
Abstract
LRRK2 is a large multidomain protein containing two functional enzymatic domains: a GTPase domain and a protein kinase domain. Dominant coding mutations in the LRRK2 protein are associated with Parkinson’s disease (PD). Among such pathogenic mutations, Gly2019Ser mutation in the LRRK2 kinase domain is the most frequent cause of familial PD in Caucasians and is also found in some apparently sporadic PD cases. This mutation results in 2- to 3-fold elevated LRRK2 kinase activity compared with wild type, providing a clear clinical hypothesis for the application of kinase inhibitors in the treatment of this disease. To date, reported screening assays for LRRK2 have been based on detection of labeled adenosine triphosphate and adenosine diphosphate or on antibody-based detection of phosphorylation events. While these assays do offer a high-throughput method of monitoring LRRK2 kinase activity, they are prone to interference from autofluorescent compounds and nonspecific events. Here we describe a label-free assay for LRRK2 kinase activity using the RapidFire mass spectrometry system. This assay format was found to be highly robust and enabled a screen of 100,000 lead-like small molecules. The assay successfully identified a number of known LRRK2 chemotypes that met stringent physicochemical criteria.
Introduction
Leucine-rich repeat kinase 2 (LRRK2), also known as dardarin from the Basque word meaning “tremor,” is a protein of 2527 amino acids encoded by the PARK8 gene. 1 It was initially identified by genomic prediction as a novel protein kinase with leucine-rich repeats and subsequently as a member of the ROCO super family. 2 These proteins are characterized by the presence of conserved Ras-like GTPase or Roc (Ras of complex proteins) domain, followed by a COR (C-terminal of Roc) domain. 3 LRRK2 itself contains 13 leucine-rich repeats, followed by the Roc, COR, and protein kinase domains as well as seven WD40 repeats.
Parkinson’s disease (PD) is the second most common neurodegenerative disorder and is characterized by tremor, rigidity, bradykinesia, and postural instability. It affects over 1% of individuals older than 65 years. 4 There is no disease-modifying treatment for PD, with current therapies such as L-DOPA serving only to abate symptoms. In 10% to 15% of PD cases, there is a familial link through autosomal dominant or recessive inheritance. 5 The prevalence of LRRK2 mutations in PD suggests a critical role for LRRK2 in disease susceptibility. The most common of these mutations is the Gly2019Ser mutation in the LRRK2 kinase domain. This mutation is found in 5% to 6% of familial PD and in 2% of sporadic PD in several European and US populations, and patients with this mutation are clinically indistinguishable from idiopathic PD. 6 The mutation causes a 2- to 3-fold increase in the intrinsic catalytic activity of the LRRK2 kinase domain compared with wild type (WT). 7 This provides a basis for the application of LRRK2 kinase inhibitors in the treatment of G2019S-linked PD to restore the aberrantly high kinase activity to normal, and indeed a number of published LRRK2 kinase inhibitors have been shown to attenuate G2019S LRRK2-related cell injury and death both in vitro and in vivo. 8 However, despite the discovery of a number of LRRK2 tool inhibitors, including some more selective compounds such as GSK2578215A, 9 clinical evaluation of LRRK2 kinase inhibitors for PD has yet to commence.
It is notoriously difficult to develop drugs for central nervous system (CNS) targets because of the challenges of crossing the blood-brain barrier, avoiding interaction with P-glycoprotein (PGP) and other efflux pumps, and optimizing binding to brain tissue and protein. The ability of a molecule to overcome these hurdles is recognized to be critically dependent on its physicochemical properties. Many multivariate models and simpler rules of thumb have been developed to try and describe a particular area of physicochemical space with an empirically optimal probability of success for CNS drug candidates. These are summarized in a recent review, 10 with recurring descriptor ranges being low molecular weight (<~350 Da), low polar surface area (PSA; <~70 Å2), and low H-bond donor (HBD) count (≤2), all of which are lower than the corresponding mean figures for marketed pharmaceuticals. This presents a particular challenge for LRRK2 inhibitor identification because traditional adenosine triphosphate (ATP)–pocket kinase inhibitors tend to have higher mean molecular weight and PSA values than the overall mean of prescribed drugs. 11
A number of assay formats have been described for identification and evaluation of LRRK2 kinase inhibitors. These include radioactivity-based assays, which rely on incorporation of 32P-labeled ATP to monitor phosphorylation of the peptides LRRKtide and Nictide, derived from the LRRK2 in vitro substrate moesin.12,13 Screens using the full-length moesin protein substrate in an AlphaScreen (PerkinElmer, Waltham, MA) format have also been described. 14 Others have exploited the formation of adenosine diphosphate (ADP) formed during the kinase reaction, through the use of kits such as ADP-Hunter (DiscoveRx Corporation, Fremont, CA) 15 or ADP-Glo (Promega Corporation, Madison, WI), to screen libraries of compounds for LRRK2 inhibitors. Recombinant cellular high-throughput assay formats have also been adopted, primarily relying on antibodies that detect phosphorylation of LRRK2 at Ser935 in time-resolved fluorescence resonance energy transfer (TR-FRET)–based readouts. 