
Abstract
Estrogens, acting through estrogen receptor α (ERα), stimulate breast cancer proliferation, making ERα an attractive drug target. Since 384-well format screens for inhibitors of proliferation can be challenging for some cells, inhibition of luciferase-based reporters is often used as a surrogate end point. To identify novel small-molecule inhibitors of 17β-estradiol (E2)–ERα-stimulated cell proliferation, we established a cell-based screen for inhibitors of E2-ERα induction of an estrogen response element (ERE)3–luciferase reporter. Seventy-five “hits” were evaluated in tiered follow-up assays to identify where hits failed to progress and evaluate their effectiveness as inhibitors of E2-ERα-induced proliferation of breast cancer cells. Only 8 of 75 hits from the luciferase screen inhibited estrogen-induced proliferation of ERα-positive MCF-7 and T47D cells but not control ERα-negative MDA-MB-231 cells. Although 12% of compounds inhibited E2-ERα-stimulated proliferation in only one of the ERα-positive cell lines, 40% of compounds were toxic and inhibited growth of all the cell lines, and ~37% exhibited little or no ability to inhibit E2-ERα-stimulated cell proliferation. Representative compounds were evaluated in more detail, and a lead ERα inhibitor was identified.
Keywords
Introduction
High-throughput screens based on luciferase reporter assays are widely used to evaluate the effects of compounds on a pathway or target of interest. 1 Reporter assays are often used as surrogate markers for a process such as cell proliferation that is more difficult to establish in 384- or 1536-well high-throughout screens. Although there have been several studies of off-target effects due to direct interaction of a compound with the luciferase protein,2,3 much less is known about the ability of a simple luciferase reporter assay to predict small-molecule effects on a complex process, such as cell proliferation. Using inhibitors of estrogen receptor α (ERα) as a model, we evaluated how effectively a primary screen based on inhibition of a luciferase reporter predicted the ability of small molecules to selectively inhibit estrogen-ERα-dependent growth of human breast cancer cells.
The complex of estrogens, such as 17β-estradiol (E2) and ERα, plays a critical role in the growth and metastases of breast cancer. The important role of estrogens in breast cancer is illustrated by the widespread therapeutic use of aromatase inhibitors that block estrogen production, as well as the selective estrogen receptor modulators tamoxifen and faslodex/fulvestrant/ICI 182,780 that work by competing with estrogens for binding to ERα. 4 The progressive development of tumors resistant to tamoxifen and other ERα antagonists, 5 as well as to aromatase inhibitors, 6 underscores the continued importance of ERα as a therapeutic target.
It is widely accepted that the ability of E2-ERα to regulate nuclear gene expression plays a key role in the ability of estrogens to stimulate proliferation of ERα-positive breast cancer cells.7,8 The E2-ERα complex regulates gene expression by direct binding to DNA sequences termed estrogen response elements (EREs) and closely related sequences and by tethering to DNA through other proteins bound at AP1 and SP1 sites.9–12 The E2-ERα complex can also act in the cytosol to rapidly activate several membrane-associated protein kinase–based signaling pathways. However, selective activation of the progrowth ERK1/2 signaling pathways by the E2-ERα complex is not sufficient to stimulate E2-dependent growth of breast cancer cells. 13
Recent reports have described assays using growth as a biological end point. 14 Although effective in 96-well plates, due to high sensitivity to small changes in the initial plating conditions, our assays for E2-ERα-stimulated proliferation of breast cancer cells did not exhibit sufficient reproducibility and precision for use in 384-well high-throughput screens. We therefore developed a screen using endogenous ERα in T47D, human breast cancer cells, stably transfected to express a luciferase reporter containing three consensus EREs ((ERE)3-luciferase). 15 The readout is the ability of small molecules to inhibit the E2-ERα induction of the (ERE)3-luciferase reporter. Although E2-ERα-regulated gene expression is essential for E2-ERα-dependent cell proliferation, we found that there was an imperfect relationship between the inhibitory potency of compounds in the luciferase reporter assay and their potency as selective inhibitors of estrogen-dependent cell proliferation. This led us to characterize in some detail the properties of 75 verified hits from the reporter assay. Around 10% of the verified hits functioned as selective inhibitors of E2-ERα-stimulated proliferation of breast cancers cells. We identified several causes for the failure of most of the hits to function as selective inhibitors. As a primary screen, HTS using luciferase reporter assays represents a viable, but imperfect, way to identify small-molecule inhibitors of E2-ERα-induced cell proliferation.
Materials and Methods
Cell Culture
Human breast cancer cell lines were maintained in the following culture media: MCF-7: phenol red–free minimal essential medium (MEM), supplemented with 5% fetal bovine serum (FBS); T47D: MEM, supplemented with 10% FBS; T47D-KBluc: RPMI-1640, supplemented with 10% FBS 16 ; and MDA-MB-231: phenol red–free MEM, supplemented with 10% FBS. Prior to experiments, ERα-positive cell lines were maintained for at least 3 days in phenol red–free charcoal-dextran (CD)–treated serum. T47D cells: 4 days in MEM containing 10% CD-FBS; MCF-7 cells: 4 days in MEM + 5% CD-FBS; and T47D-KBluc cells: 3 days in RPMI-1640 + 10% CD-FBS.
Automated High-Throughput Screening and Manual 384-Well Plate Luciferase Assays
High-throughput screening (HTS) luciferase-based assays were carried out by adding test compounds to plates using a Matrix PlateMate Plus instrument (Thermo Scientific, Waltham, MA), equipped with a 384-well pin-transfer apparatus. To reach the desired final concentration for screening with limited volume options available using the pin-transfer apparatus, 0.1 µL of each 10-mM compound stock in DMSO was transferred into 70 µL serum-free RPMI-1640. Then, 60 µL medium containing each test compound was then withdrawn from the plates using a 384-well tip cartridge, leaving 10 µL of each compound at 14.28 µM. Cells were harvested at a density of 1-million cells/mL in RPMI-1640, supplemented with 10% CD-FBS. A 1:500 dilution of 17β-estradiol (E2) in ethanol (EtOH) and a vehicle-ethanol control were added to the (+)E2 and (−)E2 cell stocks, respectively. Cells were plated at a density of 10 000 cells/well by pipetting 10 µL of cells into each well using a Matrix Wellmate dispenser. The final concentration of test compounds was 7.14 µM. The screening medium contained 0.1% (v/v) EtOH, 0.07% (v/v) DMSO, and 10 nM E2. Plates were centrifuged for 2 min at 500 rpm and incubated for 24 h (37 °C/5% CO2).
