
Abstract
Mitogen-activated protein kinase (MAPK) p38 is part of a broad and ubiquitously expressed family of MAPKs whose activity is responsible for mediating an intracellular response to extracellular stimuli through a phosphorylation cascade. p38 is central to this signaling node and is activated by upstream kinases while being responsible for activating downstream kinases and transcription factors via phosphorylation. Dysregulated p38 activity is associated with numerous autoimmune disorders and has been implicated in the progression of several types of cancer. A number of p38 inhibitors have been tested in clinical trials, with none receiving regulatory approval. One characteristic shared by all of the compounds that failed clinical trials is that they are all adenosine triphosphate (ATP)–competitive p38 inhibitors. Seeing this lack of mechanistic diversity as an opportunity, we screened ~32,000 substances in search of novel p38 inhibitors. Among the inhibitors discovered is a compound that is both non–ATP competitive and biologically active in cell-based models for p38 activity. This is the first reported discovery of a non–ATP-competitive p38 inhibitor that is active in cells and, as such, may enable new pharmacophore designs for both therapeutic and basic research to better understand and exploit non–ATP-competitive inhibitors of p38 activity.
Introduction
Mitogen-activated protein kinase (MAPK) p38 is a multi-isoform (α, β, δ, γ) member of the highly conserved MAPK family that also includes extracellular signal–regulated kinases (ERKs) and JUN N-terminal kinases (JNKs). 1 A common feature of all members of the MAPK family is their central role in integrating extracellular stimuli (including osmotic stress, ultraviolet [UV] irradiation, growth factor stimulation, and inflammatory signaling) into an intracellular response. 1 MAPKs mediate this response as part of a phospho-relay after being activated by phosphorylation on their activation loop, which for p38 occurs at threonine 180 and tyrosine 182 (T180/Y182) and is mediated by upstream kinases. 2 Historically, the kinases responsible for p38 activation are the MAPK kinases (MKKs) MKK4 and MKK6; however, an alternative activation pathway restricted to T cells and mediated by ζ-chain–associated protein kinase 70 (ZAP70) phosphorylation of p38 at tyrosine 323 (Y323) has been described.1–3 In the context of intracellular signaling, phosphorylation of p38 is required for signal propagation; however, this need not be the case for in vitro analysis of p38 activity. Several recent publications have indicated that constitutively active mutants of p38 can be generated and that these mutants are not only enzymatically active but are also present in structural conformations similar to p38 activated via phosphorylation. 4 Furthermore, one of these mutations is at the ZAP70 phosphorylation site (Y323 to threonine, Y323T) and may be acting as a phosphomimetic for that posttranslational modification. 4 Once active, p38 perpetuates the phosphorylation cascade by phosphorylating and activating a host of intracellular targets including transcription factors (such as activating transcription factor 2 [ATF2]) and additional kinases (such as mitogen-activated kinase interacting kinases [MNKs] and mitogen-activated kinase–associated protein kinases). 5 The end result of this cascade is the generation of a cellular response that varies according to the cell type and the initiating stimulus. For example, T-cell receptor (TCR) engagement generates the p38-dependent production of proinflammatory cytokines in T cells, whereas UV irradiation halts cell cycle progression due to p38-mediated modulation of the cyclin/cyclin-dependent kinase inhibitor ratio. 1
From the initial discovery of p38 as a protein kinase responsible for inflammatory cytokine production, dysregulation of the p38 signaling cascade has been closely linked to disease etiology and progression. 6 Beyond its early association with rheumatoid arthritis, p38 has since been implicated in a host of additional autoimmune diseases and cancer.7–9 Because p38 activity is associated with so many human diseases, it remains an active drug target for novel pharmaceutical discovery, including several currently open clinical trials (clinicaltrials.gov).10,11 Although research and development into the discovery of p38 inhibitors remains brisk, there are currently no U.S. Food and Drug Administration–approved p38 inhibitors for any clinical indication. 12
One explanation for the inability of p38 inhibitors to demonstrate durable efficacy in humans may be that all clinically tested p38 inhibitors act through a single adenosine triphosphate (ATP)–competitive mechanism. 10 Intracellular ATP concentrations are in the range of 1 to 10 mM, and therefore, competitive inhibitors face a considerable disadvantage. 13 The limitations of ATP-competitive inhibitors create an opportunity to better understand the enzymology of p38 activity and inhibition as well as the intracellular biology of p38 signaling through the discovery of inhibitors that are not ATP competitive. To this end, we report here the high-throughput screening (HTS) of more than 32,000 distinct chemical entities in a single-enzyme p38-specific inhibitor screen. This screen resulted in the discovery of five novel chemical scaffolds capable of inhibiting p38 activity in vitro with low-micromolar IC50’s. In addition, our lead molecule, compound 1, inhibits p38 activity in both T-cell and cancer cell–based assays for p38 bioactivity, and isothermal titration calorimetry (ITC) demonstrates that compound 1 directly binds p38 with unimolecular stoichiometry. In contrast to previous p38 inhibitors, we have determined that compound 1 is a non–ATP-competitive inhibitor.
Materials and Methods
Reagents
Unless otherwise specified, all chemical reagents were purchased from Sigma-Aldrich Corp. (St. Louis, MO). Previously published ATP-competitive p38 inhibitors, SB203580, purchased from Selleck Chemicals (Houston, TX), and BIRB-796, purchased from EMDMillipore (Billerica, MA), were used as positive controls for p38 inhibition.
Chemical Library Composition
The chemical library screened in this assay is composed of 32,064 distinct chemical entities acquired either from commercial sources or via material transfer agreements (MTAs) with individual laboratories or through collaboration with Chemical Methodologies and Library Development (CMLD; http://www.nigms.nih.gov/Research/specificareas/CMLD/Pages/default.aspx) centers. All library substances are stored in 100% DMSO at −20 °C in 384-well format at a stock concentration of 5 mM.
Cloning, Expression, and Purification of Recombinant Enzymes
p38α (wild type and mutant [Y323T]) cloning has been described previously. 2 p38α (wild type and mutant [Y323T]) and MKK6 were expressed in the bacterial strain BL21 (DE3; Life Technologies Inc., Grand Island, NY) using the vector pET15b (EMDMillipore). Point mutation for p38α (Y323T) was generated by site-directed mutagenesis using the QuikChange kit (Agilent Technologies, Santa Clara, CA). For small-scale expression of WT p38α and MKK6, after plasmid transformation, single colonies were grown in culture, and after reaching an A600 of 0.6 to 0.8, protein expression was induced with 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG). Cultures were further incubated at 14 °C overnight. Cells were resuspended in binding buffer (20 mM Tris pH 7.5 containing 0.5 M NaCl, 0.1% Triton X-100, and 1 mM phenylmethylsulfonyl fluoride), sonicated, and centrifuged at 30,000 × g at 4 °C for 20 min. His-tagged proteins were purified with cobalt-charged chelating Sepharose Fast Flow beads (GE Healthcare Life Sciences, Piscataway, NJ) and eluted with 0.35 M imidazole in binding buffer. Proteins were concentrated using Microcon YM-30 spin columns (EMDMillipore).
