
Abstract
Patients with severe lung disease may develop hypercapnia, elevation of the levels of CO2 in the lungs and blood, which is associated with increased risk of death, often from infection. To identify compounds that ameliorate the adverse effects of hypercapnia, we performed a focused screen of 8832 compounds using a CO2-responsive luciferase reporter in Drosophila S2* cells. We found that evoxine, a plant alkaloid, counteracts the CO2-induced transcriptional suppression of antimicrobial peptides in S2* cells. Strikingly, evoxine also inhibits hypercapnic suppression of interleukin-6 and the chemokine CCL2 expression in human THP-1 macrophages. Evoxine’s effects are selective, since it does not prevent hypercapnic inhibition of phagocytosis by THP-1 cells or CO2-induced activation of AMPK in rat ATII pulmonary epithelial cells. The results suggest that hypercapnia suppresses innate immune gene expression by definable pathways that are evolutionarily conserved and demonstrate for the first time that specific CO2 effects can be targeted pharmacologically.
Introduction
Hypercapnia, elevation of the level of CO2 in the lungs and blood, can occur in severe chronic obstructive pulmonary disease (COPD) and other lung disorders. Hypercapnia is a risk factor for mortality in these diseases, especially from pulmonary infection.1–3 Our previous demonstration that hypercapnia impairs lung neutrophil function, inhibits bacterial clearance, and increases mortality in a mouse model of Pseudomonas aeruginosa pneumonia suggests that hypercapnia is not merely a marker of advanced lung disease, but plays a causal role in poor clinical outcomes by inhibiting host defense against bacterial infection. 4 However, the pathways by which elevated CO2 impacts nonneuronal tissues such as the lung and cells of the immune system are not well understood, nor are the mechanisms by which nonneuronal tissues sense and respond to hypercapnia (reviewed in Cummins et al. 5 and Boron 6 ). It has been known for some time that CO2 at high concentrations can inhibit proinflammatory cytokine release,7,8 but only recently have we and others investigated this phenomenon more systematically and demonstrated that hypercapnia reversibly suppresses NF-κB-regulated innate immune gene expression by a mechanism that is independent of extra- and intracellular acidosis, other gas sensing pathways, and without cytotoxicity.9,10 Oliver et al. further showed that elevated CO2 causes reversible nuclear translocation of the noncanonical NF-κB component RelB, a negative regulator of innate immune gene expression. 11 In addition, we have shown that hypercapnia inhibits phagocytosis, autophagy, and bacterial killing in mouse and human macrophages.9,12 Interestingly, elevated CO2 has immunosuppressive effects in Drosophila, as well as in mammalian systems. We found that hypercapnia inhibits expression of antimicrobial peptides (AMPs) in Drosophila S2* cells and that it decreases bacterial clearance and increases the mortality of bacterial infections in Drosophila in vivo. 13
In addition to effects on the immune system, we have shown that hypercapnia, independently of pH, causes endocytosis of the Na,K-ATPase in both mammalian and fly cells,13,14 leading to impairment of Na+ gradient-dependent alveolar fluid reabsorption in rat lungs. In rat alveolar epithelial cells, elevated CO2 initiates a signaling cascade involving an increase in intracellular calcium, phosphorylation of adenosine monophosphate-activated protein kinase (AMPK), and activation of the atypical PKC-ζ, leading to phosphorylation of Na,K-ATPase at Ser-18. 15 Further, knockdown of JNK in both mammalian and fly cells prevents hypercapnia-induced endocytosis of the Na,K-ATPase. 16 Taken together, these findings indicate that cells of the immune system and other nonneuronal tissues have the capacity to sense and respond to elevated levels of CO2, and that such responses may be mediated by specific signaling pathways that are evolutionarily conserved. Therefore, we performed small-molecule screening in Drosophila S2* cells to identify compounds that could serve as chemical tools for probing as yet poorly defined CO2 response pathways, and that might have therapeutic potential.
Here we report the identification of several compounds that specifically upregulate expression of AMPs in S2* cells more in CO2 than in air, indicating that they likely antagonize a novel CO2 response mechanism. Further, we show that evoxine, a furoquinoline alkaloid found in plants of the Rutaceae family, 17 not only blocks CO2-mediated inhibition of immune gene expression in flies, but also counteracts hypercapnic suppression of interleukin-6 (IL-6) and the chemokine CCL2 in human THP-1 macrophages. That the inhibitory effects of elevated CO2 on innate immune gene expression can be interrupted pharmacologically has important clinical implications.