“Hits” from the primary HTS were reconfirmed by diluting 10-mM compound stocks in DMSO to 20 µM in serum-free RPMI-1640. Cells were harvested at a density of 1 million cells/mL in RPMI-1640, supplemented with 10% CD-FBS. A 1:500 dilution of 17β-estradiol (E2) in EtOH and a vehicle-ethanol control were added to the (+)E2 and (−)E2 cell stocks, respectively. Cells were plated at a density of 10 000 cells/well by pipetting 10 µL of cells into each well. The final concentration of test compounds was 10 µM, and the medium contained 0.1% (v/v) EtOH, 0.1% (v/v) DMSO, and 10 nM E2. Plates were centrifuged for 2 min at 500 rpm and incubated for 24 h (37 °C/5% CO2).
All plates were frozen at −20 °C overnight following the 24-h incubation and thawed to room temperature to promote cell lysis. Then, 10 µL Bright-Glo reagent (Promega, Madison, WI) was added to each well and allowed to incubate for 15 min on the bench before measuring luminescence.
96-Well Luciferase Assays
Five to 6 days before the experiment, T47D-KBluc cells were subcultured and plated at a high density (about 30%–40% confluence) in RPMI-1640 + 10% FBS. Two days later, the medium was changed to RPMI-1640 +10% CD-FBS. After 3 or 4 days with a medium change on day 2, the cells were harvested and counted, and 50 000 cells in 100 µL medium were added to each well of a 96-well white-wall clear-bottom plate (BD Biosciences, Franklin Lakes, NJ) in RPMI-1640 + 10% CD–calf serum (CS). The medium was replaced the next day with medium containing the test compounds with or without hormone. After 24 h, the medium was aspirated off, and 30 µL Bright-Glo reagent (Promega) was added. To help lyse the cells, the plate was placed on a shaker for 5 min. To remove any bubbles in the wells, the plate was subjected to centrifugation at 2500 rpm for 2 min.
MTS Growth Assays
Cells were harvested and plated in 96-well plates at a density of 1000 cells/well. MCF-7 cells were plated into MEM, supplemented with 10% CD-CS; T47D cells were plated into MEM, supplemented with 10% CD-FBS; and MDA-MB-231 cells were plated into MEM, supplemented with 10% FBS. The medium was replaced with treatment medium the following day, and plates were incubated at 37 °C in 5% CO2 for 3 days. Then, 20 µL CellTiter 96 Aqueous One Solution Reagent (Promega) was added to each well, and the cells were incubated at 37 °C in 5% CO2 for 1 h. A490 was then measured to assess cell viability. For each cell line, cell number was calculated from a standard curve of the number of cells plated versus A490. 16
Western Blotting
Cells were trypsinized, resuspended in MEM supplemented with 10% CD-CS, and plated into 6-well plates at a density of 300 000 cells/well. The medium was replaced with treatment medium the following day, and the cells were treated for 24 h. The cells were washed in ice-cold phosphate-buffered saline (PBS), and whole-cell extracts were prepared in lysis buffer containing 1× radioimmunoprecipitation assay buffer, 1 mM EGTA, 30 mM NaF, 2.5 mM sodium pyrophosphate, 1 mM sodium orhovanadate, 1 mM β-glycerol phosphate, 1 mM phenylmetholsulfonyl fluoride, and 1 tablet of protease inhibitor cocktail (Roche, Indianapolis, IN). Cells were collected, and debris was pelleted by centrifugation at 15 000 g for 10 min at 4 °C. The supernatants were collected and stored at −20 °C. Then, 20 µg total protein was loaded onto 10% (v/v) sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels, separated, and transferred to nitrocellulose. Membranes were incubated with monoclonal ERα [6F11] antibody (Biocare Medical, Concord, CA) or control monoclonal α-tubulin antibody (Sigma, St. Louis, MO). Bound antibodies were detected using horseradish peroxidase–conjugated secondary antibodies and chemiluminescent immunodetection with an ECL Detection Kit (GE Healthcare, Piscataway, NJ) and were visualized using a PhosphorImager (GE Healthcare, Piscataway, NJ).
qRT-PCR
pS2 mRNA levels were analyzed by quantitative real-time reverse transcriptase–polymerase chain reaction (qRT-PCR). RNAs were extracted with TRIzol reagent and purified with the RNAeasy mini-kit (QIAGEN, Valencia, CA), and 0.5 µg RNA was reverse transcribed using a DyNAmo cDNA synthesis kit (Finnzymes, Espoo, Finland). Then, 10 ng cDNA product was added to a primer mix, such that the forward and reverse primer final concentration was 50 nM. Primers used in qRT-PCR were as follows: pS2, forward (5′-ACCGGACACCTCAGACACG) and reverse (5′-CTGTGT TGTGAGCCGAGGC); 36B4, forward (5′-GTGTTCG ACAATGGCAGCAT) and reverse (5′-GACACCCTCCA GGAAGCGA). The fold change in expression of each gene was calculated using the ΔΔCt method with 36B4 as the internal control.
Transient Transfection and Dual-Luciferase Assay
T47D cells were seeded at a density of 4.5 × 104 cells in 24-well plates. Cells were transfected with 0.4 µg (ERE)4-luciferase plasmid and 2.5 ng SV40–Renilla luciferase plasmid, using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA). Transfection solutions were replaced after 24 h, and cells were treated with and without 1 nM E2 or with the appropriate small molecule in the presence of 1 nM E2. Cells were treated for 24 h prior to cell lysis and measurement of firefly and Renilla luciferase luminescence using the Stop-Glo Reagent system (Promega).