16 While each of these assay formats has its relative strengths, and many have been used to successfully screen tens of thousands of compounds against LRRK2, there are also challenges associated with them from a compound screening perspective. 17 Radioactive assays are no longer favored by screening groups due to the complexities and safety considerations of handling large volumes of 32P. Assays monitoring ADP formation are readily amenable to miniaturization, but these assays provide an indirect readout, often through the use of coupling enzymes, which can present a source of interference. These assays will also be sensitive to autophosphorylation events, which are known to occur with LRRK2, 18 thereby requiring inclusion of additional relevant controls to avoid misinterpretation of data. Cellular assays monitoring phosphorylation at Ser935 also give an indirect measure of LRRK2 kinase activity, and in some cases, hits from these screens have been shown not to inhibit LRRK2 kinase activity directly, 16 reflecting the ability of other kinases to phosphorylate LRRK2 at the pS935 residue.19,20 Both of the aforementioned fluorescent formats as well as AlphaScreen are also at risk of false positives or negatives from autofluorescent compounds in the screening collection. AlphaScreen may also suffer from interference from redox reactive compounds, which could disrupt the singlet oxygen transfer on which this technology relies. 21
Overall, the issues described above are likely to result in a number of false positives or nonspecific hits, which will artificially inflate the overall hit rate of any screen for LRRK2. This is a particular issue given that it is a challenge to isolate the required quantity of high-quality recombinant LRRK2 protein, 22 and therefore it is essential that reagent not be wasted in confirming and progressing false actives. Moreover, in our experience, preparations of purified full-length and truncated active LRRK2 protein are prone to aggregation, presumably due to the large size of even the truncated proteins, and the fact that LRRK2 is likely to form dimers and complexes in its native cellular state. 23 This adds to the challenge of screening against recombinant LRRK2 because in addition to assay format–specific false positives, there is likely to be an underlying false-positive rate due to compounds that cause inhibition through the undesirable mechanism of protein aggregation. To maximize the probability of finding true hits, one therefore needs a sensitive assay that uses a minimal necessary amount of active recombinant protein with little or no interference.
The RapidFire mass spectrometry assay platform (Agilent Technologies, Lexington, MA) has been shown to offer a high-throughput, direct, and label-free readout of enzyme activity, including measurement of phosphorylation events.24,25 The label-free, direct nature of mass spectrometry (MS)–based detection eliminates the risk of fluorescent false positives and overcomes many of the challenges of indirect detection highlighted above. Here we describe the application of RapidFire to develop a label-free assay for LRRK2 by MS detection of the phosphorylated and dephosphorylated forms of the LRRKtide peptide substrate. The assay was deployed to rapidly screen a set of 100,000 compounds, selected using strict physicochemical property criteria designed to address the particular challenges associated with identification of CNS drug candidates. A number of LRRK2 chemotypes were identified from the screen, giving confidence in the output and demonstrating that use of a direct, label-free detection method, coupled with a sharp focus on compound properties, provides a powerful platform for identifying starting points for challenging CNS targets such as LRRK2.
Materials and Methods
All reagents were purchased from Sigma-Aldrich Ltd. (Gillingham, Dorset, UK), unless otherwise stated. Assay buffer was 50 mM Hepes (pH 7.0), 10 mM MgCl2, 150 mM NaCl, 5% glycerol, 0.0025% Triton X-100, and 1 mM dithiothreitol (DTT). The buffer was stored complete at 4 °C with the exception of DTT, which was added fresh on the day of assay. The 384-well V-bottom polypropylene assay plates were from Greiner Bio-one (Stonehouse, UK). Solvents for mass spectrometry were from Fisher Scientific (Loughborough, UK). Molecular biology reagents were from Invitrogen (Waltham, MA) unless otherwise stated. Benzonase was from Novagen/Millipore (Billerica, MA).
A library of 100,000 compounds was selected to be in favorable lead-like 26 physicochemical space. The chosen compound set was characterized by the following parameters: heavy atom count (HAC), 10 < HAC < 28, –2 < clogp < 3, fsp3 > 0.3, 27 and Property Forecast Index (PFI) 3 to 7. 28 In addition, “nuisance” compounds 29 and compounds with other undesirable functional groups were removed. The compounds were obtained from a variety of sources, including commercial suppliers and the GlaxoSmithKline (GSK) Compound Collection.
Generation of 6His-Tev-LRRK2 (1326–2527)
A LRRK2 complementary DNA (cDNA) encoding residues 1326 to 2527, as described in Jaleel et al., 12 was received from Division of Signal Transduction Therapy (DSTT), University of Dundee. This gene fragment was subcloned into pFB-HTb using BamHI and NotI restriction sites. The LRRK2 plasmid was recombined into the baculovirus genome according to the BAC-to-BAC protocol described by Invitrogen. Transfection into Spodoptera frugiperda (Sf9) insect cells was performed using Cellfectin, according to the manufacturer’s protocol to generate P1 and P2 baculovirus stocks.