Materials and Methods
Exposures of Fly and Mammalian Cells to Elevated CO2
Exposure to elevated levels of CO2 was carried out in BioSpherix C-Chambers fitted with ProCO2 regulators (BioSpherix Ltd., Parish, NY) as previously described.13,18 Drosophila S2* cells were cultured in air (0.04% CO2) or 13% CO2. 13 Human THP-1 cells and rat ATII cells were cultured in 5% CO2 (normocapnia) or elevated concentrations of CO29,15 as follows: Evoxine effects on hypercapnic suppression of IL-6 and CCL2 were assessed in THP-1 cells exposed to 15% CO2 in media without added buffer. For selected experiments in which culture medium was buffered with NaOH to prevent the decrease in pH that otherwise would accompany hypercapnia, 12.5% CO2 was used, since it is possible to fully buffer the pH at this level of CO2 without adverse osmotic effects on cells. 9 The effect of evoxine on hypercapnic inhibition of phagocytosis was assessed in THP-1 cells exposed to 20% CO2, which produces a more robust effect on phagocytosis than lower CO2 concentrations. The effect of evoxine on AMPK activation was assessed in rat ATII cells exposed to 20% CO2, also because this CO2 concentration robustly activates AMPK. 15
Small-Molecule Screening
Compounds were screened at ICCB–Longwood (Harvard Medical School, Boston, MA) using the protocols of Helenius et al. as modified for small-molecule rather than dsRNA screening. 19 Briefly, primary compound screening was carried out in duplicate in 13% CO2 using the S2* Diptericin-luciferase (Dipt-luc) cell line (gift from N. Silverman). The Dipt-luc reporter consists of 2.2 kb of the Dipt promoter 20 driving firefly luciferase in the pGL3 vector (Promega, Madison, WI). 21 A Renilla luciferase-Pol III (PolIII-luc) reporter, in which a fragment of the promoter for the RNA polymerase 128 subunit drives Renilla luciferase 22 (gift of the DRSC), was transfected into the Dipt-luc cell line using Effectene (Qiagen, Valencia, CA). Cells were maintained in 1 mg/mL G418 and 200 µg/mL hygromycin in Schneider’s Insect Medium (Sigma, St. Louis, MO) containing 10% fetal bovine serum (FBS). Liquid handling was performed using a Matrix WellMate dispenser (Thermo Scientific, Waltham, MA). Ten thousand cells per well in 384-well plates were screened in a final concentration of 0.3% DMSO for ~13 h, and 10 µL of CO2-preequilibrated media was dispensed into 384-well plates, into which 100 nL of small molecule in 100% DMSO was added per well using robotic pin transfer. The concentration of small molecules varied within and between libraries, ranging from 0.1 to 10 mM and 2 to 5 mg/mL ( Table 1 ). All compounds were screened at 0.3% of the library concentration. Plates were then placed back in the CO2 chamber for 3 h to ensure media was at 13% CO2. S2* cells that had been primed 24 h prior with 1 µM 20-hydroxyecdysone (Sigma) to improve immune responsiveness were then resuspended in CO2-preequilibrated media, and 20 µL (precounted to contain 10,000 cells) was dispensed per well. Cells were then placed in a CO2 chamber for 5 h and cells were challenged with 100 ng/mL Escherichia coli peptidoglycan (PGN; E. coli 0111:B4, InvivoGen, San Diego, CA) to induce Dipt and incubated for a further 5 h in CO2. Media was neutralized with 25 mM NaOH to maintain pH at 7.0 as previously described. 13 Firefly and Renilla luminescence were measured sequentially using Dual-Glo luciferase reagent (Promega) with an Analyst GT plate reader (Molecular Devices, Sunnyvale, CA). Data were analyzed using DRSC software and Microsoft Excel. No positive control compounds were used in the screen, since no known inhibitors or enhancers of CO2-induced immune suppression were known. Instead, robustness of the screen was assessed by examining reproducibility between duplicate plates. A correlation R value of Dipt-luc/PolIII-luc ratios between representative duplicate plates was 0.75, with the percent coefficient of variation (standard deviation/mean*100) for 4 representative plates, in duplicate, from the primary screen having values