
Abstract
Posttranscriptional regulation of gene expression is an elaborate and intricate process, constituting an important mechanism for the control of protein expression. During its existence, mRNA is escorted by proteins and other RNAs, which control the maturation, transportation, localization, translational efficiency, and ultimately its degradation. Without changes at the transcription level, mRNA steady-state levels can vary dramatically by just small changes in mRNA stability. By influencing the metabolism of specific mRNAs, the abundance of specific mRNAs can be controlled in organisms from bacteria to mammals. In eukaryotic cells, the control of mRNA stability is exerted through specific cis-acting elements (sequence-specific control elements) and trans-acting factors (mRNA binding proteins and some miRNAs). mRNA stability appears to be a key regulator in controlling the expression of many proteins. Dysregulation of mRNA stability has been associated with human diseases, including cancer, inflammatory disease, and Alzheimer’s. These observations suggest that modulating the stability of specific mRNAs may represent a viable strategy for pharmaceutical intervention. The literature already describes several compounds that influence mRNA stability. Measuring mRNA stability by conventional methods is labor intensive and time-consuming. However, several systems have been described that can be used to screen for modulators of mRNA levels in a high-throughput format. Thus, these assay systems offer a novel approach for screening targets that at present appear to be poorly “drugable.” This review describes the utility of mRNA stability as a novel approach to drug discovery, focusing on assay methods and tool compounds available to monitor mRNA stability. The authors describe mRNA stability assays and issues related to this approach.
Introduction
T
The regulation of mRNA stability is complex and can involve elements in both the coding and the untranslated regions of an mRNA transcript. Regulation of mRNA stability appears to depend on the interaction between intrinsic, cis-acting elements and trans-acting factors (
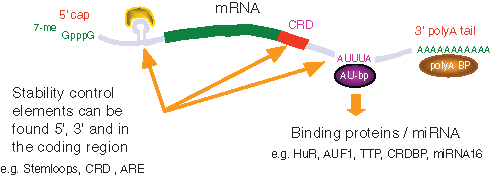
Cis-acting elements and trans-acting factors governing mRNA half-life. ARE, AU-rich elements; CRD, coding region instability determinant.
A number of trans-acting regulatory factors, including several mRNA binding proteins and some miRNAs, play a role in regulating mRNA stability. A number of these regulatory factors are ARE binding proteins; their binding to mRNA has been show to either stabilize or destabilize their RNA targets dependent on cellular conditions. 23 Based on their patterns of activity, these proteins have been tentatively assigned as stabilizers, 24,25 destabilizers, 26-28 or nucleocytoplasmic transporters of AUUUA-containing mRNAs. 29 The proto-oncogene c-fos mRNA, for instance, has a half-life of approximately 30 min, and its instability is modulated by sequences within its 3′UTR. Replacement of the 3′UTR from c-fos with the 3′UTR sequences from β-globin, which has a half-life of approximately 24 h, produces a stable chimeric transcript. Conversely, fusion of the c-fos 3′UTR to β-globin confers instability. 30
A number of signal transduction processes have been associated with changes to mRNA stability. Such changes in mRNA stability appear to be due to alterations in the affinity of RNA binding proteins for their cognate mRNA (see
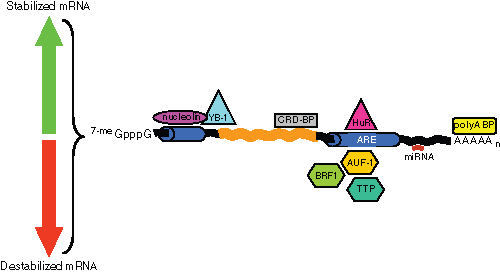
The heat shock–ubiquitin–proteasome pathway appears to be associated with AUF1-mediated mRNA regulation in another ARE-containing transcript, the granulocyte macrophage colony stimulating factor (GM-CSF). 38 AU-motif-dependent mRNA decay can thus be transiently inhibited by the activation of the JNK and p38 MAP-kinases pathways. Once the activation signal has disappeared, the AU-motif ensures rapid return to a low level of basal expression. Thus, AU-binding proteins act as mediators between the signaling cascade and the mRNA degradation machinery.