Data Analysis
Primary HTS data are representative of single assays. Percent inhibition of the E2-induced (ERE)3-luciferase or dihydrotestosterone (DHT)–induced ARE-luciferase was calculated as follows:
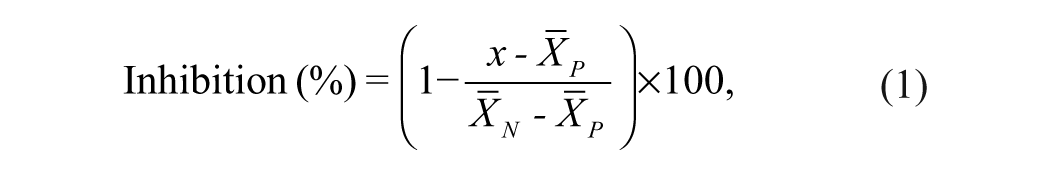
where x is the luciferase signal following small-molecule treatment,
Seventy-five small-molecule “hits” were randomly selected for further evaluation. Compounds were reconfirmed as hits in three independent experiments in quadruplicate. Small molecules were screened for their ability to inhibit E2-ERα-induced cell proliferation in ERα-positive MCF-7 cells and T47D breast cancer cells in three independent assays in triplicate. Equation (1) was used to calculate percent inhibition of E2-ERα-stimulated cell proliferation (where
Results
A Cell-Based Screen for Inhibitors of E2-ERα Induction of an (ERE)3-Luciferase Reporter Gene
Regulation of nuclear gene expression is central to the ability of estrogens bound to ERα to induce proliferation of breast cancer cells. The widely used breast cancer therapeutic tamoxifen acts by competing with estrogens for binding to ERα and interfering with recruitment of coactivators critical for ERα-mediated gene expression. To identify novel small molecules that directly or indirectly inhibit E2-ERα-mediated gene expression, a cell-based primary screen was developed using ERα-positive T47D human breast cancer cells stably transfected to express a luciferase reporter whose expression is driven by three copies of the consensus estrogen response element (ERE)3-luciferase.
15
Dose-response studies show that E2 robustly and reproducibly induces expression of the luciferase reporter (
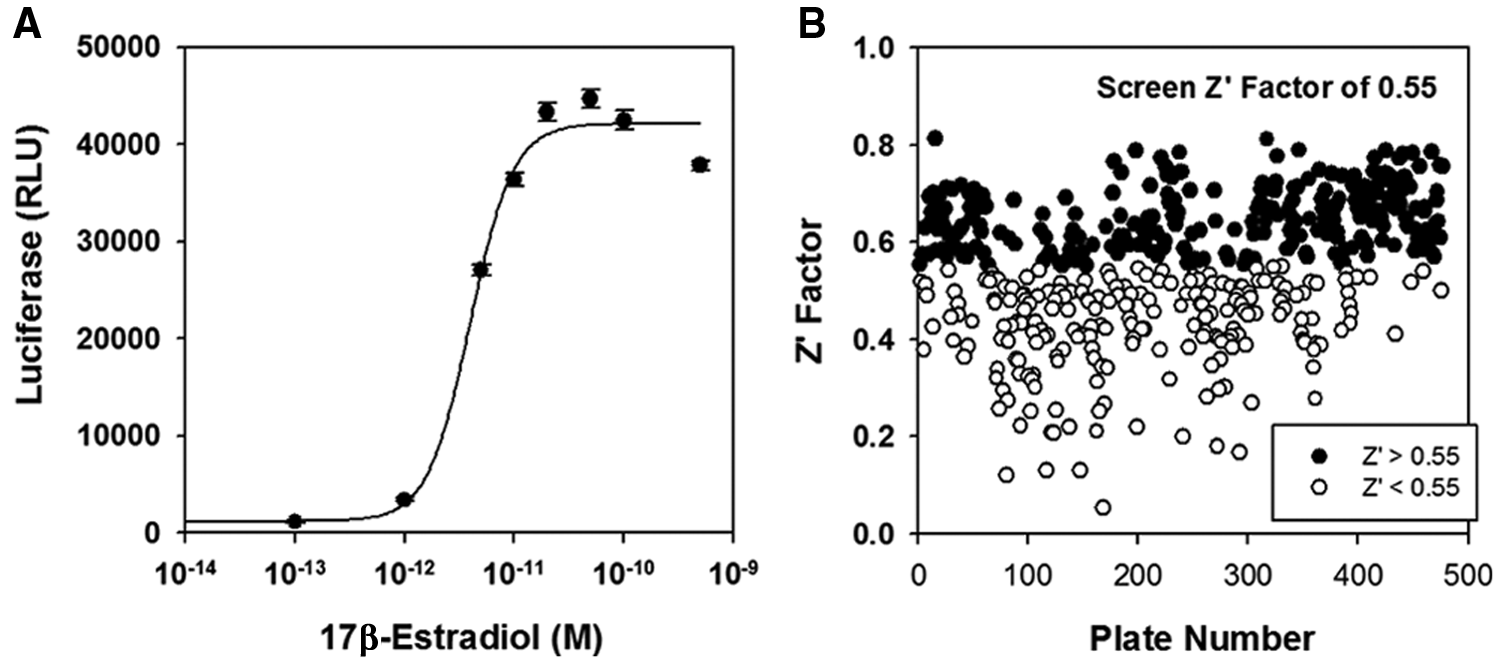
The estrogen response element (ERE)3–luciferase-based assay. (
In some screens, a constitutively active luciferase reporter can provide a useful indicator of the specificity and toxicity of potential small-molecule inhibitors. However, small-molecule inhibitors of E2-ERα-induced gene expression should also inhibit the proliferation of ERα-positive human breast cancer cells and might thereby decrease the activity of a constitutively active Renilla luciferase internal standard. To test this, we compared the effect of several well-established, specific, and nontoxic inhibitors of ERα with a mildly toxic compound identified in our follow-on assays. The well-known therapeutics tamoxifen, raloxifene, and faslodex and the toxic compound all produced similar substantial declines in expression of the constitutively active luciferase reporter gene (
Since the constitutively active luciferase could not distinguish toxic compounds from bona-fide ER inhibitors, we used tiered assays to filter out toxic compounds.
Small-molecule hits were first screened for inhibition of DHT–androgen receptor (AR) induction of a prostate-specific antigen-luciferase (ARE-luciferase) reporter in stably transfected HeLa cells. 16 This provided a way to initially flag compounds as toxic, which was later reconfirmed in subsequent toxicity assays. A second reporter also functioned as a crude method for assessing the nuclear receptor specificity of small molecules, given that ERα and AR share a high degree of structure homology and conservation in upstream signaling pathways. It also provides a way to detect inhibitors of luciferase enzyme activity. Alternatively, inhibitors of luciferase enzyme activity could have been detected by growing the T47D-kBLuc cells in the presence of estrogen alone, lysing the cells, and then adding the small molecule being tested and the luciferase reagent.
Compounds were considered hits in the primary screen if, at a concentration of 7.1 µM, they reduced luciferase units by at least 50%. All compounds reaching the 50% cutoff had reached statistical significance, as defined by ±3 SD from the negative reference. To evaluate the effective size of inhibition, we used strictly standardized mean difference as a secondary metric in lead selection.