Sf9 cells were grown in HyClone SFX (Thermo Fisher Scientific, Waltham, MA) growth media on a shaker incubator (Kuhner, Basel, Switzerland) at 27 °C, 110 rpm, until of a sufficient volume to inoculate a bioreactor. The expressions were carried out in a 25-L wave culture system (Wave Biotech, Tagelswangen, Switzerland). Sf9 cells were cultured in the same media at 27 °C, 16 to 22 rocks/min, with an 8- to 10.5-degree rock angle (using 0.2 L air/min). The cells were then infected with the baculovirus at a multiplicity of infection (MOI) of 3 to synchronously infect the culture. Cultures were infected at 6E06 viable cells/mL (high density) or 3E06 viable cells/mL (standard density). The high-density culture was run for 1 additional day to allow time for the higher density of cells to be reached prior to infection. The infected cells were cultured for a further 48 h with 50% oxygen supplementation into the wave culture system’s atmosphere to maintain the dissolved oxygen. The cells were harvested using a continuous centrifuge (CARR Centritech Viafuge, Clearwater, FL) at 2500 × g, 80 L/h. The resultant cell slurry was then recentrifuged in a Sorvall RC3BP (Thermo Fisher Scientific) (2500 × g, 20 min at 4 °C) and the supernatant discarded. The cell pellets were frozen at −80 °C for subsequent purification.
For purification, a 166-g pellet from a 20-L expression was allowed to thaw in a water bath at 27 °C with 480 mL lysis buffer/buffer A (50 mM Tris-base [pH 8.5], 300 mM NaCl, 5 mM β-mercaptoethanol, 10% [v/v] glycerol, 4 mL protease inhibitor cocktail, and 300 µL benzonase) before being dounce homogenized on ice using 20 strokes per 100 mL. The suspension was packed in ice and sonicated at 35% amplitude for 4 min, 10 s on/off using a 1-in. probe. The suspension was then centrifuged at 108,800 × g for 90 min at 4 °C.
The lysate supernatant (415 mL) was decanted from the insoluble pellet and contacted for 2 h at 4 °C with 50 mL His bind Ni NTA resin (Millipore) by end-over-end mixing. The resin was recovered by centrifugation, 200 × g, 5 min at 4 °C, and packed in an XK-26 column (GE Healthcare, Buckinghamshire, UK). The column was then washed with 10 column volumes buffer A and 10 column volumes buffer B (buffer A + 1 M NaCl). The column was then eluted with a linear gradient of buffer B to buffer C (50 mM Tris-base [pH 8.5], 1000 mM NaCl, 10% [v/v] glycerol, 5 mM β-mercaptoethanol, and 300 mM imidazole) over 1 column volume. The column was further eluted with five column volumes buffer C, collecting 5-mL fractions. All washes and elution steps were conducted at 4 mL/min.
Fractions identified by reducing sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE) as containing the protein of interest were pooled and dialyzed against 2 L buffer D (50 mM Tris-base [pH 8.5], 300 mM NaCl, 10% [v/v] glycerol, 5 mM DTT, and 0.05% [w/v] 3-((3-Cholamidopropyl)dimethylammonium)-1-propanesulfonate (CHAPS)), overnight at 4 °C, using a 10,000 molecular weight cutoff (MWCO) dialysis membrane. The dialyzed pool was concentrated by 50% using an Amicon Ultra-15 Centrifugal Filter Unit with Ultracel-100 membrane (Millipore) at 4000 × g and 4 °C.
The concentrated protein was further purified by completing two identical runs on a 320-mL SEC Superdex 200 pg column (GE Healthcare) that was preequilibrated with buffer D (as above). The column was loaded with 12 mL and eluted with 1.2 column volumes buffer D at 4 mL/min collecting 1-mL fractions. Fractions identified by reducing SDS-PAGE as containing protein of interest were tested for activity. Protein identity was confirmed by peptide mass fingerprinting.
Generation of LRRKtide Peptide
The LRRKtide peptide (sequence H-RLGRDKYKTLRQIRQ- OH, derived from moesin) was prepared as described in Jaleel et al. 12 and was supplied by Cambridge Research Biochemicals (Billingham, UK). In detail, the protected peptide was assembled on a solid-phase synthesizer using preloaded Wang resin and using standard Fmoc synthesis protocols. The crude peptide was obtained after cleavage from the resin with a mixture of trifluoroacetic acid (TFA), triisopropylsilane, and water (95:2.5:2.5 v/v) for 3 h at room temperature and was then purified using a C18 reverse-phase column using a 0.1% TFA-buffered water/acetonitrile gradient. The resulting fractions were analyzed, and fractions that were >95% pure by analytical high-performance liquid chromatography (HPLC) and giving the correct molecular weight (mw) (by matrix-assisted laser desorption ionization time-of-flight [MALDI-TOF] mass spectroscopy) were pooled and freeze dried. The final material was analyzed by HPLC and MALDI-TOF mass spectroscopy.