Recently, it has become apparent that, at least in the case of AU-mediated mRNA stability, a third player in the form of microRNA (miRNA) is at work modulating mRNA half-life. Jing et al. 39 have reported that miR16 is involved in regulating TNFα mRNA stability through interaction with RNA-induced silencing complex (RISC) and TTP. The effect of miR16 is to allow sequence-specific targeting of the RISC complex to the ARE. On the other hand, the TNFα ARE was also reported to enhance translation upon serum starvation, an effect that was coupled to two microRNP-related proteins (FXR1 and AGO2), 40 thus connecting two posttranscriptional regulatory mechanisms and perhaps explaining why under some experimental conditions, mRNA degradation appears to be linked to ongoing translation. This is further corroborated by the fact that AUF1 has been reported to be involved in regulating translation but not mRNA stability via the AU-motifs of bcl-2 mRNA. 41
miRNAs also appear to be associated with, and interact indirectly through, AGO2 and Dicer with AU-binding proteins to prevent translational initiation and induce mRNA degradation. 42 Furthermore, decapping and deadenylation are promoted by the GW182 protein, which is also involved in miRNA-mediated gene silencing. This protein is localized to Processing Bodies (P-Bodies), bringing together Argonaute (AGO)–containing RISCs and mRNA decay enzymes. miRNAs have also been implicated in directing rapid deadenylation of mRNA. 43,44 These examples demonstrate that there is growing evidence that posttranslational regulation of gene expression is highly complex, allowing a coordinated regulation by influencing mRNA stability and translational efficiency.
In various disease states, mRNA half-life and the levels of disease-related factors are altered due to aberrant mRNA stabilization. A number of human pathologies associated with altered mRNA stability have recently been reviewed in the context of ARE elements by Khabar.
11
Other pathologies associated with RNA processing have also been identified. For example, cystic fibrosis and muscular dystrophy can both be caused by premature termination codons present in their coding sequences, which leads to nonsense-mediated decay (NMD) of their mRNAs.
45,46
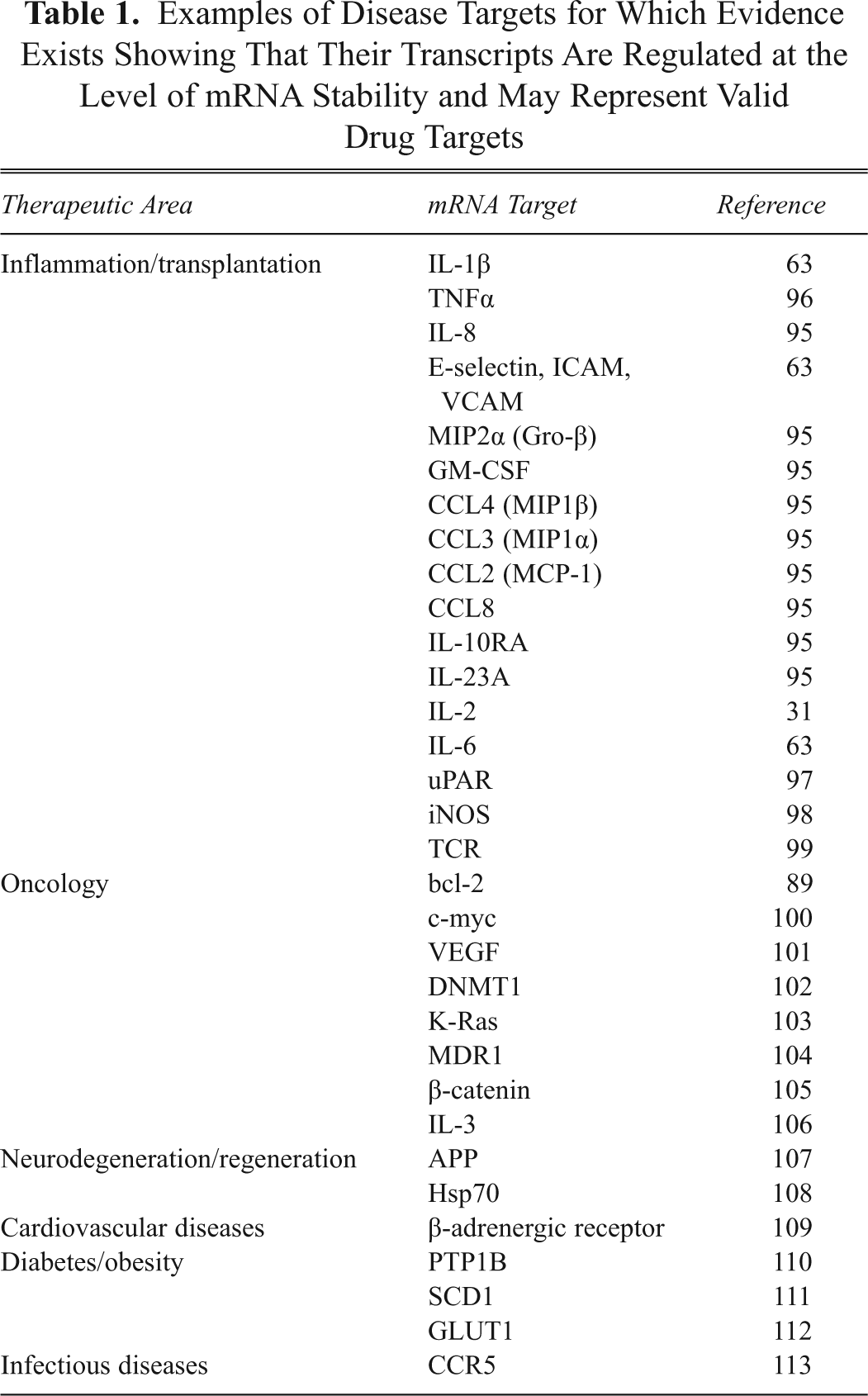
A number of examples of disease-relevant genes that are regulated at the level of mRNA stability are shown in
Examples of Disease Targets for Which Evidence Exists Showing That Their Transcripts Are Regulated at the Level of mRNA Stability and May Represent Valid Drug Targets