For HTS scale expression and purification of p38α Y323T, an overnight 50 mL starter culture was inoculated into 3 L of fresh Luria-Broth medium containing 100 µg/mL ampicillin and grown at 37 °C to A600 = 0.6 and then equilibrated to 30 °C for 30 min. Protein expression was induced with 0.2 mM IPTG supplementation for 5 h. The cells were pelleted and stored at −80 °C. Pellets were thawed on ice and resuspended in buffer containing 0.5 M NaCl, 20 mM Tris-HCl buffer (pH 8.0), 10 mM imidazole, 1% Triton X-100, and 20 U/mL Benzonase (EMDMillipore). Cells were lysed through sonification (Branson Sonifier; Branson Ultrasonics, Danbury, CT). After centrifugation (16,000 × g for 50 min), the supernatant was loaded onto a 20 mL Talon metal affinity chromatography column (Clontech, Mountain View, CA) pre-equilibrated with 0.3 M NaCl, 20 mM Tris-HCl (pH 7.5), and 10 mM imidazole buffer. The column was extensively washed, and bound protein was eluted using a linear gradient of imidazole up to 250 mM in the equilibration buffer. The protein-containing fractions were pooled, dialyzed overnight against 150 mM NaCl, 25 mM Tris-HCl (pH 7.5) supplemented with thrombin to cleave the 6x-His-tag. The dialyzed protein solution was loaded onto 1 mL of Benzamidine Sepharose resin to remove the thrombin. The thrombin-free eluted protein solution was concentrated to 15 mg/mL using a 3000 Da molecular weight cutoff centrifugal concentrator (Amicon, EMDMillipore). For size exclusion chromatography, the concentrated eluate was loaded onto a HiLoad 16/60 Superdex 75 pg column (GE Healthcare Life Sciences) equilibrated with 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 1 mM DTT. Fractions containing purified protein were pooled and concentrated to 1.4 mg/mL and stored at −80 °C.
High-Throughput Enzyme-Linked Immunosorbent Assay Screen for p38 Activity
The p38 phosphorylation target protein ATF2 was obtained as a purified recombinant protein from BPS Bioscience (No. 40520; San Diego, CA) after additional vendor-provided dephosphorylation with calf intestinal phosphatase. The kinase buffer used in all reactions had the following working composition: 25 mM Tris-HCl pH 7.5, 5 mM β-glycerophosphate, 2 mM dithiothreitol, 0.1 mM Na3VO4, and 10 mM MgCl2 and was prepared as a 10X stock solution. ATP disodium salt hydrate was dissolved to a 10 or 50 mM stock solution in 100 mM Tris-HCl pH 8.0 and stored at −20 °C until use. All screening reactions were carried out in 384-well polypropylene plates purchased from Greiner Bio-One (Monroe, NC). Reactions were quenched by the addition of an equal reaction volume of 0.5M ethylenediaminetetraacetic acid (EDTA) pH 8 or transfer to an enzyme-linked immunosorbent assay (ELISA) plate prefilled with an equal volume of EDTA. All ELISA reactions were developed using Fluotrac 600 black high-protein binding plates (Greiner Bio-One). The anti-phospho-ATF2 primary antibody (rabbit monoclonal) used at 1:5000 dilution was purchased from Cell Signaling Technology (CST; No. 5112, Lot 10; Danvers, MA). Horseradish peroxidase (HRP)–conjugated goat anti-rabbit secondary antibody, used at a dilution of 1:1000, was also purchased from CST (No. 7074, Lot 24). Bovine serum albumin used as a 1% solution (in 1X Tris-buffered saline + 0.1% Tween-20 pH 7.5 [1X-TBST]; Teknova, Hollister, CA) for plate blocking and antibody dilution was purchased as a dry powder from Fisher Scientific (No. BP-9706-100; Waltham, MA). All ELISA wash steps were carried out using 1X-TBST purchased as a 20X solution from Teknova. The HRP-dependent signal generated by the presence of the phospho-ATF2 analyte was converted into a fluorescent signal through the use of the QuantaBlu reagent purchased from Pierce Biotechnology Inc. (Rockford, IL). Solutions were dispensed into plates using a 16-channel MicroFill dispenser purchased from BioTek (Winooski, VT). All 384-well liquid-handling transfer steps were executed using a BioMek FX equipped with a 384-well micropipette head purchased from Beckman Coulter (Brea, CA). All ELISA plate-washing steps were executed using a BioTek plate washer equipped with multiplate stacking capabilities.
For HTS, substances were screened at a final concentration of 10 µM in 25 mM Tris pH 8.0 buffer containing a final ATP concentration of 50 µM. Each reaction plate also contained a high control column (0.2% DMSO and 50 µM ATP in 25 mM Tris pH 8.0 buffer) and a low control column (1 µM SB203580, 50 µM ATP, 0.2% DMSO, 25 mM Tris pH 8 buffer). Immediately before reaction initiation, each reaction plate was filled with 24 µL per well of the reaction buffer consisting of 1.25X kinase buffer, 375 nM ATF2, 1.25 nM p38, and 5 µg/mL bovine serum albumin (BSA). Reactions were initiated by the transfer of 6 µL from the dilution plate into the reaction plate initiating the reaction through the addition of ATP and bringing the final reaction conditions to 1X kinase buffer, 300 nM ATF2, 1 nM p38, 4 µg/mL BSA, 0.2% DMSO, 10 µM library compound, and 50 µM ATP. After 60 min with moderate shaking at room temperature, the reaction plates were unsealed and quenched by transferring 25 µL from all of the reaction wells to their respective wells in the ELISA plate prefilled with 25 µL 0.5M EDTA pH 8.0. The quenched plates were then heat sealed and incubated overnight on a gently rocking plate rocker.
ELISA plates were then washed and blocked for 60 min with 100 µL 1% BSA in 1X TBST. After blocking, plates were again washed and 50 µL 1:5000 dilution primary antibody solution was added to each well for a 60 min incubation period on a plate rocker. Plates were additionally washed three times, and 50 µL 1:1000 secondary antibody solution was added for a 60 min incubation period with gentle rocking. Plates were again washed three times, and 50 µL of QuantaBlu working reagent was added to each well and incubated with gentle shaking for 60 min. The QuantaBlu reaction was then quenched by the addition of 50 µL of Stop solution (provided by the manufacturer). Stopped reactions were then read using the Tecan Infinite Series Plate Reader (Tecan Ltd, Männedorf, Switzerland) with an excitation of 320 nm and an emission of 420 nm. Relative fluorescence units for each substance well were then normalized as a percentage of the average high control reading for the same plate. The hit threshold was set at percentage normalized activity ≤35%. Assay performance was evaluated by calculating the Z-factor for each plate according to the method of Zhang et al. 14 Quadruplicate confirmational screening was carried out essentially identically to that of the primary screen. Using the eight independent channel head on the BioMek-FX, each primary hit was cherry-picked from the stock plate and rescreened as described above. Those substances whose percentage normalized activity reconfirmed with a 95% confidence interval were designated as confirmed hits, and every attempt was made to acquire additional fresh material for further analysis.
Steady-State Michaelis-Menten Analysis
To determine the KMATP for the recombinant enzyme preparation, a 12-point ATP dose response was set up using ATP concentrations from 25 to 3000 µM over a time frame of up to 80 min. Aliquots of 300 µL of 3X each ATP concentration were prepared in a reaction buffer containing 3X ATP, 2% DMSO, 5 µg/mL casein, and 100 mM Tris pH 8. A master mix of reaction buffer was prepared containing 1.5X kinase buffer, 0.75 nM p38, 5 µg/mL casein, and 0.75 µM ATF2. Sixty microliters of reaction buffer was added to each well of a 96-well polypropylene reaction plate. To the top row of the plate was added 90 µL of 0.5M EDTA (this is the T = 0 control). The 30 µL of each 3X ATP solution was added to each row of the reaction plate (bottom to top) using an electronic repeating pipettor so that all reactions were essentially initiated simultaneously. At intervals of 5, 10, 20, 30, 45, 60, and 80 min, 90 µL of 0.5M EDTA was added to subsequent rows on the plate, quenching the reaction at the indicated time point. After the time course, the entire reaction plate was transferred in triplicate to a high-binding ELISA plate and developed as described above for the primary screening assay. The raw data were then imported into GraphPad Prism (San Diego, CA) for subsequent analysis. For each ATP concentration, a linear regression analysis of the reaction progress curves was performed to ensure that linearity was maintained at each concentration (guarding against substrate depletion) and to generate a reaction rate. For this data set, the r2 value of the best fit line was 0.9834 or higher, indicating minimal deviation from linearity (data not shown). The slopes (reaction velocities) of the best-fit lines were then directly plotted against the ATP concentrations, and the Michaelis-Menten constant for ATP was calculated by nonlinear regression analysis using the following equation:
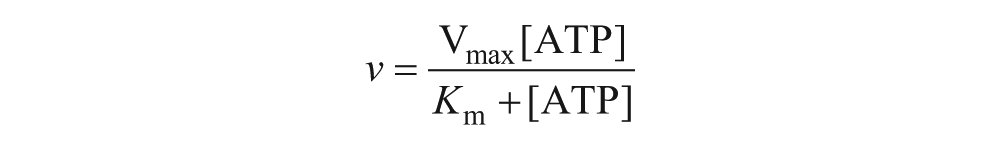
The KmATF2 value was executed in very similar manner to that of ATP using a 12-point ATF2 dose response for ATF2 concentrations between 25 and 1000 nM. The time points over which the reaction progress was monitored were 0, 5, 10, 15, 20, 30, 45, and 60 min. The linear regressions of the reaction progress curves had a minimum r2 value of 0.9537, again demonstrating very little deviation from linearity. The plot of reaction velocity was carried out by nonlinear regression using the above equation (with [ATF2] substituted for [ATP]).