RapidFire Mass Spectrometry Assay
The assay for LRRK2 inhibition was based on the direct measurement of the LRRKtide peptide and also of LRRKtide phosphorylated at the threonine residue highlighted in bold (H-RLGRDKYK
The assay was performed in 384-well V-bottom polypropylene plates with a final reaction volume of 10 µL. Test compounds were added to plates as a 100-nL solution in DMSO using an Echo 555 acoustic dispenser (Labcyte, Sunnyvale, CA) prior to the addition of assay components. All wells of columns 6 and 18 of assay plates contained 100 nL DMSO to give 100% activity and 100% inhibition controls, respectively (column 18 was a no-enzyme control). Single-concentration testing was at 10 µM final compound concentration. For pIC50 determination, compounds were tested in duplicate using an 11-point, 4-fold dilution series from 100 µM final assay concentration prepared using an Agilent Bravo (Agilent Technologies).
To these compound plates, 5 µL of an enzyme solution containing 120 nM 6His-Tev-LRRK2 (1326–2527) in assay buffer was added using a Multidrop Combi dispenser (Thermo Fisher Scientific), giving a final assay concentration of 60 nM LRRK2. Assay buffer was added to column 18 in place of LRRK2 protein, as a 100% inhibition control. Plates were then incubated for 30 min at room temperature. Following incubation, 5 µL of a substrate solution containing 50 µM LRRKtide peptide substrate and 40 µM ATP in assay buffer was added to all wells of the plate using the Multidrop Combi, to give final assay concentrations of 25 µM LRRKtide and 20 µM ATP. Plates were then incubated for 1 h at room temperature. After 1 h, 50 µL of 1% formic acid solution in water was added to all wells using the Multidrop Combi to quench the reaction, and plates were centrifuged at 3000 rpm for 10 min. Once quenched, plates were stable and could either be stored at −20 °C or analyzed immediately.
For ATP Km determination, ATP was titrated 2-fold from a 2-mM final assay concentration at fixed concentrations of 100 µM LRRKtide and 120 nM LRRK2 protein. For LRRKtide Km determination, LRRKtide was titrated 1.5-fold from a 1.4-mM final assay concentration at a fixed concentration of 1 mM ATP and 120 nM LRRK2 protein. For the reaction time course, columns of a 384-well plate were quenched with formic acid at intervals up to 60 min.
For analysis, plates were transferred onto a RapidFire high-throughput solid-phase extraction (SPE) system (Agilent Technologies). Sample was aspirated directly from each well of quenched assay plates for 500 ms and loaded onto the RapidFire micro-scale solid-phase C4 extraction cartridge (Agilent Technologies) to remove buffer salts with HPLC-grade water containing 0.1% formic acid (RapidFire buffer A) in a 3-s wash cycle at a flow rate of 1.5 mL/min. Analytes were then coeluted into the mass spectrometer in a 3-s elution cycle using 80% acetonitrile containing 0.1% formic acid (RapidFire buffer B) at a flow rate of 0.8 mL/min. This was followed by a 500-ms reequilibration step. LRRKtide and Phospho-LRRKtide were monitored on a Sciex API 4000 triple quadrupole mass spectrometer (Applied Biosystems, Concord, Ontario, Canada) in positive electrospray ionization (ESI) mode following multiple-reaction monitoring (MRM) transitions at (Q1 mass/Q3 mass) 644.8/638.8 Da and 671.4/638.8 Da, respectively. These masses relate to the triply charged peptide species.
Data Analysis
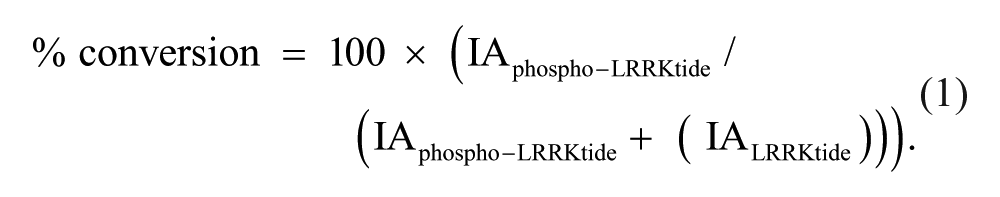
Peaks were integrated using the RapidFire integrator software, and percent conversion from substrate to product was calculated according to equation (1), where IA is the integrated peak area for the respective analyte.
Percent conversion values were then further analyzed in ActivityBase (IDBS, Surrey, UK) to generate percent inhibition data through normalization to 0% and 100% inhibition control wells in columns 6 and 18, respectively. For pIC50 determinations, curves were then fitted to equation (2):
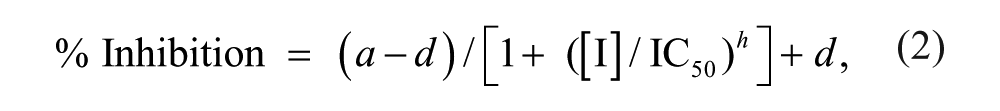
where a is the uninhibited response, d is the fully inhibited response, [I] is the inhibitor concentration, and h is the Hill slope.