Therapeutic approaches for the modulation of mRNA stability may occur at a number of different levels. The possibility of directly altering protein-RNA interaction, although conceptually the simplest way to alter mRNA stability, seems the most unlikely mechanism by which a small molecule would be able to influence such a system. The reason for this is that protein-protein interactions have proven difficult to inhibit by small molecules because of the tendency for such interactions to occur over large surfaces of the proteins with generally hydrophobic and van der Waals interactions. It might be expected that similar interactions will drive RNA-protein interactions. 47 There are a number of examples of small molecules that alter the interaction of proteins with an RNA molecule by altering the conformation of the RNA such that the protein of interest can no longer recognize the RNA molecule. Examples of small molecules binding RNA and altering its biological activity include tRNA and rRNA with aminoglycosides, 48 small-molecule interaction with iron response elements (IRE) sequences altering structure, 49 and studies from the Dervan group 50 with molecules able to interact with helical nucleic acid sequences. However, we believe that the most likely mechanism for small molecules to modulate the stability of specific mRNAs is by altering the activity of proteins that interact with target mRNA or by interfering with signal transduction pathways governing mRNA stability or translatability. A number of such compounds that have been described having such an effect will be discussed.
The primary aim of this review is to discuss the methods available for detecting mRNA levels and thus screening for such molecules.
Current Screening Methods to Monitor mRNA Stability
A number of methods are available for the detection and quantification of mRNA present in a cell sample. Traditional methods such as Northern blots, RNase protection, or slot blots have the disadvantage that they only measure either one or only a limited number of transcripts at a time and are very difficult to automate. Newer techniques for analysis of gene expression include: (a) unbiased, open systems such as serial analysis of gene expression (SAGE), 51 differential display (DD) analysis, 52 RNA arbitrarily primer (RAP)–PCR, 53 restriction endonucleolytic analysis of differentially expressed sequences (READS), 54 amplified restriction fragment-length polymorphism (AFLP), 55 total gene expression analysis (TOGA), 56 and the quantitative multiplex RT-PCR method, Standardized RT-PCR (StaRT-PCR) 52 ; and (b) focused, closed systems such as high-density cDNA filter hybridization analysis: (HDFCA), suppression subtractive hybridization (SSH), differential screening (DS), cDNA arrays, or oligonucleotide chips and tissue microarrays, methods that have been reviewed previously. 52
Although closed systems are excellent for the initial screening of a large number of gene transcripts, the value of the information generated is biased because the techniques are generally limited to an often arbitrarily chosen set of known sequences. Only an open system or platform has the potential to evaluate the expression patterns of tens of thousands of genes that have not yet been cloned or partially sequenced in a quantitative and unbiased manner.
