MET signaling (oncogene addiction), also required for proliferation in 2D. It has been shown for GTL16, another MET-amplified cell line, that MET signaling mainly stimulated PI3-K and ras-mediated signaling in 2D cultures. 30 Other pathways (e.g., JNK and NFkB) reported to be engaged by MET31,32 may be activated in more complex settings only.
No reference cell lines were found that endogenously expressed MET mutants and would allow for an adequate comparison, although such mutations are described in the literature (e.g., for renal papillary carcinoma patients). 33 This observation revealed another advantage of this system: the potential to rapidly generate cell lines expressing mutant RTKs in an otherwise equal genetic background, especially if no tool cell lines are available.
A potential flaw of the present assay system might reside in the permanent activation state of the TMD-activated kinases, potentially preventing Type II (inactive-state binders) from effective interaction with the kinase domain. Type I MET inhibitors such as PHA665752 should depend on the presence of tyrosine 1230 (Y1230)—inducing an active kinase conformation—for binding, whereas Type II MET inhibitors such as BMS777607, which bind to the kinase in its inactive conformation, may show reduced binding and thus reduced inhibitory activity. This preclusion did not hold true, however. All of the different types of MET inhibitors were ranked correctly by our Rat-1 recombinant cell lines according to the predicted MET inhibition mechanism. PHA665752, JNJ-38877605, and PF231066 inhibited wild-type TMD-MET as well as the M1250T variant with high potency, but were inactive when aspartic acid 1228 or tyrosine 1230 was mutated. This confirms PHA665752 and PF231066 as ATP-competitive Type I MET inhibitors, which require π stacking interactions with Y1230 for effective binding to MET. 24 Accordingly, our data set would identify JNJ-38877605 as a Type I inhibitor as well, as confirmed by Underiner et al. 34
In contrast, inhibitors XL184, BMS777607, XL880, and MGCD-265 showed similar potent inhibitory activity even against cell lines harboring altered amino acids at position 1228 or 1230. Indeed, BMS77607 and XL880 have been characterized as Type II MET inhibitors whose binding mode is not dependent on Y1230. As suggested by their similar potency when comparing the D1228 and Y1230 mutants against other MET variants, inhibitors XL184 and MGCD-265 would be expected to fall into the same class.
Taken together, the TMD RTK system is a valuable and reliable new assay system. A universal activation principle permits the development of cellular test systems containing a large panel of activated TMD RTKs in an identical cellular background without the need for ligand-induced activation. Using the same cellular background enables side-by-side comparison of results obtained for different RTKs. Ligand-independent activation has the advantage to activate the RTK even when the ligand is unknown or difficult to express in a format capable of activating the respective RTK. The next step will be to expand the portfolio of TMD-activated RTK harboring cell lines.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.