of (28.6% and 27.1%), (30.9% and 30.4%), (33.9% and 33.2%), and (28.6% and 28.6%). Consistent with the R value, percent coefficient of variation values for duplicate plates are very similar, suggesting that most of the variation is due to biological effects of small molecules rather than technical factors. The most likely source of false positives is from liquid handling errors resulting in low PolIII-luc signal. The most likely source of false negatives is a small dynamic range caused by low induction of Dipt-luc by PGN, a subsequent relatively low amount of CO2 suppression, and modest rescue by small-molecule treatment.
Summary of Primary Screen Results.
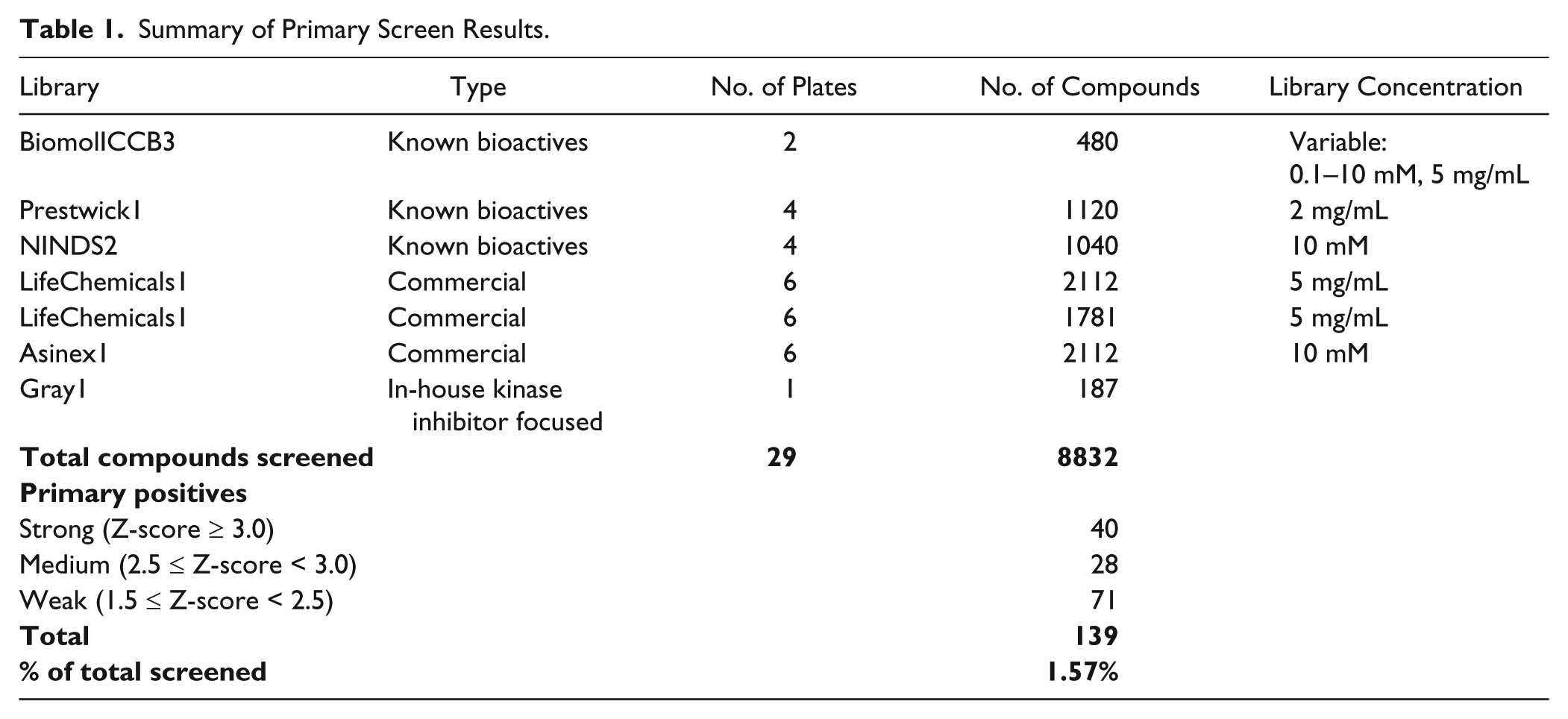
Compounds of interest were identified based on their Z-score, which represents the number of standard deviations the signal from one well on a plate is above or below the plate mean. Z-scores were calculated by normalizing the Dipt-luc signal from each well to the polIII-luc signal from the same well, calculating the average Dipt-luc/polIII-luc ratio on a plate, and determining the deviation of each well from the plate average. Secondary screening was performed in air and 13% CO2 in triplicate wells filled using multichannel pipettes.
Analysis of Evoxine from Second Supplier (Latoxan, France)
1H nuclear magnetic resonance (NMR) (500 MHz, DMSO-d6): δ 7.99 (d, J = 2.8 Hz, 1H), 7.93 (d, J = 9.4 Hz, 1H), 7.43 (d, J = 2.8 Hz, 1H), 7.42 (d, J = 9.4 Hz, 1H), 5.08 (d, J = 5.9 Hz, 1H), 4.46 (s, 1H), 4.43 (s, 3H), 4.41 (d, J = 2.2 Hz, 1H), 4.02 (dd, J = 10.0, 8.1 Hz, 1H), 3.93 (s, 3H), 3.62 (ddd, J = 8.0, 5.9, 2.1 Hz, 1H), 1.17 (s, 3H), 1.11 (s, 3H)
13C NMR (126 MHz, DMSO-d6): δ 163.8, 156.7, 151.6, 143.7, 141.6, 140.7, 117.6, 114.0, 113.7, 105.4, 101.7, 76.1, 71.4, 70.9, 60.7, 59.4, 27.5, 24.2
MS (m/z): [M + 1]+ calculated for C18H21NO6, 348.14; found, 348.10
High-performance liquid chromatography (HPLC) purity = 96.9% using a Zorbax C8 column (4.6 × 150 mm), gradient elution 0%–95% CH3CN (with 0.05% trifluoroacetic acid [TFA]) in H2O (with 0.05% TFA) over 30 min, 1.0 mL/min, UV @ 254 nm, RT = 11.30 min
Drosophila Cell Culture and Assays
Drosophila S2* cells were cultured in air (0.04% CO2) or 13% CO2, as previously described. 13 RNA was extracted using Trizol and qPCR was performed on RNA extractions from three separate samples using protocols and primers as described 13 using iScript cDNA synthesis kit and iQ SYBR Green Supermix (Bio-Rad, Hercules, CA). Antimicrobial peptide mRNA levels were normalized to ribosomal protein 49 (RP49) mRNA levels.
Mammalian Cell Culture and Assays
Human THP-1 macrophages were cultured as described in Wang et al. 9 In brief, cells were differentiated with 5 nM phorbol myristate acetate (PMA) for 48 h, starved overnight in 0.5% FBS media, and then stimulated with E. coli K12 lipopolysaccharide (LPS) (1 ng/mL final concentration) for 3 h in air or elevated CO2. In selected experiments, 25 mM NaOH was added to the culture medium of cells exposed to 12.5% CO2 to prevent a decrease in pH. Cells were treated with 48 µM evoxine for 30 min before exposure to hypercapnia and for the duration of the experiment. RNA was extracted using Trizol from three to five separate samples and qPCR performed using primers: IL-6 forward, 5′-GGT ACATCCTCGACGGCATCT -3′; IL-6 reverse, 5′-GTGCCT CTTTGCTGCTTTCAC; β-actin forward, 5′-ACGGGGTCA CCCACACTGTGC-3′; β-actin reverse, 5′-CTAGAAGCA TTTGCGGTGGACGATG-3′. For each experiment, all values on a plate were normalized to the control (5% CO2 with DMSO), and then values from replicates were averaged.
The effect of evoxine on phagocytosis was determined using the assay of Wang et al. 9
Rat alveolar type II (ATII) cells were isolated and cultured as previously described. 15 ATII cells were treated with 48 µM evoxine for 30 min prior to 10 min exposure to 20% CO2 in buffered medium. Western blotting for AMPK and p-AMPK was also as described. 15
Statistical Analysis
Two-tailed Student t test on normalized values were used for all comparisons.