IC50 Determination for Confirmed Hits
Compounds 1 and 5 were obtained from Sigma-Aldrich (Nos. S706868 and B5774). Compounds 2 and 3 were obtained as part of an MTA with Dr. Gang Liu (Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China).15,16 Compound 4 was obtained as part of an MTA with Dr. Peter Wipf (Department of Chemistry, University of Pittsburgh, Pittsburgh, PA). 17 The experimental setup was identical for each compound: there were only subtle differences in the concentration ranges over which the compounds were tested. Compound 1 was tested using a 12-point dose response over a range of concentrations from 0 (DMSO control) to 25 µM. For each compound concentration, 150 µL aliquots of 3X the final compound concentration were prepared in a reaction buffer containing 3X final compound concentration, 2% DMSO, 1.5 mM ATP, and 25 mM Tris pH 8. A master mix of reaction buffer was prepared containing 1.5X kinase buffer, 0.15 nM p38, and 0.45 µM ATF2. Sixty microliters of reaction buffer was added to 12 wells of a 96-well polypropylene reaction plate. The reaction was then initiated by the dilution of 30 µL of the compound/ATP mixture into the reaction wells. As a background control, 30 µL 0.5M EDTA was used. Reactions were allowed to progress before being quenched by the addition of 0.5M EDTA. Quenched reactions were then transferred in triplicate to the ELISA read plate and developed as described above for primary screening. The raw data were then imported into GraphPad Prism, where it was first background corrected and then all fluorescent measurements were normalized as a percentage of the vehicle control. The normalized data were then plotted as a dose-response plot (percentage activity vs log[I], where [I] is the inhibitor concentration) and analyzed by nonlinear regression using the following equation:
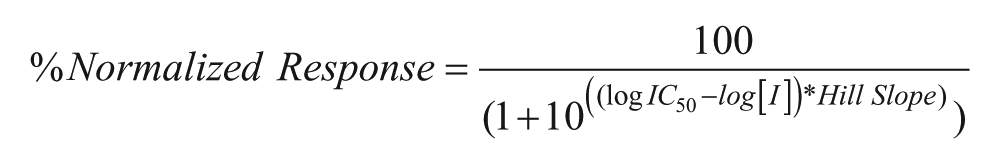
IC50 analyses for compounds 2 to 5 were carried out identically to compound 1 over the following concentration ranges (in addition to vehicle and background controls): compounds 2 and 5: 0 to 100 µM, compound 3: 0 to 200 µM, compound 4: 0 to µM.
Breast Cancer Cell-Based Assay to Verify Intracellular p38 Inhibition
All cell culture–related media were purchased from Quality Biological (Gaithersburg, MD). Anisomycin was purchased from Sigma-Aldrich. The breast cancer carcinoma cell line MDA-MB-231 was cultured in RPMI 1640 media supplemented with 10% fetal bovine serum and 10 U/mL penicillin-streptomycin in 37 °C tissue culture incubator with 5% CO2. Forty-eight hours prior to lysate harvesting, 3 × 106 cells were plated in 10 cm tissue culture–treated Petri dishes purchased from BD Biosciences (San Jose, CA). On the morning of harvesting, complete culture media containing 0.25% DMSO and concentrations of compound 1 (1.25X the final concentration), up to 250 µM, were prepared and sterile filtered. The previous plating culture media was aspirated off and replaced with 4 mL of media containing vehicle control, compound 1 or 12.5 µM BIRB-796, a published ATP-competitive bioactive p38 inhibitor. 8 The cells were incubated in their respective media for 60 min. After 60 min, 1 mL of 50 µg/mL anisomycin (0.25% DMSO) was added to all but one of the culture plates (to which 1 mL of vehicle media was added) for an additional incubation period of 45 min. After this period, the media were aspirated, the plate was briefly washed with 1X-Dulbecco’s phosphate-buffered saline, and 500 µL of ice-cold lysis buffer (20 mM Tris HCl pH 7.4, 150 mM NaCl, 1 % NP-40, 10 % glycerol, 1 mM EDTA, 1 mM EGTA, and 1X protease/phosphatase inhibitor cocktail [CST, No. 5872]) was added to each plate. The plates were then placed on a rocker for 30 min at 4 °C and were then scrapped and lysates transferred to chilled microcentrifuge tubes. DNA was then sheared using a small-bore needle and 1 mL syringe, the lysate was centrifuged for 10 min at 14,000 × g, and the supernatant was retained for further analysis. Lysate protein was quantitated against a buffer-matched BSA standard using the Pierce 660 nm protein quantitation assay (Pierce Biotechnology).
Triplicate sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels were prepared for subsequent Western blot analysis. Lysates were diluted into Laemmli sample buffer and denatured by heating for 10 min at 95 °C. Thirty micrograms of lysate was loaded per lane into NuPAGE Novex 4-12% Bis-Tris gradient gels (Life Technologies, Grand Island, NY), and gels were electrophoresed for 1.5 h at 150 V using Novex MOPS-based running buffer. Gels were then transferred to nitrocellulose membrane by electrophoresis in Novex transfer buffer for 1 h at 100 V. Membranes were then blocked for 1 h in Licor blocking buffer (Licor Technologies, Omaha, NE) and incubated overnight with the following antibody combinations: (1) phospho-MNK1 (CST, rabbit polyclonal No. 2111), (2) total p38 (CST, rabbit polyclonal No. 9212)/tubulin (CST, mouse monoclonal No. 3873), (3) phospho-ERK (CST, rabbit polyclonal No. 9101)/tubulin. Following extensive washing in 1X TBST, the blots were then probed with both anti-rabbit and anti-mouse secondary antibodies conjugated with IR active fluorophores (680 nm and 800 nm, respectively; Licor Technologies) for 1 h at room temperature. After additional washing, the Western blots were developed using the Licor Odyssey IR scanner. Blot 1 was then reprobed for 2 h at room temperature with a phospho-p38–specific antibody (CST No. 9211) as well as the tubulin loading control antibody and developed as described above. Additional antibodies used include phospho–heat shock protein 27 (HSP27; Ser82; CST, rabbit monoclonal No. 2401) and phospho–stress-activated protein kinase (SAPK)/JNK (Thr183/Tyr185; CST, rabbit monoclonal, No. 4668).