Low-throughput methods
Methods to measure abundance of specific mRNA sequences are essential prerequisites to monitor stability of a specific transcript. The first method used for quantifying mRNA levels was with Northern blots. In such experiments, the level of an RNA transcript could be quantified either when the RNA is in a steady state or after the inhibition of RNA synthesis, by actinomycin D or alpha-amanitin, to measure the half-life of specific mRNAs.
The development of reverse transcription–PCR (RT-PCR) has generally become accepted as the new “gold standard” for measuring the relative abundance of RNA transcripts, with the availability of kits, automated PCR machines, and the ability to multiplex, allowing the convenient normalization of transcript numbers to internal controls (usually a housekeeping gene such as GAPDH or absolute quantification using an amplicon). Although Northern blots and RT-PCR allow the abundance of specific transcripts to be measured, they do not allow a global assessment of the effect of a treatment on mRNA levels in a cell. Methods that allow a global assessment of transcript abundance include array methods where oligonucleotides or cDNAs are immobilized on a solid support, and SAGE. Array methods require amplification and labeling of the transcripts, allowing relative/comparative assessments of transcript abundance. Array methods, however, retain some bias as they depend on design, number of genes selected, controls, and gene sequences. SAGE offers a completely unbiased analysis of the entire genome but is very work intensive, time-consuming, and expensive.
The strength of these methods lies in their ability to monitor, in a comprehensive manner, the changes in mRNA abundance to as wide a number of transcripts as possible. Such a transcriptome-wide analysis then allows assessment of the global effects of a treatment on a cell, be it by agents that stimulate the cells such as growth factors, antigens, stress, or low molecular weight compounds. Technology development in the field of “deep sequencing” means that it is now possible to use such technology for the sequencing and even quantification of a comprehensive portion of the mRNA transcriptome. 57 Recently, a method to allow comparison of the effects of treatments on the transcriptome to be made has been published. 58
Higher throughput assay methods
Biochemical approaches
Such biochemical systems must all include: (a) in vitro transcription of the target mRNA; (b) a cell-free extract that provides the proteins necessary for translation, an important factor controlling mRNA stability, as well as other factors required for mRNA degradation or stabilization; and (c) a means to measure the levels of RNA, even those in a specific conformation. Such biochemical methods allow faster and thus higher throughput analysis using standard biochemical readouts used in high-throughput screening (HTS) labs (such as fluorescence polarization [FP] or time-resolved fluorescence resonance energy transfer [TR-FRET]). An example of a cell-free system was recently described by Meisner et al. 59
Here we describe a minimal primary consensus sequence together with the conformation-determining flanking sequence of an mRNA stability region by first defining a single-stranded mRNA accessibility sequence. By elongating the oligonucleotide up- and downstream of the core, they reduced the accessibility of the RNA binding site by modulating the secondary structure of the binding motif without changing the core sequence. It has been possible to define “opener” and “closer” oligonucleotides for manipulating the accessibility of the mRNA binding protein HuR. These oligonucleotides result in a change in conformation, either allowing or preventing the protein from binding, thereby mimicking the events in mRNA stability regulation. The cell-free system thus described could potentially also be used to screen for small molecular weight compounds that mimic the effect of these openers or closers, allowing one to target a large number of disease-relevant genes by using a common assay format.
The throughput of such biochemical assays is usually not limited, as the assays can readily be miniaturized to at least 384- or 1536-well formats such that reader capacity becomes the limiting step (with throughput of approximately 200, 1536-well plates per day being possible). The real limiting factor with such assays is the provision of assay reagents and the stability of RNA as a reagent (due to the ubiquitous nature of RNases in the environment).
Cell-based approaches
The most common way to determine if mRNA sequences present in a transcript are responsible for conferring mRNA stability is to create chimeric gene sequences, commonly fused to a stable gene such as β-globin, and to then monitor transcript levels after inhibition of transcription. 22 The abundance of the RNA transcripts is then commonly monitored by Northern blots 60 or by RT-PCR 61 following the inhibition of transcription. However, such analysis of RNA levels is not suitable for rapid HTS.