Access to Research Materials
All plasmids used in these experiments are available upon request.
Results
To identify antagonists of CO2-induced immune suppression, we screened a total of 8832 compounds from seven different libraries containing natural and synthetic compounds of known and unknown bioactivity ( Table 1 ). The screen was performed using Drosophila S2* cells because a CO2-responsive reporter suitable for high-throughput screening has not yet been identified in mammalian cells, and we had previously used S2* cells to perform a genomewide double-stranded RNA screen to identify Drosophila genes that mediate hypercapnic immune suppression. 18 The S2* cell line is commonly used for in vitro immunological experiments and most likely represents a macrophage lineage. 23 A dual-luciferase reporter assay was adapted for high-throughput screening by stably cotransfecting S2* cells with a CO2-regulatable firefly luciferase driven by 2.2 kb of the Diptericin promoter (Dipt-luc)13,20,21 and a CO2-insensitive RNA polymerase III promoter driving Renilla luciferase (PolIII-luc). 18 PolIII-luc was used for internal normalization to control for effects of compounds on cell toxicity and general transcription, and screening was performed in neutralized culture medium to avoid confounding effects of acidosis (see Materials and Methods). Extensive prescreen optimization indicated that the Dipt-luc reporter expression closely paralleled endogenous Dipt expression under all tested concentrations and durations of CO2 exposure. 18 Thirteen percent CO2 was determined to be an optimal treatment for robust reporter responses in high-throughput screening conditions, but still within the range observed in patients with severe pulmonary dysfunction. The screen was performed in duplicate in 384-well plates using 100 ng/mL E. coli PGN to induce Dipt-luc.
The primary screen identified 139 compounds (
Table 1
,
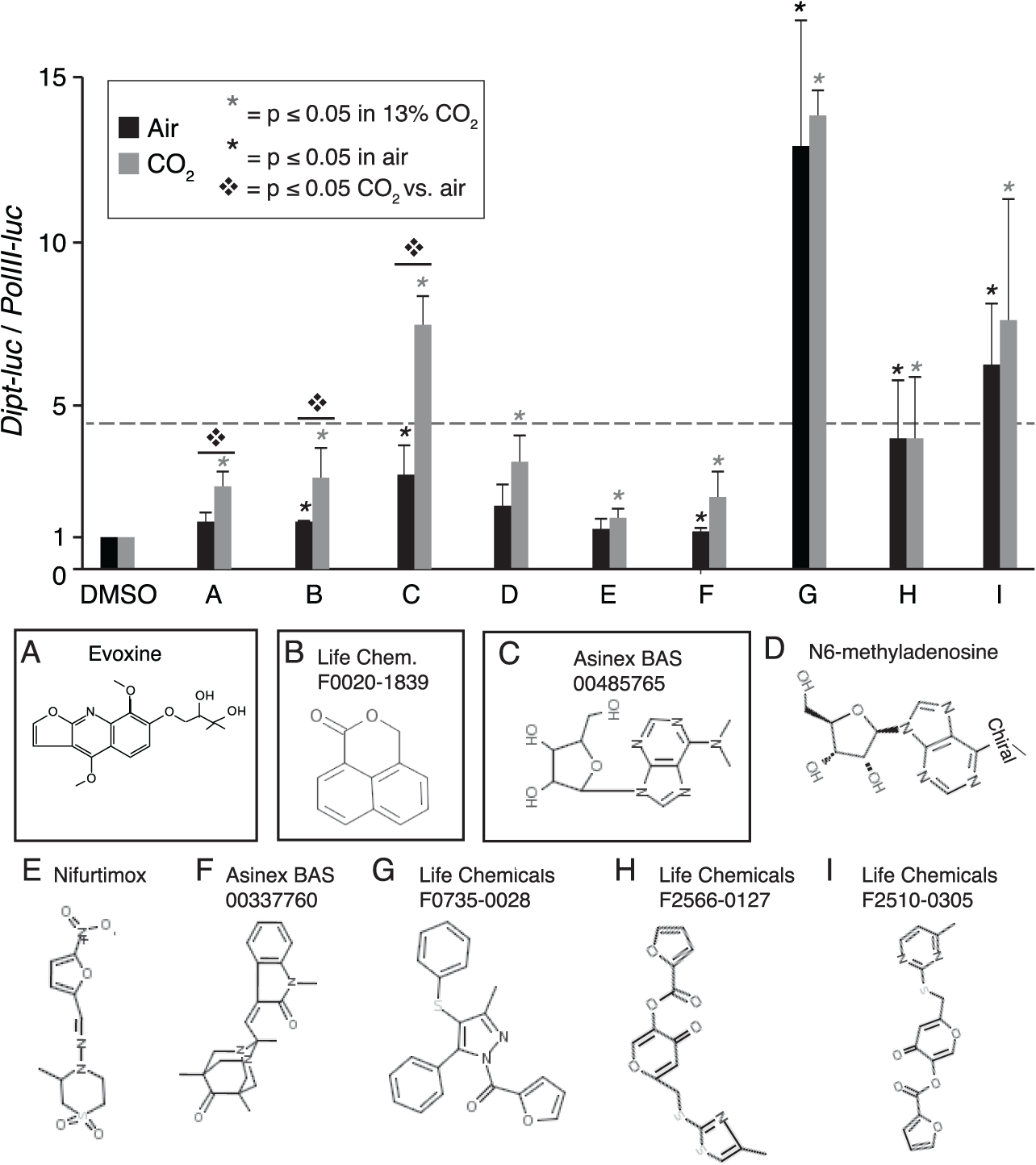
Identification of three small molecules that counteract CO2-induced immune suppression. Secondary assays confirm 9 of 27 tested compounds increase normalized Diptericin-luciferase reporter activity, of which 3 compounds have a stronger effect in CO2 than in air (boxed compounds). Air and CO2 bars (black and gray) are normalized to their DMSO controls shown as first results on the X axis. Black and gray asterisks, significant (p < 0.05) upregulation of Dipt-luc in air and CO2, respectively, by compound vs. DMSO; ❖, significant difference (p < 0.05) between fold increase in CO2 and in air; dashed line, a Dipt-luc/polIII-luc ratio of 4.5 over DMSO that for CO2 bars would indicate that a compound restored Dipt-luc expression to levels in air (i.e., average fold suppression of Dipt-luc by 13% CO2 is 4.5); error bars, standard deviation.