T-Cell–Based Proliferation and Cytokine Secretion Assays for Bioactivity
Assays were executed as previously described. 3 Briefly, primary mouse splenocytes were harvested and T cells were isolated and enriched using a mouse CD4+ T-cell enrichment column (R&D Systems, Minneapolis, MN). In a 96-well format, 5 × 104 CD4+ T cells per well were stimulated with either plate-bound anti-CD3 (2 µg/mL) and anti-CD28 (2 µg/mL) or 12-O-tetradecanoylphorbol 13-acetate (PMA; 5 ng/mL) and ionomycin (1 µg/mL) for 48 h in the presence of the indicated concentrations of compound 1, 10 µM SB203580, or vehicle control. Cells were then pulsed with 1 µCi [3H]thymidine (American Radiolabeled Chemicals, St. Louis, MO) for 14 h before harvesting. [3H]thymidine uptake was determined with a Wallac 1450 MicroBeta Liquid Scintillation Counter.
Tumor necrosis factor–α (TNF-α) secretion was quantified through the use of the Mouse Ready Set Go! TNFα kit (eBioscience, San Diego, CA). A total of 5 × 104 cells were stimulated with plate-bound α-CD3 (2 µg/mL) and α-CD28 (2 µg/mL) or PMA (5 ng/mL) and ionomycin (1 µg/mL) for 48 h in the presence of the indicated concentrations of compound 1, 10 µM SB203580, or vehicle control in 96-well flat-bottomed plates. Supernatants were collected, and TNF-α concentration was measured according to kit protocol.
ITC for Direct Binding of Compound 1
The calorimetric titration experiment of compound 1 with p38 was performed on a VP-ITC Titration Calorimeter from Microcal, Inc. (Northampton, MA). In the experiment, 5 µL aliquots of compound 1 (0.26 mM) were injected into a rapidly mixing (300 rpm) solution of p38 (0.026 mM) within the calorimeter cell (cell volume = 1.4426 mL). A total of 55 injections were made during the course of the experiment. A control experiment injecting identical amounts of compound 1 into buffer in the absence of p38 was conducted. Titrations were carried out at 30 °C in a 25 mM K+/PO4 pH 7.4 buffer supplemented with 150 mM KCl, 1 mM MgCl2, and 0.26% ethanol. The isotherm, corrected for dilution/buffer effects (0.56 kcal/mole), was fit to a nonlinear least-squares curve-fitting model (for a 1-set of identical sites) using Microcal Origin 5.0 analysis software. From the binding curve, values for enthalpy, binding affinity, and stoichiometry were extracted. Free energy and entropy of interaction were calculated using the following equations:
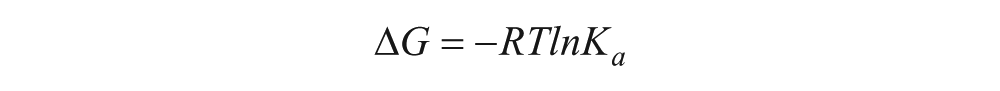
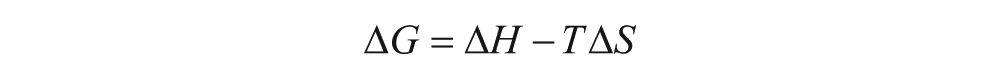
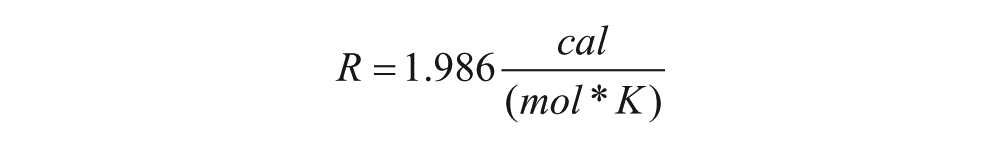
Mechanism of Inhibition Analysis for Compound 1
This experiment was executed in a manner similar to that described above for Michaelis-Menten analysis in the absence of an inhibitor. For all concentrations of ATF2, additional reactions containing a final concentration of 0.001, 0.0075, and 0.01 µM compound 1 were also carried out. For all concentrations of ATP, additional reactions containing a final concentration of 0.005, 0.05, and 0.1 µM. For the linear regression of the reaction progression curves, the only data point that had an r2 value less than 0.90 was for the lowest ATP concentration (25 µM) at the highest compound 1 concentration (which has the lowest overall signal), indicating that for the data set as a whole, there was excellent linearity associated with the reaction progress curves, instilling high confidence in the reaction velocities generated using these regressions. The data were analyzed using the best fit model (mixed model inhibition), a modified form of the Michaelis-Menten equation depicted below:
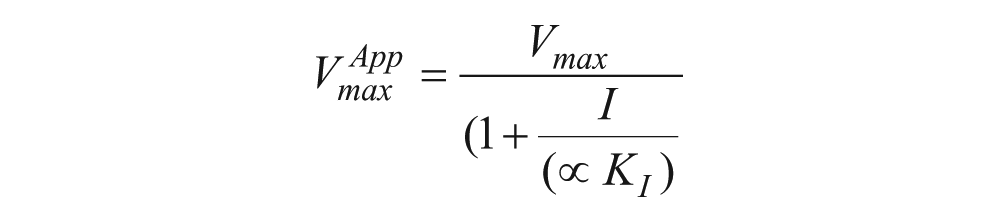
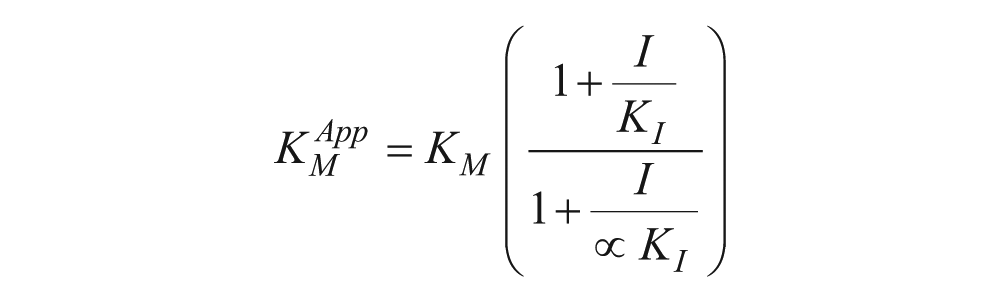
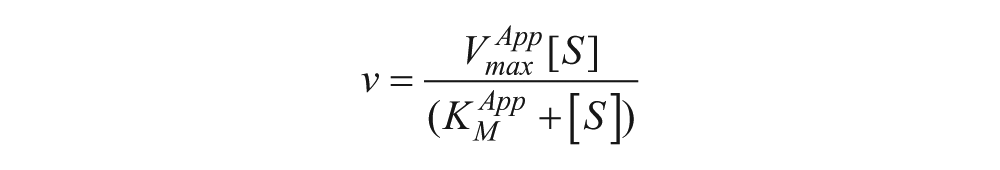
Results
Expression and Characterization of Recombinant p38α Kinase
Poly-histidine–tagged human p38α kinase bearing the activating mutation tyrosine 323 to threonine (Y323T, hereafter referred to as p38Y323T) was purified from Escherichia coli lysates using affinity chromatography. Further purification was achieved by size exclusion chromatography. Fractions collected from a single peak of the appropriate relative mass (~41 kDa) were pooled, concentrated, and tested for kinase activity. Kinase activity was validated by measuring the p38Y323T-dependent phosphorylation of the well-characterized p38 phosphorylation target ATF2 in an ELISA format.
18
Following verification of kinase activity, the ELISA conditions were optimized with regard to enzyme concentration, primary and secondary antibody concentration, and wash stringency. As shown in
HTS for Novel p38 Inhibitors
We screened ~32,000 pure compounds in a 384-well format at a final concentration of 10 µM for the ability to reduce p38Y323T-catalyzed phosphorylation of ATF2. Each substance well was normalized to the average high control for that plate (vehicle control). For the purposes of primary screening, a positive hit was defined as any substance well with a percentage normalized activity measurement ≤35%. Each plate also contained a low-control column, 1 µM SB 203580, a well-characterized p38 inhibitor. The inclusion of this control allowed us to monitor the variance in reagent delivery during liquid handling and, more importantly, to quantitate assay quality in the form of the calculation of a Z-factor value for each plate. 14 Over the course of 148 plates, the average Z-factor value was 0.75, with 88% of plates having a value of 0.7 or higher ( Fig. 1A ), all of which reflects excellent assay performance. 14 As shown in Figure 1B , there was very good separation between nonhits (average reading 91.3 ± 18.0 percentage activity, 31,581 substances) and preliminary hits (average reading 11.7 ± 11.7 percentage activity, 483 substances).