A number of reporter gene-based assays have been developed to address this limitation and can be used for the discovery of novel compounds affecting mRNA stability/translatability. One of them is described by Kastelic and Cheneval. 62 The objective of this assay system is to exploit earlier findings that it is possible to modulate mRNA levels with low molecular weight compounds. 63 Because AREs and other mRNA stability elements appear in a number of mRNAs known to be relevant in several human diseases, it allows this approach to be expanded into a platform technology that exploits the presence of stability determining elements. This approach was also recently suggested by Benjamin et al. 64 and shown to be suitable for screening.
The throughput of such assay systems in a well-equipped HTS lab, even using a workstation approach, in our experience, is limited to approximately 200 plates per day per full-time equivalent (FTE), 3 times a week. The use of frozen cells 65 can increase the number of times a week an assay can be run. Miniaturization to 1536-well formats is usually possible, and so the bottleneck for performing screening campaigns with this assay format is often that of cell line generation.
These systems allow screening for compounds that modulate reporter gene expression through mRNA sequence motifs in a high-throughput format. The stability (half-life) of a target mRNA sequence motif in the assay is monitored through the expression and activity of a reporter gene. The reporter construct, having been designed to include stability sequences of the target mRNA present in appropriate regions of the reporter construct, is stably transfected into a disease-relevant cell line. Because the aim of this system is to find compounds affecting mRNA stability, or indeed translation, through stability regulatory motifs, a control is built into the same cell. The control, a second identical expression plasmid that expresses a different reporter gene but does not contain any mRNA stability motifs, is simultaneously introduced into the cell. Thus, control and motif-specific measurements can be carried out at the same time in the same cell. This enables specific selection of compounds with effects dependent only on the motif introduced.
Other groups 66-70 have described similar systems with the aim of facilitating compound characterization. For example, the GEMS (Gene Expression Modulation by Small Molecules) system has been described by PTC Therapeutics. 71
Paillusson et al. 72 have developed a reporter system that mimics nonsense-mediated mRNA decay by constructing a T cell receptor minigene in which the green fluorescent protein–open reading frame (GFP-ORF) was inserted such that the stop codon acts as a premature stop codon (PTC). The authors then show that eliminating essential NMD factors resulted in an increase in GFP fluorescence, so that the system faithfully reflects NMD. The system, however, does not rule out other effects such as enhanced transcription or translation, or inhibition of general mRNA turnover.
Boelz et al. 73 describe a cell-based chemiluminescence reporter system to monitor NMD. Using a dual-luciferase approach, the authors introduced a PTC into the 3′ region of a renilla luciferase/β-globin fusion gene. Co-transfection of this construct with a control firefly luciferase expression vector allows for normalization and overcomes some of the drawbacks of using co-reporters such as CAT, β-gal, and GFP, which have different chemistry, handling, or instrumentation needs. Furthermore, this study showed that the luminescence measured faithfully reflected mRNA levels.
The Quanti Gene™ nucleic acid quantification kit as used by Warrior et al. 74 employs a branched DNA (bDNA) technology to measure mRNA directly from cells. Unlike PCR, which employs target amplification, this method relies on signal amplification for detection. Using a “sandwich” nucleic acid hybridization procedure, direct measurement of mRNA from crude cell lysates is possible. Target-specific capture extenders and label extenders are added to cell lysates, which allow the tethering of the target mRNA to the assay plate and subsequent hybridization of the bDNA amplifier. The branches of the DNA amplifier are then labeled with alkaline phosphatase, providing the amplification step for the assay. This approach has the advantage that it is easy and fast to develop compared to reporter gene assays, which optimally require stably transfected cell lines, result in more physiologically relevant data, and can be used with any cell type, including primary cell lines. However, the assay itself is a multistep, complex, and non- homogenous process with a lower throughput than for standard reporter gene assays. In the experience of the authors, such assay formats are limited to 384-well plate formats (because of the need for liquid transfer of the cell lysate to the capture plate). Because of the multiple assay steps, the throughput is limited to processing one hundred to two hundred 384-well plates a day, for one researcher. As with reporter gene assays, the need to seed and treat cells in the plates limits the number of days such an assay can be run to 3 to 4 days a week.