We selected evoxine for further analysis because of its low toxicity and because it did not increase Dipt-luc expression in air. For tertiary testing, evoxine was obtained from a different supplier than the one that supplied compound for primary and secondary screening. Mass spectrometry, NMR, and HPLC analyses performed by the Northwestern Center for Molecular Innovation and Drug Discovery (CMIDD) confirmed the structure and purity (96.9%) of evoxine from the second supplier (see Materials and Methods). Quantitative PCR analysis of Dipt expression in S2* cells confirmed and extended the results observed with the Dipt-luc reporter, demonstrating that over a 1000-fold concentration range, evoxine increased Dipt expression more in elevated CO2 than in air ( Fig. 2a ). Notably, even at the highest concentration tested, 4.8 mM, evoxine did not affect S2* cell viability, as assessed by polII-luc levels. The greatest increase in Dipt-luc expression in 13% CO2 was observed at 48 µM, with higher concentrations reducing induction of Dipt-luc not only in elevated CO2 compared to 48 µM, but also in air compared to vehicle-treated controls. This activity profile may result from evoxine having off-target effects that reduce Dipt expression at higher evoxine concentrations and/or may result from the target of evoxine having multiple functions such that moderate inhibition blocks hypercapnic immune suppression, but complete inhibition reduces Dipt expression irrespective of inhibition of hypercapnic immune suppression. Interestingly, evoxine was previously reported to have antiplasmodial activity with an IC50 of 24.5 µM, 17 similar to the optimal 48 µM concentration observed in our assays.
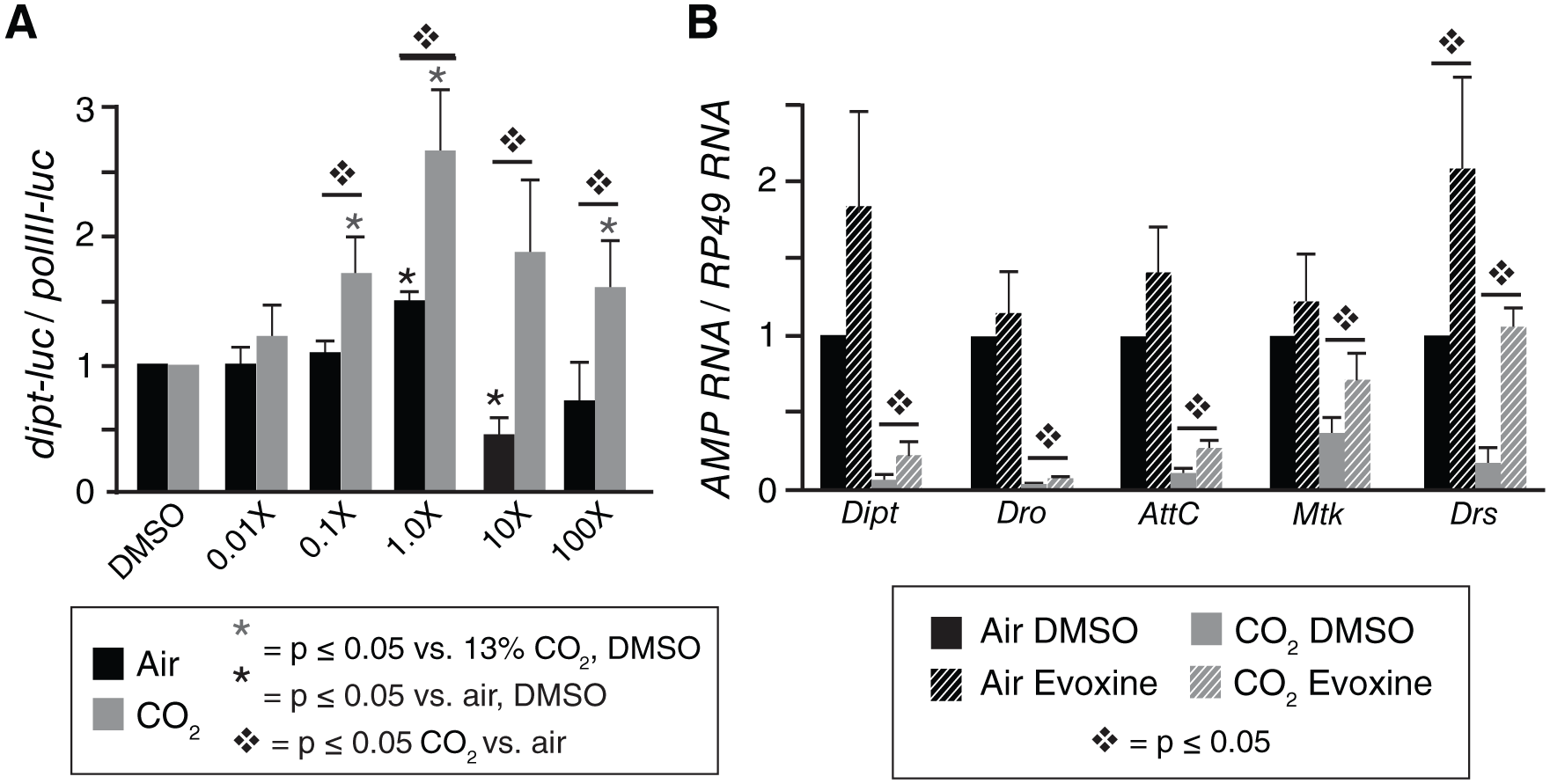
Evoxine counteracts suppression by elevated CO2 of multiple antimicrobial peptides in Drosophila S2* cells. (
To determine whether the effects of evoxine were specific for Dipt induction, or whether evoxine can prevent the CO 2 -induced immune suppression of multiple Drosophila AMPs in S2* cells, we used qPCR to assay mRNA levels of other AMP genes, including Drosocin (Dro), AttacinC (AttC), Metchnikowin (Mtk), and Drosomycin (Drs). For these experiments, we used the 48 µM concentration of evoxine that optimally blocked CO2-induced suppression of Dipt induction ( Fig. 2a ). In 13% CO2, evoxine increased the PGN-induced mRNA levels of all AMPs (p < 0.05), restoring the levels of Mtk and Drs to near unsuppressed levels ( Fig. 2b ). Evoxine at this concentration also modestly increased the induced levels of AMPs in air, but the change was statistically significant only for Drs, which evoxine increased ~2-fold in air versus 5-fold in 13% CO2. These results indicate that evoxine preferentially affects components of a putative hypercapnia response pathway that regulates many AMP genes. However, the varying degree to which evoxine increased expression of the different AMPs (e.g., compare Dipt and Drs in Fig. 2b ) suggests that rather than acting directly on each AMP, CO2 regulation of AMP expression may act asymmetrically on the IMD and Toll pathways that convergently regulate AMP gene expression.