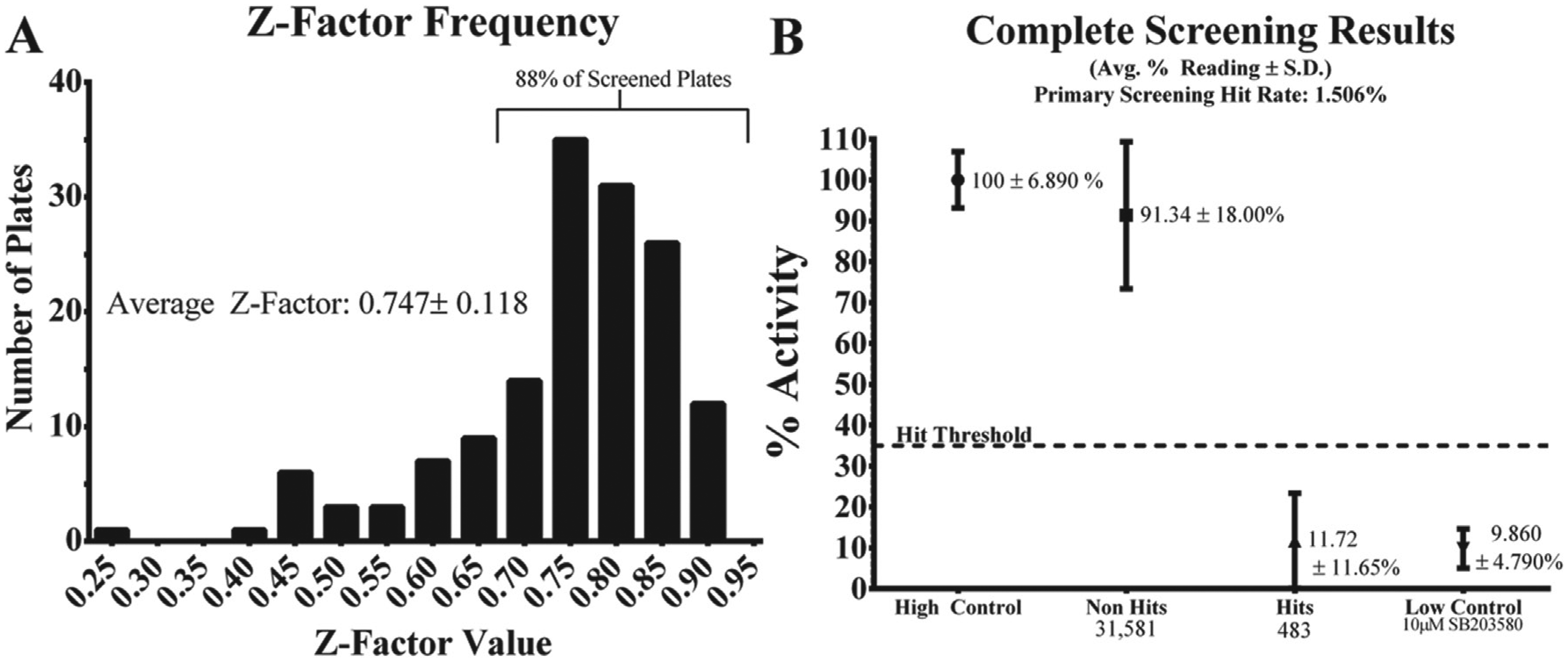
Key high-throughput screening quantitative metrics. (
All 483 preliminary hits were rescreened in quadruplicate, which yielded 99 substances with at least 50% inhibition, resulting in a confirmed hit rate of 0.31%. Forty-two confirmed hits were resourced and subjected to a 10-point dose-response testing to determine the IC50 for each compound. Each IC50 was then evaluated for potency (defined as an IC50 ≤50 µM) and cooperativity (defined as a Hill coefficient substantially divergent from 1; see
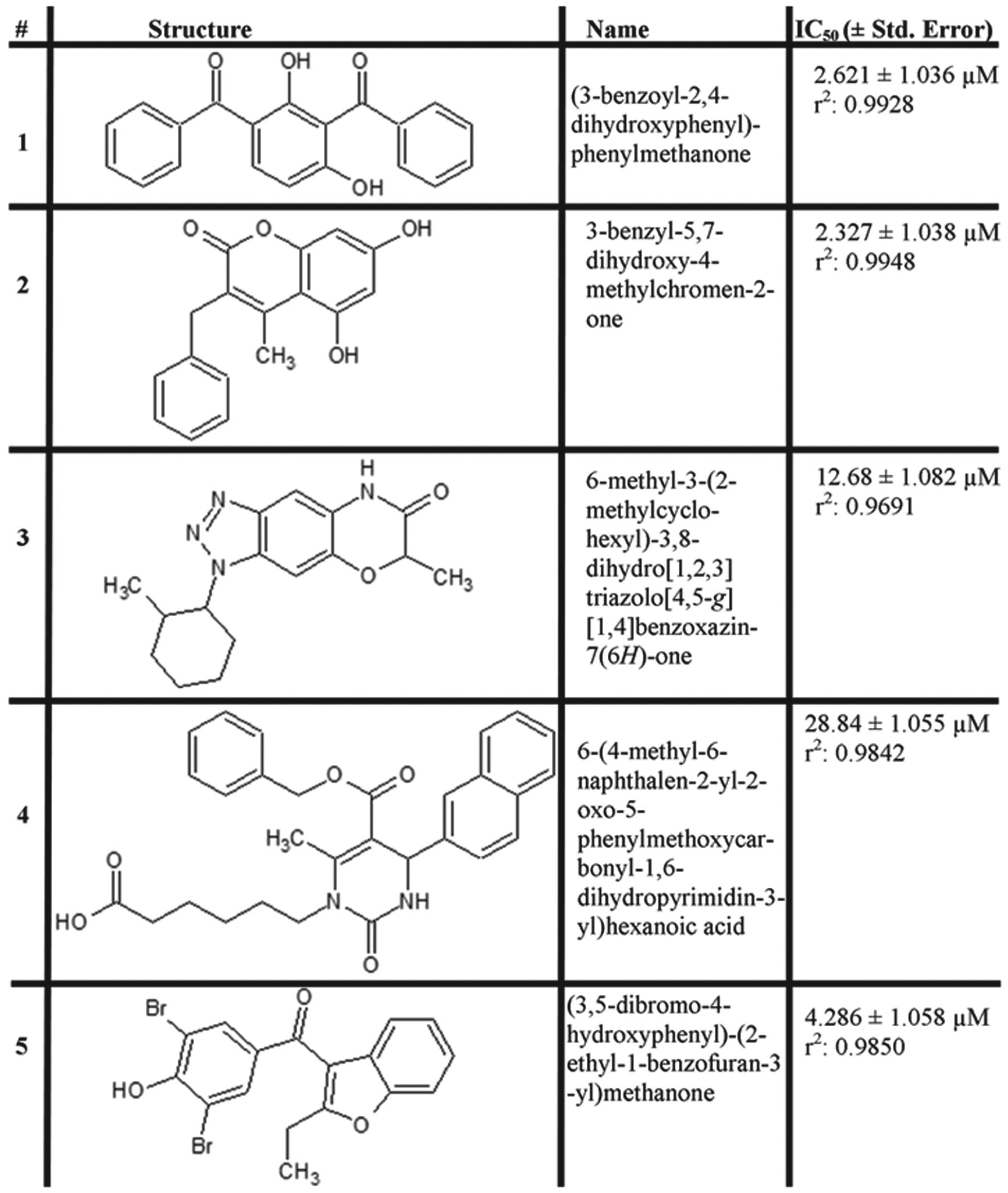
Novel p38 kinase inhibitors discovered during the screening campaign. Numbered as referenced within the article text: chemical structures, IUPAC name, and measured p38 IC50 value.
p38 Inhibitors Demonstrate Intracellular Activity
After narrowing the potential p38 inhibitors to five compounds with acceptable in vitro activity, we determined if any of the compounds exhibited the ability to specifically inhibit p38 activity within cells. To our knowledge, there are no stable culturable cell lines that exhibit constitutive p38 kinase activity; therefore, it was necessary to stimulate p38 activity in the presence of our putative inhibitor to determine cell-based efficacy.