Evaluation of 75 Verified Inhibitors of E2-ERα Induction of the (ERE)3-Luciferase Reporter Gene
To evaluate the effectiveness of our luciferase-based assays in identifying useful lead inhibitors that selectively inhibit E2-ERα-induced proliferation of ERα-positive human breast cancer cells, we characterized a randomly selected set of 75 hits from a subset of approximately 16 000 compounds. Data from the primary HTS screen for each compound screened at 7.1 µM are shown for inhibition of (ERE)3-luciferase and ARE-luciferase in
Figure 2
and in terms of SSMD in
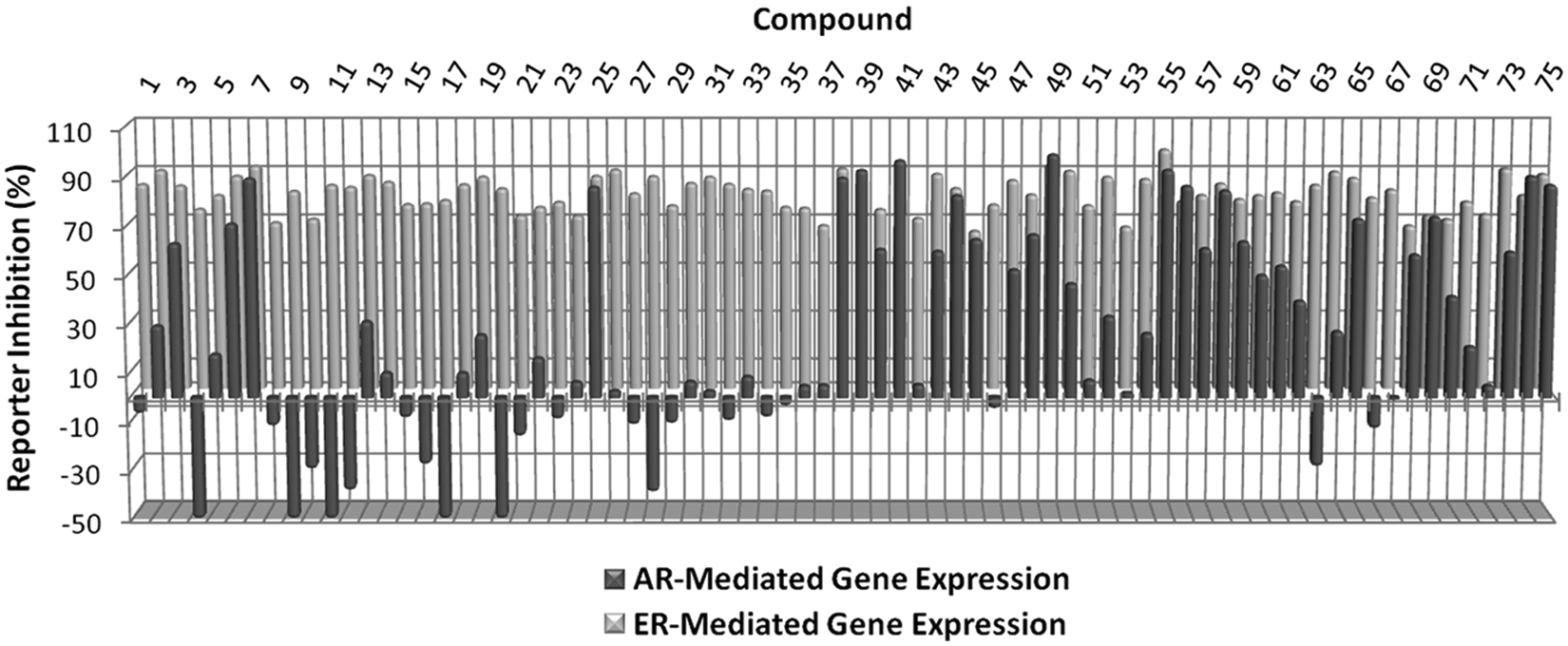
Primary screening data for the 75 representative compounds selected for further characterization. For each compound, percent inhibition of dihydrotestosterone (DHT)–androgen receptor (AR)–stimulated ARE-luciferase activity (black bars) and 17β-estradiol (E2)–estrogen receptor α (ERα)–stimulated estrogen response element (ERE)3–luciferase activity (gray bars) is shown. The strictly standardized mean difference (SSMD) score for each small molecule is shown in
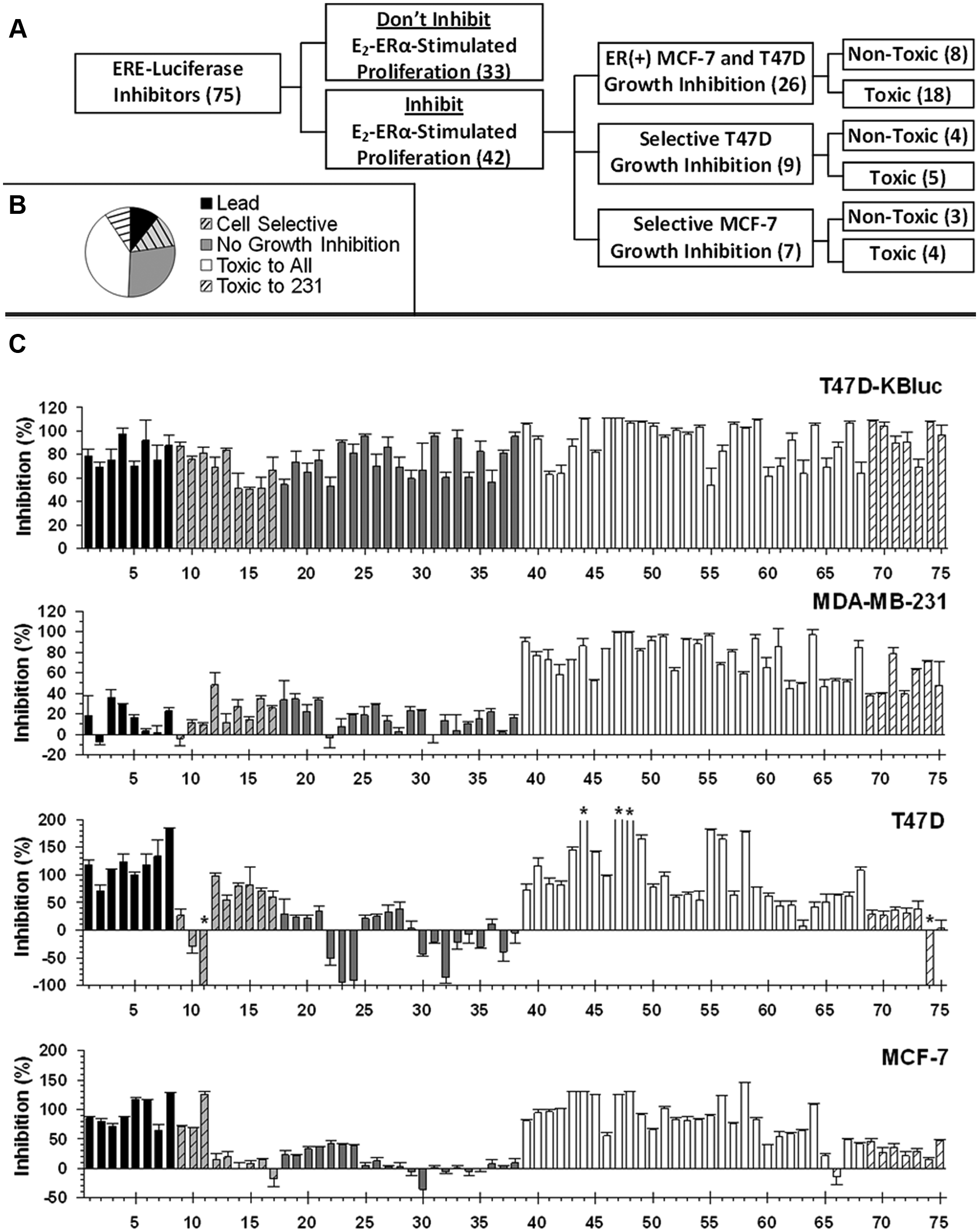
Summary of assays used to evaluate inhibitors of estrogen receptor α (ERα)–mediated gene expression as a surrogate marker for inhibitors of 17β-estradiol (E2)–dependent growth. (
Detailed Characterization of Selected Compounds
We examined the properties of four structurally unrelated representative compounds in more detail ( Fig. 4A ). For the four compounds that were selected, we performed dose-response studies of inhibition of E2-ERα-mediated expression of the (ERE)3-luciferase reporter ( Fig. 4B ) and of E2-ERα-stimulated proliferation ( Fig. 5 ).