We have previously provided evidence that the pathways by which hypercapnia suppresses innate immune effectors may be conserved.9,13 To investigate whether evoxine could alter the effects of elevated CO2 in mammalian immune cells, we tested whether evoxine could prevent hypercapnic suppression of LPS-induced expression of clinically important cytokines in human THP-1 macrophages. Treatment of THP-1 cells with 48 µM evoxine under normocapnic culture conditions of 5% CO2 (corresponding to a normal mammalian blood CO2 level) decreased the induction of IL-6 and CCL2 mRNA by ~30%–40% and ~60%, respectively (Fig. 3a–c), similar to the decreased induction of dipt-luc in S2* cells exposed to higher concentrations of evoxine ( Fig. 2a ).
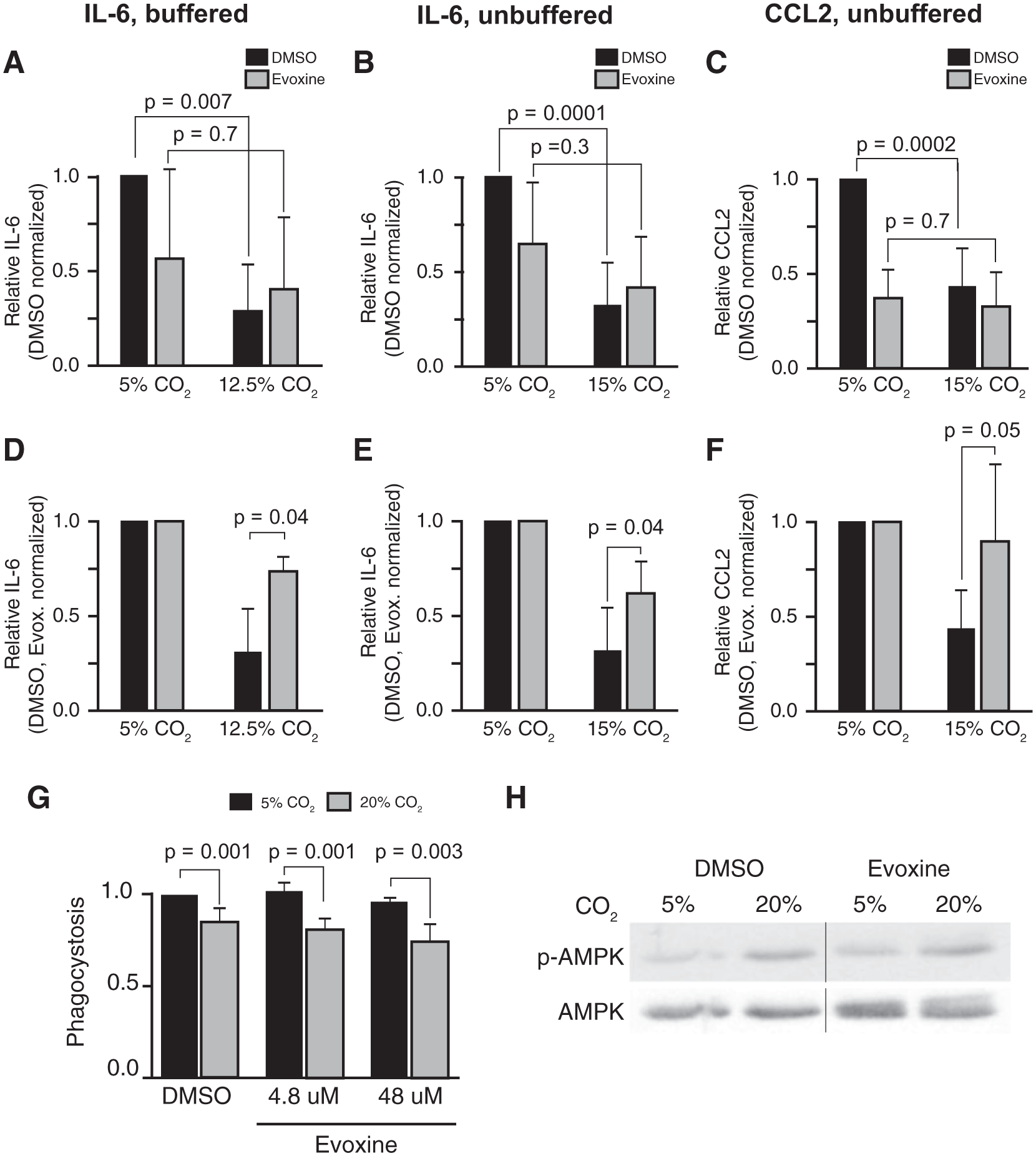
Evoxine counteracts specific effects of hypercapnia in mammalian cells. (
Critically, even though evoxine reduced cytokine induction under normocapnic conditions, it nonetheless blocked the three- to fourfold reduction in IL-6 and CCL2 mRNA levels caused by exposure to elevated CO2 (Fig. 3a–c and Wang et al. 9 ). Evoxine blocked hypercapnia’s immune suppressive effects both in buffered media ( Fig. 3a , 12.5% CO2, pH 7.4) and in unbuffered media with lower pH ( Fig. 3b , c , 15% CO2, pH 7.2). To better visualize these effects, we normalized the values of IL-6 and CCL2 in evoxine-treated cells in 5% CO2 (normocapnia) to a value of 1.0, and applied this correction to their levels in hypercapnia (Fig. 3d–f). Represented this way, LPS-induced IL-6 and CCL2 mRNA levels were ~60% and ~100% higher, respectively, in 15% CO2 in the presence versus absence of evoxine. Thus, as in Drosophila S2* cells, evoxine has CO2-specific effects and mitigates hypercapnic suppression of innate immune gene expression in human macrophages.
We next sought to determine whether evoxine could modulate other effects of hypercapnia on nonneuronal cells, or whether its effects are specific to transcriptional regulation of innate immune genes. We tested whether evoxine can block hypercapnic inhibition of phagocytosis in human THP-1 macrophages 9 and whether it can prevent hypercapnic activation of AMPK in rat alveolar epithelial cells. 