11
As our model cell line, we chose the breast cancer carcinoma cell line MDA-MB-231.
23
Whereas p38 activity has been demonstrated to be important in many tumor types, it has recently been noted to be particularly important in the survival of breast cancer metastases, which is the site of origin for this cell line.9,24 Cells were preincubated with a range of concentrations of each of the five p38 inhibitors for 1 h prior to a 45-min stimulation with 10 µg/mL anisomycin. The cells were then lysed in situ and Western blots performed to evaluate the effect that pretreatment had on p38-dependent phosphorylation of a downstream target, MAP kinase–interacting kinase (MNK1).
5
As shown in
Figure 3
, compound 1 was able to achieve greater than 50% reduction in the phosphorylation of MNK1 in response to anisomycin stimulation without reducing the level of phosphorylated/activated p38 or ERK1/2, a closely related MAPK family member. In addition, we observed a reduction in the phosphorylation level of a canonical p38 phosphorylation target, HSP27, whereas no reduction in the phosphorylation status of SAPK/JNK was observed (

Compound 1 inhibits p38-dependent phosphorylation of MNK1. Anisomycin was used to stimulate p38 activity in the presence of compound 1. Compound 1 was able to achieve intracellular p38 inhibition without reducing ERK phosphorylation or phosphorylation of p38 itself.
The immunologic activity of p38 has been best characterized in the context of TCR signaling.1–3 TCR engagement generates a p38-dependent signaling cascade that results in both cell proliferation and the production of proinflammatory cytokines, such as TNF-α. 1 We therefore determined the effect of compound 1 treatment on primary murine T cells. We measured p38-dependent T-cell proliferation and TNF-α production following TCR engagement (anti-CD3/CD28 stimulation) and cell stress (PMA + ionomycin) in the presence of compound 1 in dose response. As shown in Figure 4 , compound 1 was able to reduce both p38-dependent proliferation (measured by thymidine incorporation) and TNF-α secretion in a dose-dependent manner. Figures 3 and 4 indicate that compound 1 is pharmacologically active in two different cell-based models of p38 bioactivity.
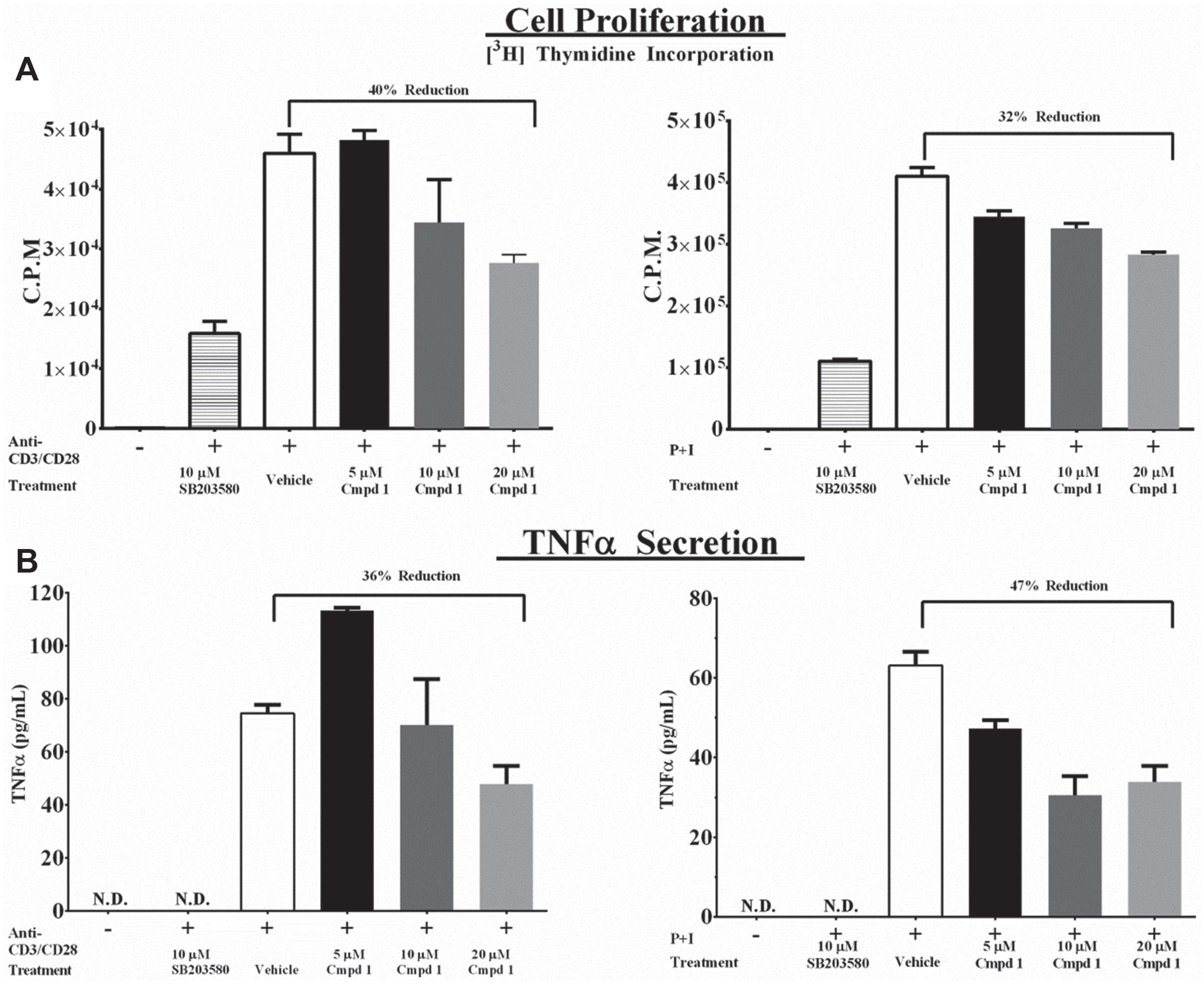
Compound 1 reduces p38-dependent T-cell proliferation and cytokine secretion. (
ITC Indicates Direct Stoichiometric Binding of Compound 1 to p38
ITC was employed to assess the thermodynamic parameters of compound 1 binding to p38 and further evaluate its mode of binding. The heat flow associated with binding is directly related to both the enthalpy (ΔH) and extent of the interaction. A progressive decrease in the heats of binding was observed with each successive injection of compound 1 into a solution of p38 ( Fig. 5 , upper panel). The exothermic heats of interaction (ΔH; Fig. 5 , upper panel) suggest that polar/electrostatic contacts are mediated between compound 1 and p38, presumably involving one or more of the functional groups of compound 1 ( Fig. 2 ). The measured binding affinity for compound 1 was 1.76 µM, a moderate affinity given the small size (318 Da) of the inhibitor. The overall interaction was enthalpically driven (–ΔH contributing to the –ΔG), but the binding entropy (+TΔS) also favorably contributes to the interaction. The positive entropy of binding suggests that the conformational freedoms of both compound 1 and p38 were not limited upon binding and that there likely was no energetically disfavored trapping of water molecules at the binding interface. The measured equilibrium stoichiometry of binding was 1 ( Fig. 5 , lower panel), indicating a 1:1 molecular complex between compound 1 and p38.