Martel et al. 75 have taken RNA analysis one step further by introducing the multiplexed Array Plate mRNA assay. In this approach, cells grown and subjected to compound treatment in 96-well plates were subjected to an in situ nuclease protection assay against a set of predefined targets. Processed cell lysates were then transferred to a microplate that contained a predefined oligonucleotide array that separated and immobilized the assay probes. Quantitative detection of array-bound probes was by enzyme-mediated chemiluminescence. In this way, effects of compounds on the mRNA levels of several genes can be determined. Although this represents a step up compared to reporter gene assays with respect to higher content, the method remains biased to the choice of the predefined targets and, with multiple assay steps, will remain a challenge to automate for true HTS.
DNA microarray assays provide a further step in the direction of transcriptome-wide quantitative analysis of mRNA levels and the understanding of gene expression. Canales et al. 76 compared microarray results with other quantitative gene expression platforms. The methods used were TaqMan, an assay using RT-quantitative real-time PCR, 77-79 which has also been used to validate microarray data earlier, 80-82 and standardized (Sta)RT-PCR, as described by Willey et al., 83 which is a competitive PCR-based method that measures the amount of a PCR end-product. After conversion of mRNA to cDNA using reverse transcriptase and either random or gene-specific oligonucleotides, hexamers, or oligo dT, the cDNA is added to standardized mixtures of competitive templates and dispensed into microplate wells containing gene-specific primers. The end-products are separated and quantified by high-throughput microfluidic electrophoresis. This method has also been used to validate microarray data 84 and the QuantiGene System described above by Warrior et al. 74 They observed a high correlation between quantitative gene expression values and microarray results, with only few differences among all platforms. The microarray systems were less sensitive, and detection levels varied only for lower expressed genes. Overall, there was an excellent correlation between each method tested, and it was concluded that the microarray data were validated by alternative quantitative gene expression platforms, supporting the use of microarray platforms for the quantitative characterization of gene expression.
Application to other targets such as miRNA
Genetic screens to identify genes modulated by miRNA activity have also been reported and are conceptually similar to the some of the assay systems reported for monitoring mRNA stability. By introducing the miRNA binding site from the UTR of the targeted gene into a suitable reporter gene, it is possible to monitor the effect of miRNAs whether they act at the level of mRNA stability or to alter the level of translational efficiency. 85,86 In fact, the availability of such assay systems has already allowed the description of small molecules interacting to modulate the activity of miRNAs. 87,88
Tool Compounds Described in the Literature
A number of compounds have already been described as modulating mRNA stability and thus can act as possible control compounds for developing assays to monitor this process. Possibly the first example of such an effect was the fungal metabolite radicicol analog A (RAA) that was shown to induce a rapid degradation of IL-1β, TNFα, IL-6, and Cox-2 transcripts in stimulated THP-1 cells mediated by the presence of AREs in the target mRNA’s 3′UTR.
63
This discovery demonstrated that mRNA stability can be influenced with small molecular weight compounds. Other molecules also reported to influence mRNA stability include paclitaxel,
89
cyclosporin A,
90
thalidomide,
91
and glucocorticoids
92
(see
Compounds Known to Influence mRNA Stability

Specificity of the assay
Compound specificity is a key consideration when identifying molecules that can modulate a specific target mRNA stability. In addressing this subject, however, it is important to distinguish between two different issues: (1) the specificity of the cellular regulatory mechanism that governs modulation of a target mRNA (thus leading to construction of an assay) and (2) the inherent specificity of compounds identified through such an assay (which is an issue for the pharmaceutical development of any compound).
Regulation of mRNA stability is also cell type specific. Certain mRNAs are unstable in one cell type but not in another. For example, cell-specific posttranscriptional regulation of CFTR (cystic fibrosis transmembrane conductance regulator) mRNA has been reported. 94 In a 3′UTR-dependent manner, TNFα decreased CFTR mRNA in human HT-29 colon cells but not in pulmonary Calu-3 cells, confirming that in this example, regulation of mRNA stability is cell type specific. Therefore, as mRNA regulation is cell type specific, it is possible for small molecular weight compounds to act differently on the same transcript depending on the cell type in which it is expressed.