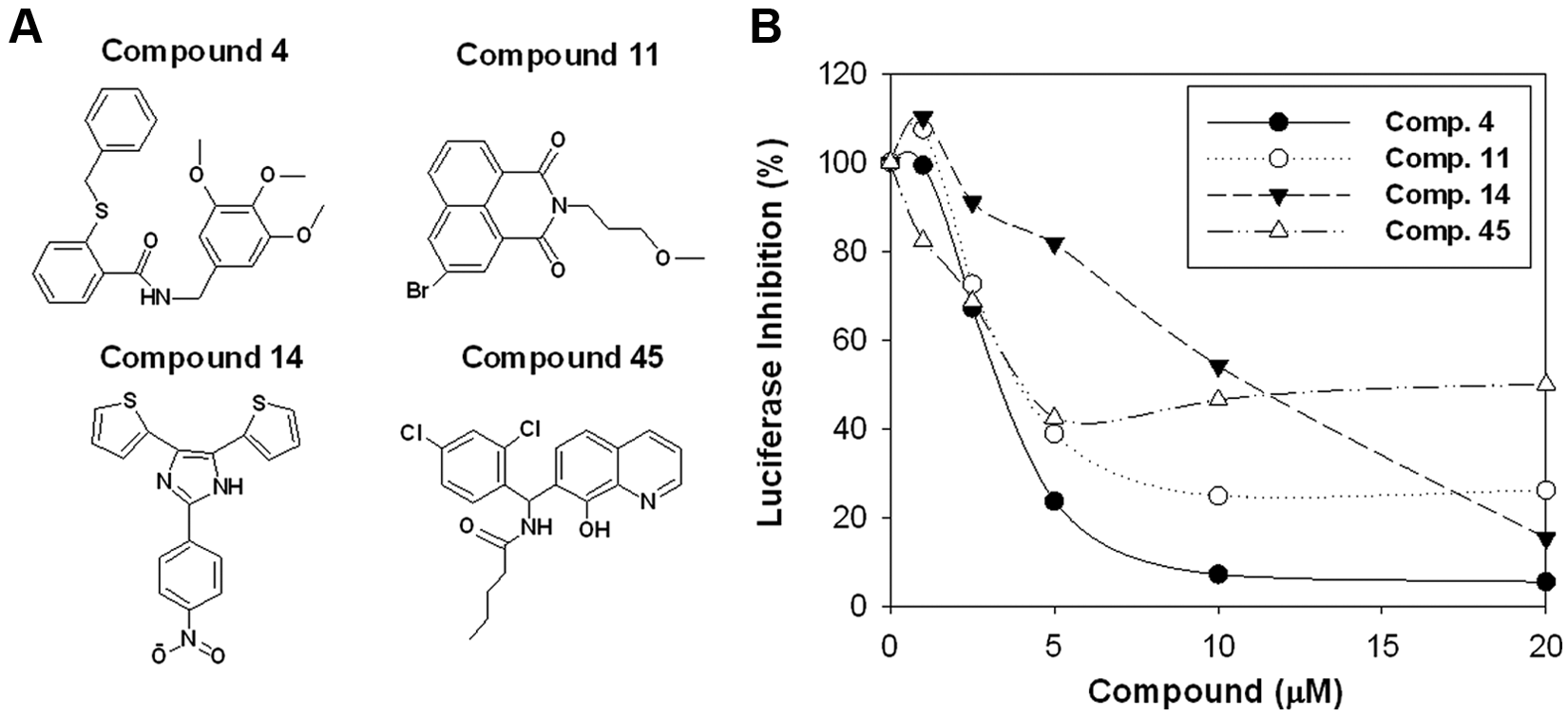
Characterization of representative inhibitors. (
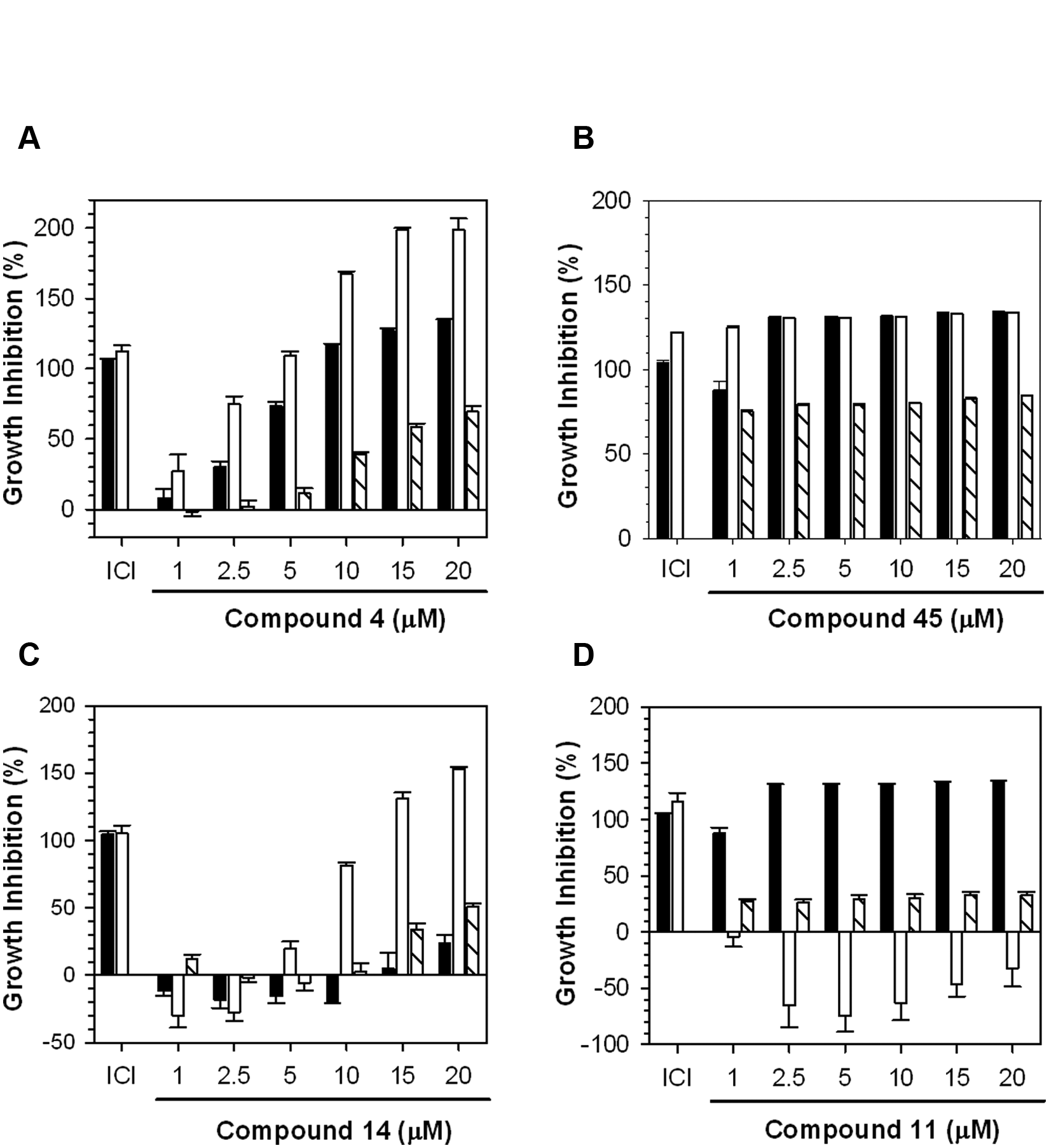
Dose-response studies of the effects of the inhibitors on cell proliferation. Dose-response studies were used to evaluate the effect on the 17β-estradiol (E2)–estrogen receptor α (ERα)–dependent proliferation of MCF-7 and T47D cells and on the growth of ERα-negative MDA-MB-231 for (
At all concentrations tested, compound 45 strongly inhibited proliferation of both the ERα-positive MCF-7 and T47D cells and the ERα-negative MDA-MB-231 cells ( Fig. 5B ). Its overall toxicity is responsible for the ability of compound 45 to inhibit expression of the (ERE)3-luciferase reporter ( Fig. 4B ). The shorter time cells were exposed to compounds in the reporter assay, 1 day as opposed to 3 days, likely accounts for the more limited inhibition seen in the reporter assay. Since compound 45 was toxic to all the cells across a broad range of concentrations ( Fig. 5B ), it was not analyzed further. Compound 4 was identified as a potential selective inhibitor of ERα action ( Fig. 3 ). Compound 4 elicited a dose-dependent inhibition of E2-ERα-induced (ERE)3-luciferase with an IC50 of ~4.0 µM ( Fig. 4B ). From 2.5 to 10 µM, compound 4 selectively inhibited proliferation of the ERα-positive cells compared with the ERα-negative MDA-MB-231 cells ( Fig. 5A ). The IC50s for inhibition of the proliferation of the ERα-positive cells were 3.7 and 1.7 µM for the MCF-7 and T47D cells, respectively, and 12.8 µM for the ERα-negative MDA-MB-231 cells ( Fig. 5A ). We also