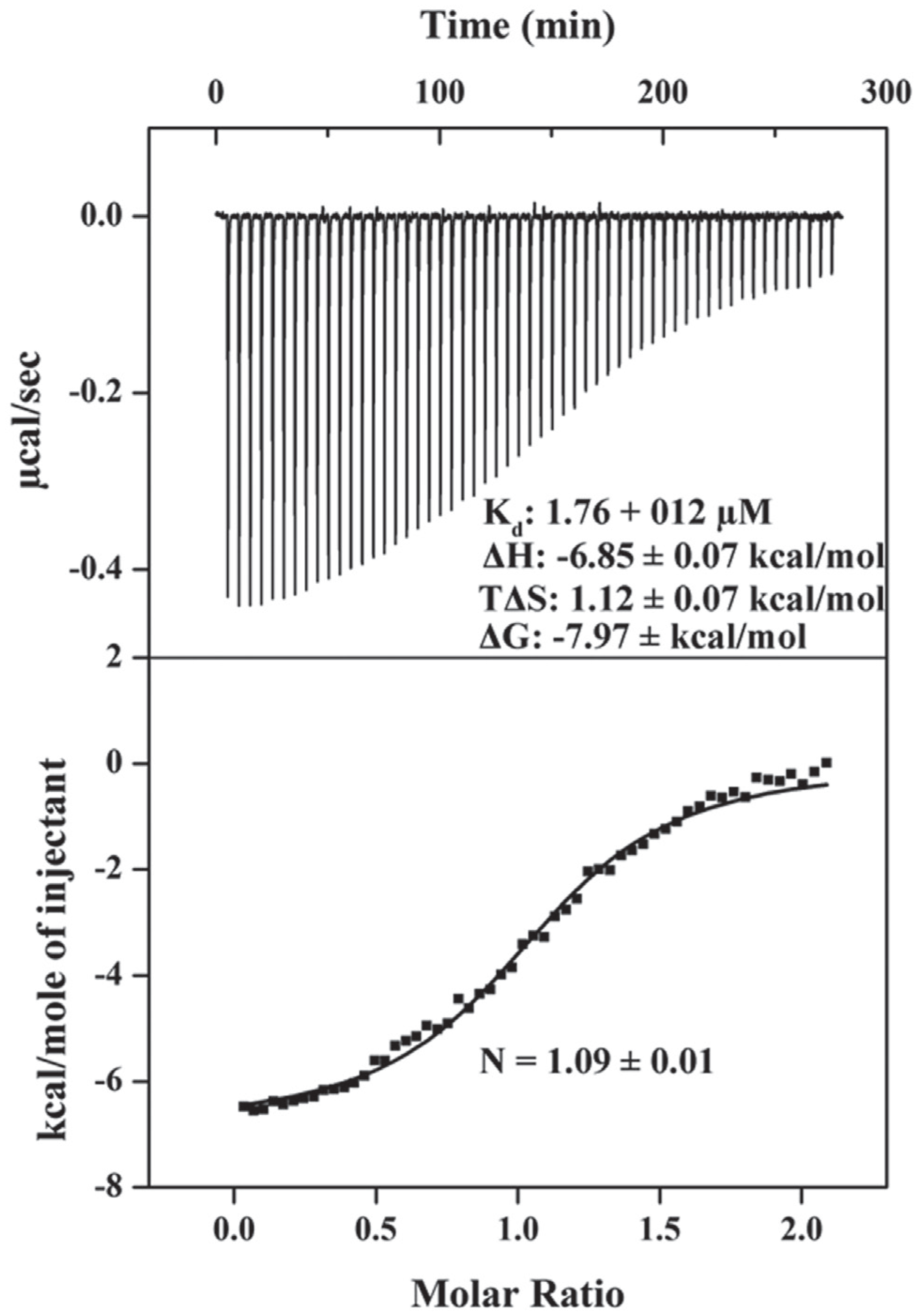
Isothermal titration calorimetry (ITC) indicates direct binding of compound 1 to p38. (
Non–ATP-Competitive Mechanism of p38 Inhibition
Having demonstrated that compound 1 is an effective p38 inhibitor in both biochemical and cell-based assays and that it exhibited favorable thermodynamic binding parameters, we sought to determine its mechanism of inhibition. To do so, we measured the steady-state Michaelis-Menten constant at three concentrations of compound 1 over a broad range of substrate concentrations for both ATP and ATF2. Based on nonlinear regression analysis, as shown in
Figure 6
, compound 1 exerts its effect in a noncompetitive manner for ATP. A mixed model of noncompetitive inhibition best fits the data for ATP, with r2 = 0.9391, Ki = 47.34 ± 5.053 nM, α = 1.603 ± 0.2458, and KmATPGlobal = 85.06 ± 3.630 µM. Because of the poor fit of the ATF2 data to the vehicle controls (
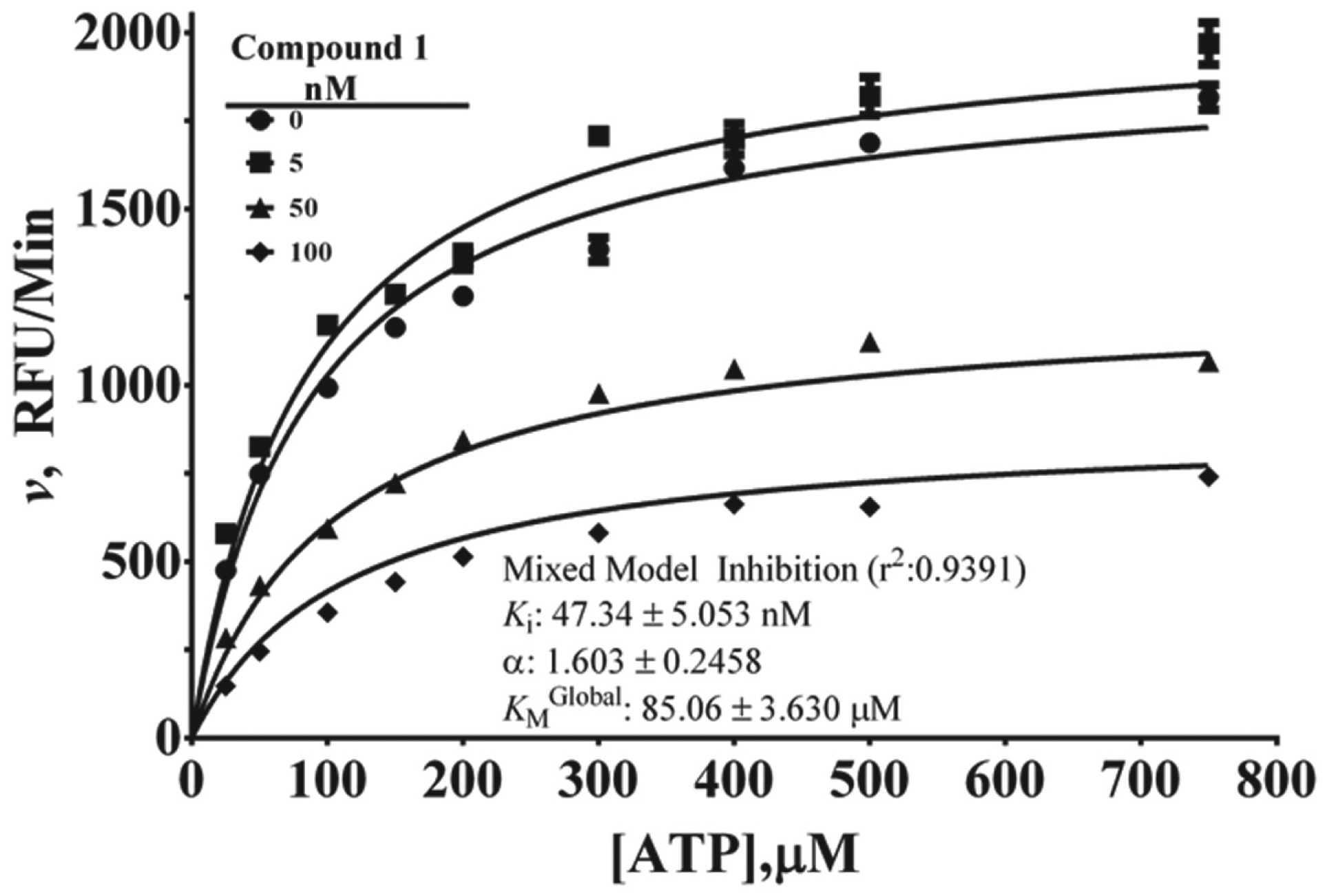
Non–adenosine triphosphate (ATP)–competitive mechanism of inhibition determination for compound 1. Steady-state Michaelis-Menten analysis was carried out to evaluate the impact of three concentrations of compound 1 on enzyme activity in the presence of increasing concentrations of ATP, with ATF2 held constant at 500 nM. Nonlinear regression analysis indicates that compound 1 is a mixed-mode non–ATP-competitive inhibitor.