Specificity of the compounds
Examples of molecules able to selectively modulate specific mRNAs exist. Mak et al. 95 reported RAA to be highly selective for altering the stability of mRNAs from a family of cytokines. In researching the mechanism of action of RAA, a SAGE analysis showed that of approximately 18,000 genes reviewed, only 34 were downregulated by RAA, and of these, only 11, all having an ARE in their 3′UTR, were destabilized when validated by RT-PCR. All 11 of these confirmed mRNAs were related proinflammatory chemokines. Of note is the observation that other mRNAs, also containing AUUUA motifs, were not affected. This suggests that RAA was highly selective for the related mRNAs and was able to discriminate between transcripts having a similar instability motif.
Further evidence that specificity of mRNA destabilizing compounds identified is dependent on the class of compound, as well as the cell type or disease model being studied, has been provided by Nair et al. 90 Another example is cyclosporin A, which has been reported to destabilize interleukin-3 (IL-3) mRNA in an autocrine tumor cell line by acting through the AUUUA instability motif in its mRNA. However, it shows no effect on destabilizing IL-1β mRNA in monocytes that also has the AUUUA motifs. RAA, on the other hand, destabilized IL-1β mRNA in monocytes acting through AUUUA but had no effect on IL-3 mRNA in mast cells or macrophages (unpublished results). This further demonstrates that low molecular weight compounds from different classes, although affecting mRNA stability through the same minimal stability regulatory mechanism, are dependent on the compound class as well as the cell type or disease. Clearly, different compounds affect the regulation of mRNA stability differently, and the choice of cell line for assay construction is important for a given target and should be disease relevant and express the target of interest—points crucial for ensuring the identification of disease-relevant and selective compounds using an mRNA stability screen.
Considerations for Assay Design
Assay construction involves the introduction of a reporter gene construct containing mRNA stability control elements (SCE) of the selected target into an appropriate cell line. Construction of each assay thus should make use of the unique SCE, its configuration, and the use of a relevant cell type, so that each assay is specific and unique, with respect to the genome, for each mRNA to be targeted.
To capture the complexity of mRNA modulation, cell-based assays should be developed in a cell line relevant for the disease of interest as cell type, cellular stimulation, the presence or absence of mRNA binding proteins, miRNA, and other factors influence mRNA profiles. Such cell-based assays have clear advantages above cell-free assay systems. A significant advantage of such functional assays is that they can identify any molecule that has a desired effect on the target of interest irrespective of mechanism of action.
However, to assess the specificity of hits coming from an initial screen against a given target in a given cell line, these hits should undergo further evaluation, including comparison to a co-transfected control plasmid, transfection of the target into multiple pertinent cell lines for cross-comparison, and transfection of multiple target constructs into one cell line for cross-comparison and determination of selectivity. The latter two comparison steps, together with the initial screening assay, result in compounds that are selective for one target and do not affect other targets or cells not relevant to the target of interest. Selected compounds should then undergo cytotoxic assays, measurement of their effect on endogenous mRNA levels, protein-level expression, and DNA microarray to assess potential genome-wide effects on mRNA levels and provide further assurance on specificity.
Cell-based mRNA stability assays have the advantage that they are able to identify compounds that modulate mRNA levels through a number of possible mechanisms. These could include direct interaction of the compound with the mRNA, interference of the compound with protein-mRNA interaction, and/or influencing processes that govern the modulation of mRNA stability such as signal transduction, enzymatic mRNA degradation, or cotranslational mRNA degradation. Although all “effective” regulatory domains engineered into mRNA stability assays are unique, further steps should be taken to ensure that selected compounds will be specific for the intended mRNA target. A built-in control allows compounds that do not work through the SCE to be identified and eliminated as false positives. Only compounds specifically influencing the reporter containing the SCE are considered because the activity of the compound is dependent on the presence of the SCE introduced.
The use of counterscreens will further confirm specificity and identify compounds that are highly selective for the mRNA of interest, but have no activity against other mRNAs screened. Hits identified with one mRNA stability screen are counterscreened against other mRNA stability assays. In this way, the identification of compounds that may affect targets other than the one of interest is minimized. As more screening campaigns are run using a given compound library, compounds repeatedly identified as active, and therefore not specific, can be eliminated from future screens. In this way, identification of highly selective compounds becomes faster as the information generated for each compound in a library becomes cumulative.