15 However, unlike its effect on hypercapnic suppression of IL-6 and CCL2, evoxine failed to overcome the reduction of phagocytosis caused by elevated CO2 ( Fig. 3g ). Evoxine also failed to prevent the hypercapnia-induced phosphorylation of AMPK ( Fig. 3h ). Thus, evoxine is able to block some, but not other, responses to hypercapnia. Based on these observations, we postulate that there are at least two separable conserved nonneuronal CO2 signaling pathways ( Fig. 4 ), one of which may mediate rapid posttranslational responses such as Na,K-ATPase internalization via AMPK and JNK phosphorylation, and another that mediates longer-term transcriptional responses such as production of AMPs and cytokines. This is supported by our finding that knockdown of AMPK or JNK does not relieve CO2-induced suppression of Dipt-luc in S2* cells. 18 Such a response architecture would be analogous to responses to varying oxygen levels, where hypoxia-inducible factor (HIF)–regulated transcriptional changes are seen within minutes to hours, but guanylate cyclases respond to hypoxia within seconds. 26
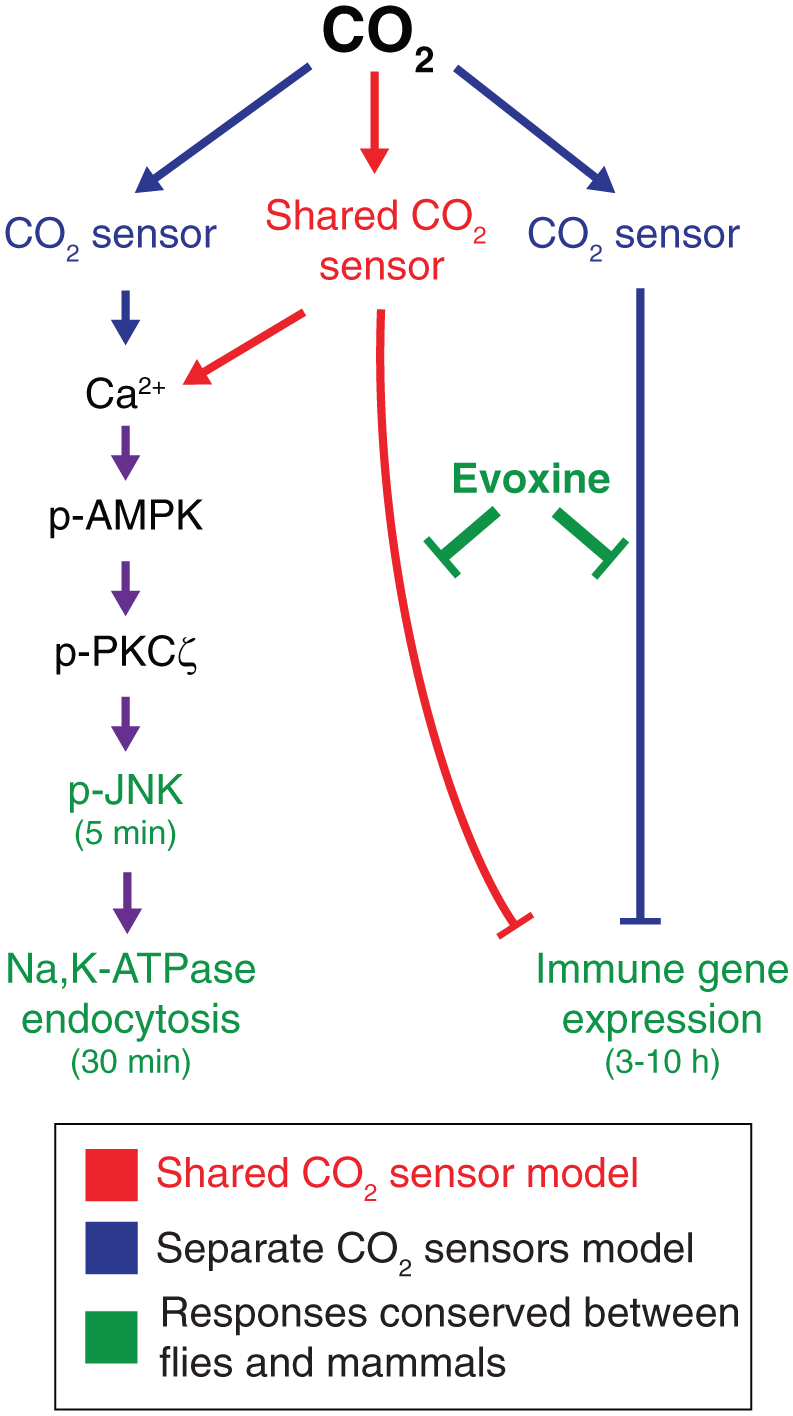
Models for organization of CO2 signaling pathways. Two alternative models for architecture of CO2 response pathways are shown. A model with distinct nonneuronal CO2 sensors that each modulate immune and other downstream effects, such as Na,K-ATPase endocytosis, is shown in blue. A model in which a single CO2 sensor leads to a branched pathway is shown in red. Genes and responses known to be conserved in CO2 signaling between mammals and Drosophila are shown in green.
Discussion
A critical finding of this screen is that it provides the first evidence that a small molecule can overcome suppression of innate immune gene expression caused by elevated CO2. It is encouraging that the screen identified several compounds that selectively improved the expression of AMPs by Drosophila S2* cells during hypercapnia. Furthermore, one of these, evoxine, also blocks hypercapnic suppression of LPS-induced IL-6 and CCL2 expression in human macrophages. It is also promising that although evoxine has been previously reported to inhibit cytochrome P450 (CYP1A2), 27 it is not toxic to Drosophila cells even at concentrations 100 times those that had optimal effects on hypercapnic responses. Given that there seems to be a strong correlation of compound toxicity between Drosophila and mammals, 28 these results suggest that evoxine-based compounds could be well tolerated by mammals. Consistent with this, the LD50 for evoxine in rats is 370 mg/kg, 29 and plant extracts containing significant amounts of evoxine are used in traditional African medicine. 30 Thus, it is plausible that therapeutics based on evoxine might be developed to target CO2-responsive pathways without major off-target effects. This work has also identified three structurally similar small molecules that are strong general enhancers of Dipt-luc expression, but further work is needed to determine whether they could be used to boost immune responses in mammalian cells.