For Km determinations, data were fitted to the nonlinear curve-fitting programs of GraFit version 7.0.3 (Erithacus Software Ltd., Surrey, UK). Saturation curves were fitted to equation (3):
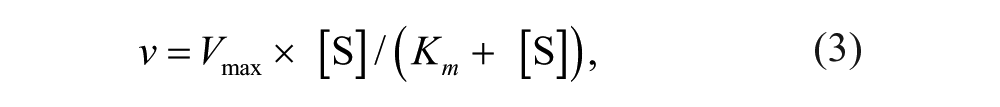
where Vmax is the maximum velocity (apparent kcat), [S] is the substrate concentration, and Km is the Michaelis constant.
Results and Discussion
LRRK2 kinase inhibition is an attractive target for the treatment of Parkinson’s disease, with a clear association between the most frequent pathogenic mutation in the LRRK2 kinase active site, G2019S, and elevated protein kinase activity compared with WT. However, despite a decade of research, no LRRK2 kinase inhibitor has entered the clinic for treatment of a CNS disease. As such, an unmet need remains for identification of LRRK2 kinase inhibitors with suitable activity profile and physicochemical properties for development as CNS clinical candidates. Such needs could be met with robust high-throughput assay formats with which to identify suitable starting points. RapidFire mass spectrometry uses online solid-phase trapping and cleanup of the target analytes with MRM to provide a high degree of specificity as well as a direct, label-free measure of peptide substrate phosphorylation, free from many of the artifacts that might contribute to false-positive hits in other assay formats. This platform can be readily applied to screening sets of around 100,000 molecules. In this instance, such a set was selected specifically to target the stringent physicochemical properties required for a lead against a CNS target.
Assay Development
The LRRKtide peptide (RLGRDKYKTLRQIRQ) was selected as the LRRK2 substrate for use in the RapidFire assay. This peptide encompasses the Thr567/Thr564/Thr558 of ezrin/radixin/moesin and was identified previously in an in vitro screen of rat brain extracts for proteins phosphorylated by recombinant LRRK2.
12
Other peptide substrates for LRRK2 are available, such as Nictide,
13
which has been further optimized to give a lower Km and slightly higher Vmax than LRRKtide. A lower Km, and therefore lower concentrations of substrate, may not necessarily confer an advantage for a RapidFire screen, where the substrate and product are directly detected by MS, although the Km for Nictide is still 10 µM, which should be easily detected. In this case, however, LRRKtide was employed as this was already available in house from previous assay work. Initially, the mass spectrometer was tuned and MRM transitions established at (Q1 mass/Q3 mass) m/z 644.8/638.8 and m/z 671.4/638.8 for the LRRKtide peptide substrate and phosphorylated LRRKtide peptide product (p-LRRKtide), respectively. RapidFire methods were set up using generic loading/elution buffers based on acetonitrile/water mixtures containing 0.1% (v/v) formic acid to facilitate ionization in ESI mode. Solid-phase cartridges of C4 (type A), C8 (type E), and C18 (type C) were evaluated, with C4 giving the best performance based on signal intensity and peak shape. Common RapidFire flow rates are 1.5 mL/min for loading buffer and 1.25 mL/min for elution buffer. In this case, a better peak shape was observed when the elution buffer flow rate was reduced to 0.8 mL/min, and this condition was therefore selected (data not shown). Once RapidFire and MS methods were finalized, the LRRKtide peptide substrate and p-LRRKtide peptide product were titrated to determine the linear dynamic range of detection. The assay was linear from 100 µM to 15 nM for both analytes. Limits of detection and quantification were determined by visual inspection of calibration data and were approximately 15 nM and 60 nM, respectively, and very little crosstalk was observed between the LRRKtide and p-LRRKtide masses (
LRRK2 assays have also been reported with moesin protein substrates, for example, in AlphaLisa format. 14 Detection of protein substrate phosphorylation is feasible by RapidFire if the RapidFire instrument is coupled to a time-of-flight mass spectrometer with the ability to detect intact proteins. In this case, the RapidFire was coupled to a triple quadrupole mass spectrometer, limiting us to a peptide substrate. It is not known if moesin is a true physiological substrate of LRRK2, and therefore any additional value of screening the protein over the peptide substrate is unclear. In addition, assays with peptide substrates are well defined for LRRK2 and have been used to identify and characterize many of the known tool inhibitors.12,13
Having established the analytical methods for LRRKtide and p-LRRKtide detection, the Km for LRRKtide and ATP could be determined in the RapidFire format. For the LRRKtide Km, LRRKtide was titrated from 1.4 mM and ATP held constant at a saturating concentration of 1 mM. For the ATP Km, the ATP was titrated from 2 mM and LRRKtide peptide held constant at 100 µM. This was chosen as the highest concentration possible within the linear range of the detection. Although at this concentration, LRRKtide is not saturating, the percent conversion of substrate to product observed even at the highest ATP concentrations and longest time point was not sufficient to cause peptide substrate depletion (~ 20 µM p-LRRKtide formed at 60 min at 2 mM ATP). In both cases, a reaction time course was performed over an hour, with 120 nM LRRK2 protein, and the rate of reaction at each substrate concentration used to plot the Km. Due to the need to titrate LRRKtide beyond the top of its linear dynamic range, determined as 100 µM, for the Km experiment, only product peak area was used to plot the data for the LRRKtide Km and was compared with a standard curve of p-LRRKtide run in the same experiment. The LRRKtide Km was determined as 216 µM ( Fig. 1A ), which is consistent with values previously reported for this peptide in a radioactive assay format.12,30 The Km for ATP was determined as 110 µM ( Fig. 1B ), also within 2-fold of previous reports using truncated WT LRRK2 protein. 30 The apparent kcat value was calculated to be 0.15 s−1. For further assay work, both ATP and LRRKtide concentrations below Km, at 20 µM and 25 µM, respectively, were employed. This was in part to avoid the need to synthesize huge quantities of peptide for the screening campaign and, in the case of the ATP, for consistency with other assay formats previously run in house (data not shown). This was based on the expectation that the majority of hits identified from the screen would likely be ATP competitive, and pharmacology should therefore be only minimally affected by screening below Km.