The outcome of a primary screen and counterscreens can be displayed by so-called heat maps, which compare the activities of a compound on all assays. Data from these heat maps can identify compound hits that show activity against only one specific SCE. A schematic hypothetical heat map (
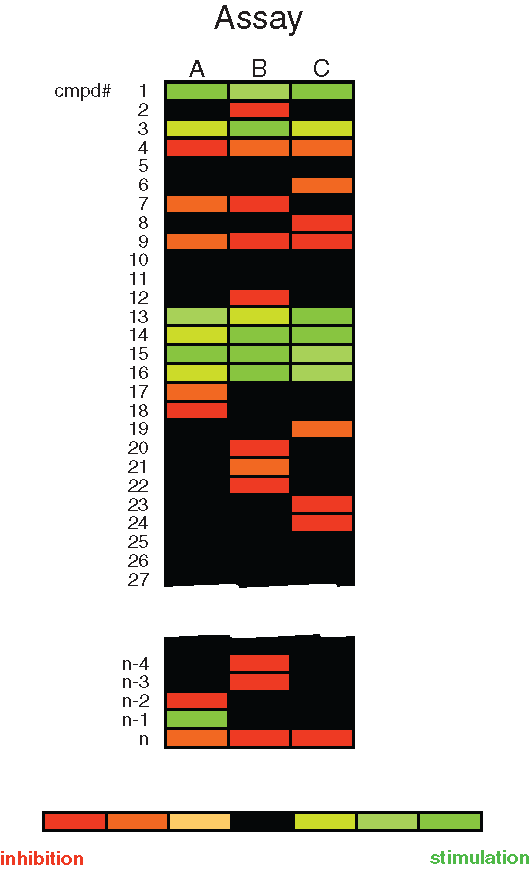
Comparison of compounds screened against 3 different mRNA stability motifs target A, B, or C in different cell lines. The heat map readout shows compounds that either inhibit (red) or stimulate (green) the target. Compounds that show stimulation regardless of the mRNA motif (e.g., compound 1) would be of no further interest. Similarly, compounds that show inhibition regardless of the motif (e.g., compound 4) would also be of no further interest. n is the number of compounds screened.
Conclusion
Selectivity and specificity is a critical issue when targeting mRNA. However, a wealth of evidence exists showing that it is possible to identify small molecules that can specifically modulate the stability of selective mRNA transcripts. Specificity concerns are common for all compounds, not just those related to mRNA stabilization. Concern with respect to off-target effects is common for all compounds no matter how they are discovered, and thus effects may be inherent to the structure of the molecule rather than any mRNA modulating action. This is a general pharmaceutical development issue. Even though the precise molecular target and mechanism of action may be known, unpredicted side effects may still occur. Of greater concern may be “on-target” side-effects if the molecular target plays multiple roles. These potential side effects can be evaluated early on through standard parallel screens that can be incorporated into lead selection. The fact that compounds of known pharmacological relevance have been shown to affect the stability of selective mRNAs suggests that even compounds targeting other aspect of cell physiology might have effects on transcript stability.
For these reasons, a number of different companies have started discovery efforts to identify compounds modulating mRNA stability (e.g., Novation Pharmaceuticals and PTC Therapeutics Inc.), and other companies are selling reagents that allow the effect of compounds on the stability of specific mRNAs to be monitored (e.g., SwitchGear Genomics [www.switchgeargenomics.com]). In addition, companies have started to sell reporter constructs using the presence of ARE elements to destabilize the reporter RNA so as to make the reporter gene more responsive to the chosen inducer used in these reporter constructs (e.g., Promega and New England Biolabs). Although these are obviously an interesting application of these RNA stability motifs, they do introduce the possibility that reporter constructs designed to monitor effects on a given promoter will now also detect the influence of compounds or miRNAs that modulate mRNA through these ARE elements.
In conclusion, there are now a number of methods for monitoring mRNA levels, some of which are suitable for HTS. The availability of compounds reported to alter mRNA metabolism will also aid in the development of assays to identify treatments capable of altering mRNA levels.