evaluated two small molecules that selectively inhibited one of the two ERα-positive cell lines. Compound 14 was a moderately effective inhibitor of (ERE)3-luciferase with an IC50 of ~11.1 µM ( Fig. 4B ). Although compound 14 had little effect on cell proliferation at 1 to 5 µM, at concentrations above 10 µM, it robustly inhibited proliferation of the T47D cells with little effect on the MCF-7 cells ( Fig. 5C ). Compound 11 inhibited (ERE)3-luciferase with an IC50 of ~3.4 µM ( Fig. 4B ). Compound 11 stimulated growth of the T47D cells but strongly inhibited growth of the MCF-7 cells and exhibited moderate dose-independent inhibition of the MDA-MB-231 cells ( Fig. 5D ). Thus, this unusual small molecule has opposite effects on the proliferation of the two ERα-positive cell lines.
Effect of Selected Compounds on the Level of ERα and on Estrogen Induction of pS2 mRNA
Several ERα inhibitors act in part by decreasing the level of ERα.16,20 We therefore examined whether the lead inhibitor, compound 4, and the two cell-type selective inhibitors, compounds 11 and 14, influence levels of ERα in T47D and MCF-7 cells. ICI 182,780/fulvestrant/faslodex and TPSF are competitive and noncompetitive ERα inhibitors known to function in part by reducing ERα levels.16,20,21 As expected, ICI 182,780 and TPSF dramatically reduced ERα levels in the MCF-7 and T47D cells ( Fig. 6A , B ). The lead ERα inhibitor, compound 4, strongly reduced ERα levels in both cell lines. In contrast, the two cell line selective inhibitors either had no effect (compound 14) or elicited a small decline in ERα levels (compound 11) ( Fig. 6A , B ).