In addition to being the first small molecule to be identified that can overcome suppression of innate immune gene expression caused by elevated CO2, evoxine has proved to be an important tool for probing hypercapnia response pathways. First, because of the ability of a single compound to selectively block the effects of elevated CO2 on AMP and cytokine expression in Drosophila and mammalian cells, it can be argued that hypercapnia suppresses innate immune gene expression by acting on one or a small number of signaling pathways, rather than by nonspecific mechanisms affecting many targets. Second, in mammalian cells, the ability of evoxine to selectively target a pathway controlling cytokine expression, without affecting other tested hypercapnia-regulated activities, suggests the existence of at least two nonneuronal CO2 response pathways. Such pathways could share a common CO2 sensor and diverge in downstream signaling, or might be entirely distinct ( Fig. 4 ). At present, the nature of these pathways is unclear, but in both flies and mammals, hypercapnic immune suppression appears to result from an effect downstream of nuclear translocation of NF-κB proteins.9,13 Regardless of the precise organization of the CO2-responsive pathways, the ability of evoxine to prevent hypercapnic suppression of AMP and cytokine/chemokine expression in Drosophila and mammalian immune cells strongly suggests evolutionary conservation of CO2 response pathways regulating immune genes, as we have shown for pathways regulating Na,K-ATPase endocytosis. 16 Ultimately, this conservation should allow both systems to be used in defining the molecular components that mediate the effects of elevated CO2 on nonneuronal cells.
Footnotes
Acknowledgements
We thank Andrew Rennekamp for comments on the manuscript; the members of the ICCB–Longwood screening facility, especially Su Chiang and Dave Wrobel; the members of the Harvard Drosophila RNAi Screening Center (DRSC); and the members of the Northwestern Center for Molecular Innovation and Drug Discovery (CMIDD), especially Gary Schiltz and Karl Scheidt. We also thank the DRSC for plasmids and Lynn Welch for operational support.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the NIH to GJB and PHSS (NHLBI/R01-HL107629), to JIS (NHLBI/R01-HL-85534), to ICCB–Longwood (NERCE/NSRB U54-AI057159), and to DRSC (NIGMS/R01-GM067761); from the American Heart Association to GJB (grant-in-aid award, 0855686G) and to ITH (predoctoral fellowship, 0715562Z); from the Chicago Biomedical Consortium with support from The Searle Funds at The Chicago Community Trust to NU CMIDD; and from the Northwestern University Robert H. Lurie Comprehensive Cancer Center to the NU HTA core facility.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.