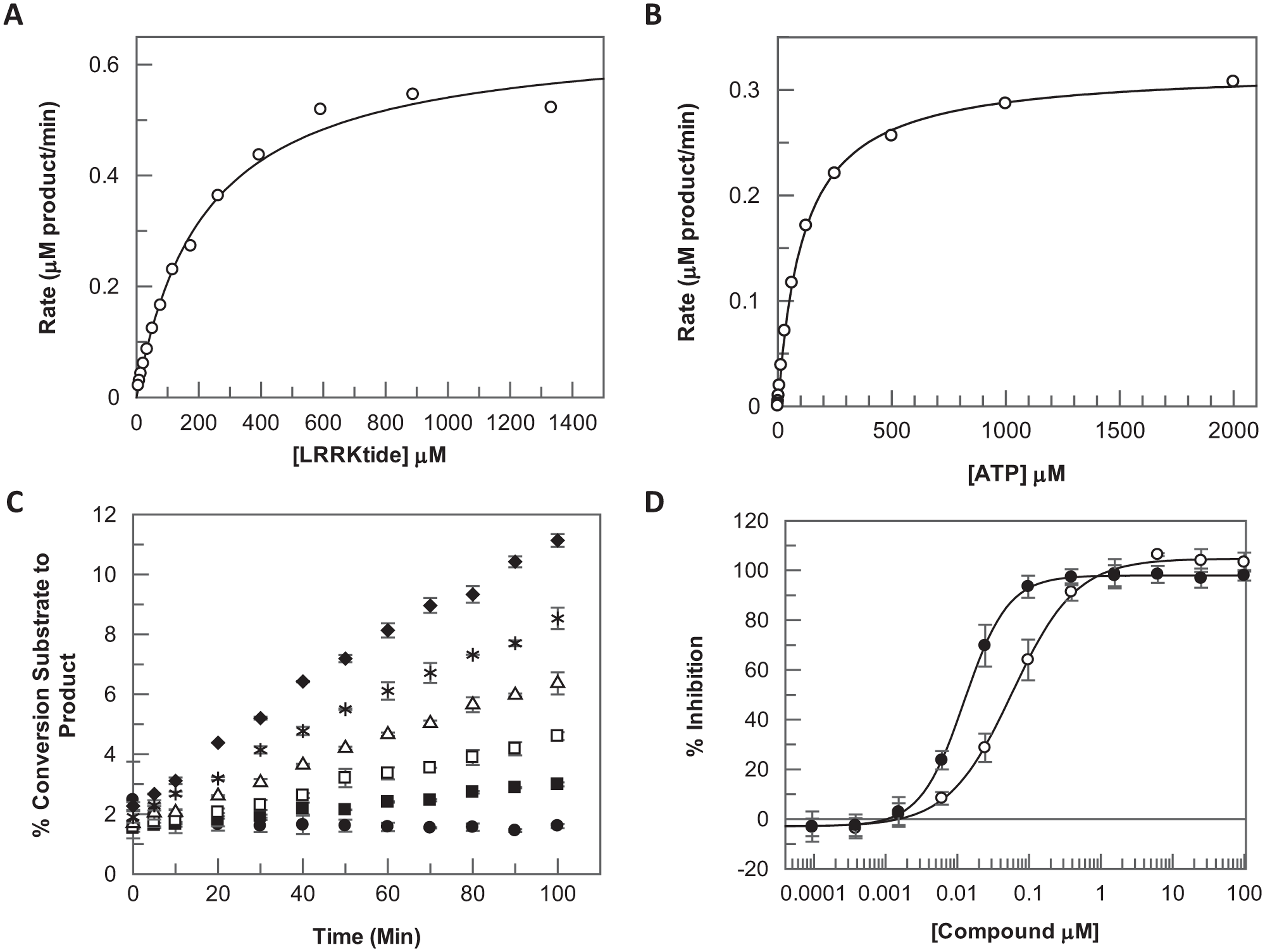
Development of the RapidFire mass spectrometry assay for LRRK2. (
Having established substrate concentrations for use in the assay, a reaction time course was completed using these final concentrations of ATP and LRRKtide, at a range of LRRK2 protein concentrations ( Fig. 1C ). The lowest LRRK2 concentration at which sufficient signal to background could be achieved to give a robust assay (at least 2:1) was 60 nM, and the reaction was linear for at least 1 h at this concentration. These conditions were taken forward for screening. The ability to robustly detect substrate phosphorylation at nM concentrations of LRRK2 was very encouraging, since LRRK2 protein is challenging to produce at scale. At the assay concentrations enabled here, just 30 mg of LRRK2 was required to complete the screening campaign.