Discussion
The therapeutic importance of p38 kinase activity in supporting the initiation and progression of a wide variety of diseases is apparent in the >200 clinical trials for which “p38” is a searchable term on the U.S. National Institutes of Health website clinicaltrials.gov. Unfortunately, for the tens of p38 inhibitors that have reached the clinical trial stage, therapeutic efficacy has remained only an aspirational goal. The reasons for these failures include on-target hepatoxicity in some cases and unexplained treatment-dependent resistance in others.10,12 Regardless of the explanation, there is one commonality that unites all of the p38 inhibitors tested in clinical trials so far; they are all ATP-competitive inhibitors. A previously identified non–ATP-competitive p38 inhibitor has not demonstrated intracellular activity, and because of its substrate-selective mechanism of inhibition, it has been modeled to be poorly suited to biologic activity in the presence of multiple p38 substrates within the cellular context.26–28 To this end, we believe that our study represents an interesting alternative approach for p38 inhibitor exploitation because of the discovery of the first non–ATP-competitive p38 inhibitor active in cells.
Generally, kinase-dependent inhibitor screens have often been carried out through the use of [32P]-labeled ATP, which, although providing exquisite sensitivity, also introduces the handling, analysis, and disposal of radioactive material. The ELISA-based HTS kinase assay used here may offer a different and cost-effective means for kinase screening. A unique aspect of our assay is the use of a constitutively active p38Y323T mutant. This has removed the need for a preactivation step and the variation inherent in the requirement to frequently activate a fresh kinase preparation for screening. Although it is not universally true that all kinases are susceptible to activating mutations, there is a growing body of literature both in the form of disease-associated activating mutations and in vitro mutational screening that indicates that this phenomenon may be true for many enzymes.29,30 One obvious limitation of an ELISA-based assay is that it is not a direct measure of product formation, unlike the measurement of [32P] incorporation into an analyte. However, we have used a known ATP-competitive inhibitor, SB203580, to validate that our assay has the sensitivity to identify and characterize biochemical inhibitors of p38 (
A potential limitation of our assay became apparent when characterizing the kinetic parameters associated with the reaction velocity of the peptide substrate, ATF2. As shown in
Beyond the characterization of compound 1, there are interesting observations to be made about the other four compounds. Compound 2 is reported on the PubChem website (pubchem.ncbi.nlm.nih.gov) to have demonstrable activity in 13 bioassays, including an additional kinase assay, glycogen synthase kinase 3 beta, as well as being cited in the literature as inhibiting testis-specific serine/threonine kinase 1. 15 This suggests that compound 2 may be binding a structural motif common to serine/threonine kinases. Although compound 3 has no previously reported biological activity and low-micromolar in vitro potency, in our cell-based assay for p38 inhibition, it appears to reduce the phosphorylation of ERK1/2 in addition to MNK1, indicating that it does not have specificity for p38 (data not shown). Although compound 4 does not show intracellular activity against p38 in our hands, it has been shown to inhibit cytochrome p450 activity and had some activity in reducing RAR-related orphan receptor gamma (RORγ) transcriptional activity (PubChem). This last observation is particularly interesting because both p38 and RORγ have been implicated as playing a role in a variety of autoimmune disorders, so there may be positive synergistic effects to reducing the activation of both pathways. 7 Finally, compound 5 (benzbromarone) has been used in humans as a uricosuric agent for patients suffering from gout. 32 However, its use has been largely restricted due to rare but serious treatment-induced hepatotoxicity. 32 The monosodium urate crystals associated with gout have been reported to induce p38 activity, so despite the rare hepatotoxicity observed during treatment with benzbromarone (~1:17,000), it is possible that there could be an additional p38 inhibition–dependent benefit to benzbromarone treatment in patients with gout. 33 For our lead compound, compound 1, the only previously published activity is in vitro inhibition of ovine cyclooxygenase-1 (COX-1) activity discovered through the use of in silico docking studies. 34 Although confirmation of this reported activity is beyond the scope of this article, it is interesting to note that with regard to anti-inflammatory activity, COX-1 inhibition would be considered synergistic with p38 inhibition, further supporting the exploration of this compound in the context of inflammatory disease models.
Given the modest cellular potency of these molecules in two different cell-based assays for p38 bioactivity, we do not believe that they themselves are directly suitable candidates for further investigations beyond in vitro characterization and cell-based assays; however, many of these compounds are chemically amenable to further medicinal chemistry optimization and structure-activity relationship studies.15–17 Such modifications could result in a better correspondence between biochemical and cellular potency. Furthermore, the measured ITC data indicate that there is a discrete unimolecular binding site for compound 1, providing further support for crystallography studies, rational drug design, and structural-activity relationship studies. The potential utility of a non–ATP-competitive inhibitor for p38 is apparent in the behavior of compound 1 in cells relative to an ATP-competitive inhibitor, BIRB-796 ( Fig. 3 , lanes 6 vs 9). BIRB-796 has a reported IC50 for p38 in the low-nanomolar range, and yet, to achieve demonstrable cell-based activity, it is necessary to use higher concentrations. 35 At higher concentrations, our own data as well as others indicate that BIRB-796 reduces the phosphorylation, thus activation, of p38 in addition to reducing its enzymatic activity, indicating that it may be inhibiting other upstream kinases in the MAPK cascade.25,35 We have also observed a similar phenomenon with a different ATP-competitive p38 inhibitor, SB203580 (data not shown). Compound 1, on the other hand, is able to exert a demonstrable reduction in p38 activity in cells without reducing the level of p38 phosphorylation or the phosphorylation state of related kinases JNK and ERK. We believe that this is due to its novel noncompetitive mechanism and demonstrates the potential benefits of the discovery and use of noncompetitive p38 inhibitors. In addition to the potential therapeutic value of noncompetitive p38 inhibitors, it is also quite likely that there is valuable structural information to be obtained through crystallographic analysis of these inhibitors in complex with p38 MAP kinase.
Footnotes
Acknowledgements
The authors would like to thank Cheryl L. Thomas and Antony M. Wamiru for assistance in HTS, John A. Beutler for substance procurement, and James B. McMahon and Curtis J. Henrich for their critical constructive evaluation of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by funds from the National Institutes of Health (NIH) Intramural AIDS Targeted Antiviral Program (B.R.O. and M.J.). This project has been funded in whole or in part with federal funds from the National Cancer Institute, NIH, under contract HHSN26120080001E (S.R.S.). This project has also been supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.