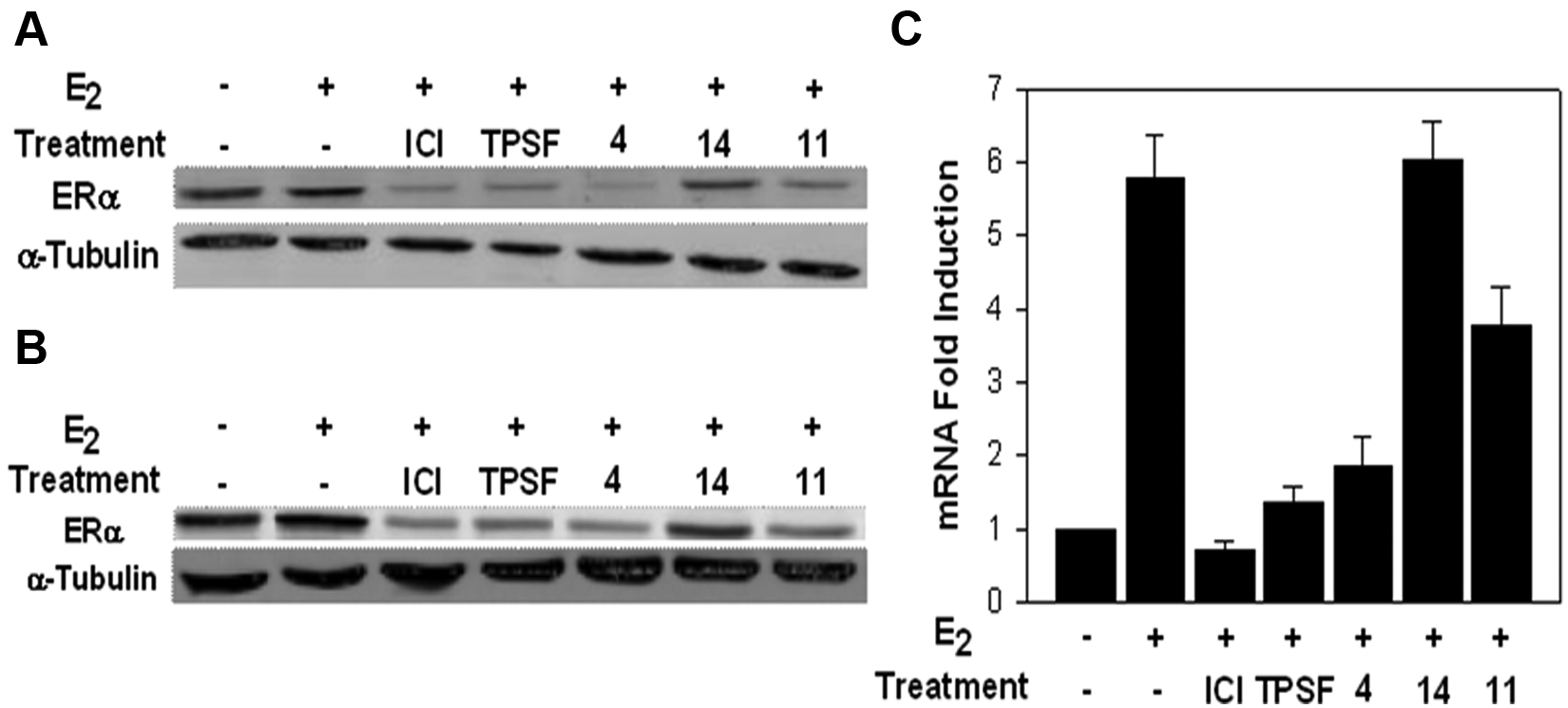
Effect of the three inhibitors on estrogen receptor α (ERα) levels and on the induction of pS2 mRNA. The effects of the three compounds on the level of ERα were evaluated in Western blots of extracts from (
We carried out our initial assays for inhibition of E2-ERα-mediated gene expression using a stably transfected (ERE)3-luciferase reporter. Although this stably transfected reporter gene will exhibit a more nearly native chromatin structure than a transiently transfected reporter gene, it is still likely different from the chromatin structure of a true endogenous gene. We therefore examined the ability of the three small molecules to inhibit E2-ERα induction of the widely studied endogenous pS2 gene in the same cell line in which we performed the (ERE)3-luciferase assays.22,23 We used quantitative RT-PCR to measure pS2 mRNA levels (
Discussion
In evaluating a single reporter luciferase-based assay as a surrogate assay for estrogen-induced proliferation of cancer cells, two potential sources of off-target effects stand out: small molecules that inhibit because they exhibit direct interaction with luciferase and small molecules that are broadly toxic. Both types of small molecule can reduce luciferase activity and be scored as hits in the primary screen.2,3
Approximately 37% (28 of 75, Fig. 3 ) of the hits identified in the luciferase assay did not reach the cutoff for inhibition of E2-ERα-stimulated proliferation in both MCF-7 and T47D human breast cancer cells. This included the 21 compounds in the “no-growth inhibition” category ( Fig. 3 , 18–37) and the 7 compounds in the “toxic” category that only inhibited cell growth in MDA-MB-231 cells ( Fig. 3 , 69–75). Nearly all of the latter compounds (6 of 7) inhibited cell growth modestly (>25%) in both ERα-positive breast cancer cell lines. The more pronounced effects on cell growth in the ERα-negative MDA-MB-231 cells can be attributed to the higher sensitivity of this cell line to damage. 16 The no-growth inhibitors could be further subclassified as (1) compounds that modestly inhibited cell proliferation but did not reach the 50% cutoff ( Fig. 3 , 18–28) or (2) compounds that exhibited little or no ability to inhibit E2-ERα-stimulated proliferation of the breast cancer cells ( Fig. 3 , 29–38). Partial growth inhibition may reflect different dose-response curves for inhibition of gene expression and cell proliferation. Consistent with this idea, the concentration of the noncompetitive ERα inhibitor TPSF required to inhibit cell proliferation is ~3-fold higher than is required to inhibit the luciferase reporter. 16 Compounds that showed no ability to inhibit E2-ERα-stimulated proliferation of the breast cancer cells might be acting as direct inhibitors of luciferase enzyme activity. However, 8 of 9 compounds that failed to significantly inhibit growth ( Fig. 3 , 29–38) also failed to significantly inhibit the ARE-luciferase reporter ( Fig. 2 , 29–38). This suggests that these compounds are not functioning as luciferase inhibitors.