Assay Pharmacology and Robustness Testing
To investigate the performance of the RapidFire assay format, we assessed the pharmacology of the published LRRK2 inhibitors LRRK2-IN-1 and H1152 ( Fig. 1D ). LRRK2-IN-1 gave an IC50 of 12.5 nM, which is highly consistent with the value of 13 nM reported in the literature. 31 H1152 gave an IC50 of 58 nM, which is more potent than that originally reported, of 240 nM against WT LRRK2, albeit in a radioactivity-based assay format. 13 IC50 values were generated after normalization of data to DMSO and “no-enzyme” control wells. While a no-enzyme control was used in this case, one could also employ a published inhibitor at a suitable concentration as a low control in the assay. However, IC50 curves for both compounds were of good quality and plateaued at 100%, suggesting that the no-enzyme low control was suitably representative of compound inhibition, and this condition was therefore carried forward for logistical reasons.
To further validate the assay for single-concentration screening, a set of 1400 diverse compounds (4 × 384-well plates) was screened under the final assay conditions at 10 µM final compound concentration on two independent test occasions. A plate containing only 100 nL DMSO in each well (1% DMSO final assay concentration) was also included in each run. The assay gave high-quality data with a mean Z′ of 0.88 and 0.89 for the two replicates, respectively. Hits correlated well between replicates with a correlation coefficient of 0.93 ( Fig. 2A ). The average response on the DMSO plates was close to zero, as expected, at −0.36% and 0.6% for the two replicates, respectively, and just two wells flagged as active across both replicates, giving a low false-positive rate of 0.2% and suggesting that there were no artifacts or plate patterns present in the data ( Fig. 2B ).
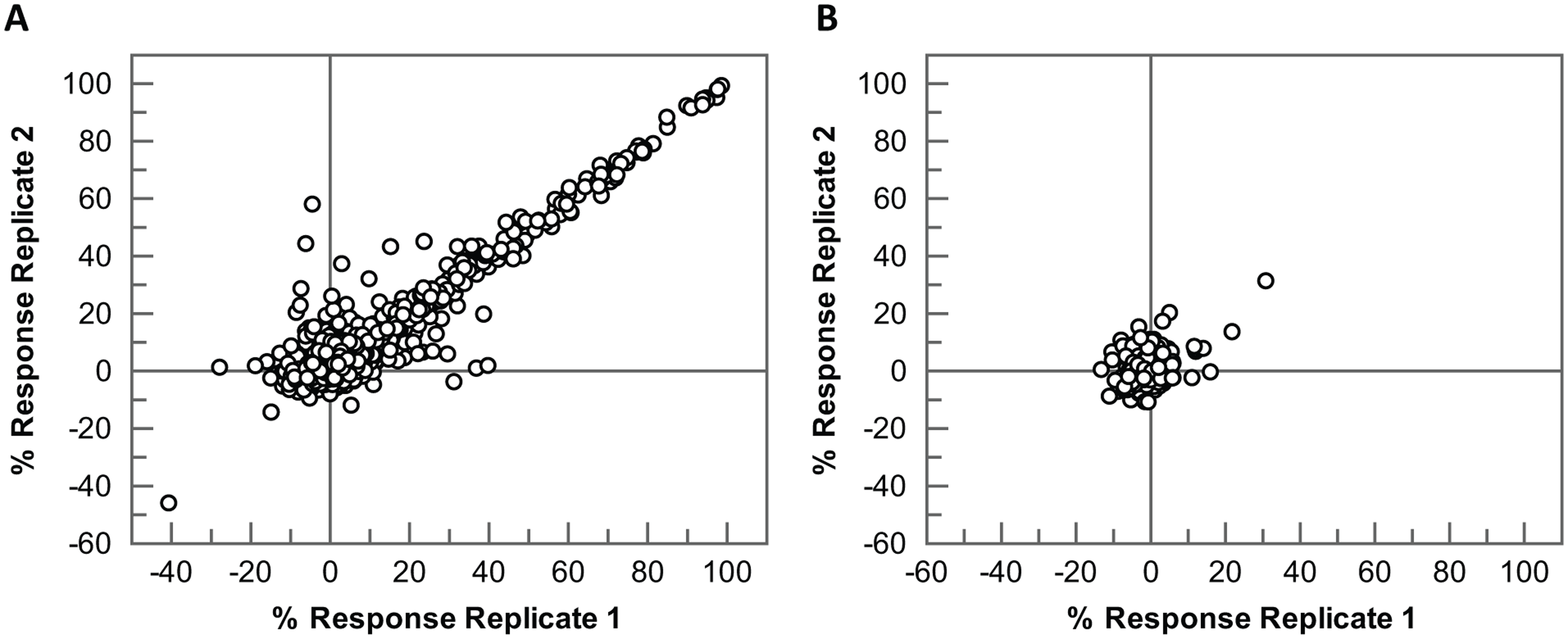
Robustness testing of the LRRK2 RapidFire mass spectrometry assay for single-concentration screening. In total, 1400 compounds (4 × 384-well plates) were screened at a single concentration of 10 µM on two independent test occasions, along with one 384-well DMSO plate on each occasion. (
Compound Screening
Having fully validated the LRRK2 RapidFire assay for single-concentration screening, the platform was used to screen a library of 100,000 compounds, selected according to the criteria described in the Materials and Methods.