Approximately 49% (37 of 75,
Fig. 3
) of the compounds were toxic, as a result of their ability to inhibit cell growth in ERα-negative MDA-MB-231 cells. Approximately 81% of the toxic compounds displayed global toxicity (
Fig. 3
, 39–68), whereas 19% selectively inhibited growth of the MDA-MB-231 cells (
Fig. 3
, 69–75). The large number of toxic compounds is likely due to the high sensitivity of MDA-MB-231 cells to damage,
16
the rigorous 4-day treatment interval used to assess for growth effects, and the more stringent growth inhibition cutoffs used to classify compounds as toxic (30% cutoff vs. 50% cutoff for ERα-positive cell lines). Our data suggest that use of an alternative reporter system in parallel with the primary screen has predictive value in flagging potentially toxic compounds. Approximately 70% of the compounds considered toxic inhibited the ARE-luciferase reporter by >30% (
Fig. 2
, 39–75). These compounds all displayed SSMD scores <–2 (
Perhaps most surprising was our finding that some small-molecule hits from the (ERE)3-luciferase reporter assay inhibited proliferation in either T47D cells or MCF-7 cells but not in both cell lines. MCF-7 and T47D cells are the most widely used lines of ERα-positive breast cancer cells. These cell lines were independently derived from different tumors. Since the hits were identified using a primary screen for inhibitors of (ERE)3-luciferase expression in stably transfected T47D cells, and T47D cells contain less ERα than MCF-7 cells, 24 we anticipated we might identify small molecules that were more effective in T47D cells than in MCF-7 cells. Instead, we identified some small molecules that were toxic in MDA-MB-231 cells but only damaged one of the two cell lines and a few small molecules that preferentially inhibited proliferation of each of the cell lines without damaging MDA-MB-231 cells. Compound 14 exhibited a dose-dependent inhibition of the proliferation of T47D cells with little effect on proliferation of MCF-7 cells. At 10 µM, compound 14 inhibited proliferation of the T47D cells by ~81% with no effect on proliferation of the MCF-7 cells or MDA-MB-231 cells ( Fig. 5C ). In contrast, compound 11 killed all the MCF-7 cells and actually stimulated estrogen-dependent growth of the T47D cells from 2.5 to 20 µM ( Fig. 5D ). The surprising properties of compound 11 demonstrate that effects on cell proliferation can be entirely dissociated from effects on reporter gene expression. It is unlikely that compounds 11 and 14 directly target ERα action as they had little or no ability to inhibit estrogen induction of pS2 mRNA in the same T47D line used for the luciferase assays ( Fig. 6C ). These data illustrate the importance of early evaluation of primary hits using more than one cell line. Since T47D cells proliferate more rapidly in full serum compared with stripped serum supplemented with estrogen, small molecules such as compound 11 can also exhibit off-target effects on pathways that lead to increased cell proliferation. The higher frequency of small molecules that activate growth in T47D cells may reflect the fact that MCF-7 proliferation rates in the presence of estrogen are near maximum and are higher than those of T47D cells, making MCF-7 cells much less susceptible to off-target effects that increase rates of cell proliferation.
About 10% of the primary hits from the luciferase screen represented leads that inhibited estrogen-dependent proliferation of both the MCF-7 and T47D cells with minimal effects on proliferation of the ERα-negative MDA-MB-231 cells ( Fig. 3 , 1–8). From 1 to 5 µM, compound 4 elicited progressive inhibition of the proliferation of MCF-7 and T47D cells with little effect on proliferation of MDA-MB-231 cells ( Fig. 5A ). Since compound 4 showed some toxicity in MDA-MB-231 cells at 10 µM, we used 5 µM to test its effect on estrogen induction of pS2 mRNA. Consistent with its effect on the (ERE)3-luciferase reporter and on E2-ERα-stimulated cell proliferation, compound 4 effectively inhibited induction of pS2 mRNA and is a lead compound for further development.
The application of cell-based assays in HTS remains inherently challenging. The large number of biological targets and high degree of crosstalk between integrated signaling pathways predispose cell-based assays to higher “hit” frequencies, lower confirmation rates, and higher numbers of toxic compounds in follow-up assays.25,26 Approximately 10% of the verified hits from the luciferase-based screen ultimately met our end point for selective inhibition of estrogen-dependent proliferation of ERα-positive cell lines. Although this might seem to argue for a cell proliferation–based screen, a primary screen based on inhibition of cell proliferation would have exposed many of the same issues. Since single-cell proliferation assays do not usually distinguish between toxic compounds and those targeting the pathway of interest, many of the hits from a proliferation-based screen would have emerged as toxic when tested in a control cell line. Our finding that some compounds exhibit cell line–specific inhibition of estrogen-dependent proliferation means that compounds identified in a single cell line may still fail in other cell lines because they do not target ERα. In our hands, assays for estrogen-dependent cell proliferation assays in 384-well plates exhibited less precision and reproducibility than luciferase-based assays and require several days compared to one day for luciferase assays. Although luciferase-based assays offer the advantage of improved efficiency in HTS, assay interference remains an important obstacle to generating high-quality leads. Although the role of assay interference in our screen is largely unknown, the coupling of multiple cell proliferation assays to the primary screen provided an effective filter for rapidly eliminating compounds that lack target specificity. Alternatively, this empirically driven approach of asking cells to tell us what small molecules are capable of inhibiting E2-ERα-stimulated proliferation provides a way to select for bioactive compounds, independent of their ability to elicit assay interference in the primary screen. Recent studies have shown that several important small molecules function as dual inhibitors of adenosine triphosphate (ATP)–dependent kinases and luciferases, including resveratrol and the MEK1/2 inhibitor, PD090859.27–29 Since the role of E2-ERα in gene expression is essential to stimulating breast cancer cell proliferation, and a number of pathways can modulate ERα transcriptional activity through posttranslational modifications, 30 our screen would not preclude identification of such bioactive compounds. Small molecules generally target several sites in cells and, through signal integration and unintended crosstalk, can elicit toxicity that is readily observed in downstream readouts such as E2-ERα-mediated gene expression. Carrying out multiple cell proliferation assays, using cells with and without the target, provides a very effective toxicity filter. This is reflected by the large numbers of toxic compounds we identified. With appropriate follow-on verification assays carried out in multiple cell lines, a luciferase-based primary screen represents a useful surrogate assay for a more complex process, such as hormone-stimulated cell proliferation.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institutes of Health, grant RO1 DK-071909.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.