The screen was completed as 15 assay runs of 18 plates each and took just 2 weeks to complete using two RapidFire systems. The assay performance was excellent throughout, with all plates giving Z′ > 0.4 and an average Z′ of 0.77 ± 0.11 for the screen as a whole ( Fig. 3A ). The coefficient of variation based on DMSO control wells was ~3% within each run and ~5% between runs. The robust statistical cutoff was calculated for each run as the average sample response plus 3× the standard deviation of the sample response. This varied on a run-by-run basis according to the compounds screened but was consistently below 50% inhibition ( Fig. 3B ). In total, 1985 compounds displaying activity above the robust statistical cutoff were selected for further study, generating a hit rate of 1.99%. Of these, 576 compounds gave greater than 50% inhibition at the 10-µM test concentration. Compounds were then further evaluated to prioritize those that met more stringent physicochemical property criteria thought to be beneficial for a CNS drug candidate. 10 The most active compounds (n = 920) with one or no H-bond donors, two or fewer aromatic rings, and polar surface area less than 100 Å2 were selected for screening as 11-point IC50 curves (4-fold serial dilution from 100 µM top final assay concentration). Overall, the correlation of pIC50 with percent response from single-concentration testing was good ( Fig. 4 ).
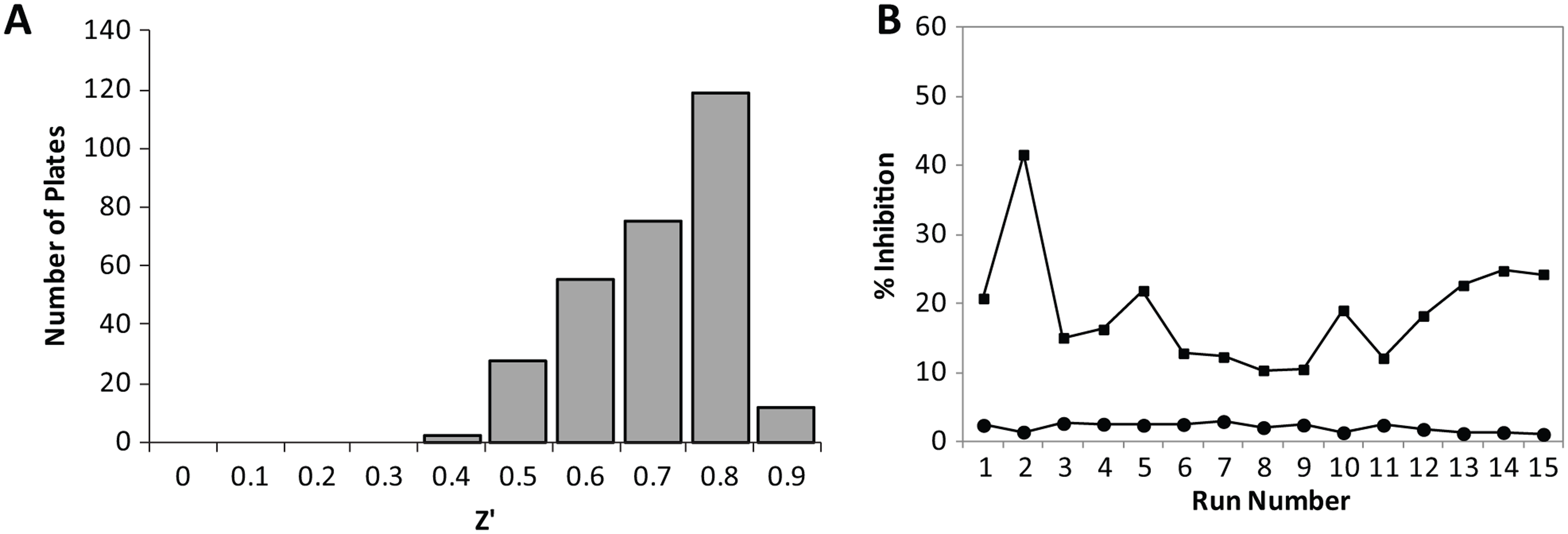
LRRK2 RapidFire screen performance. (
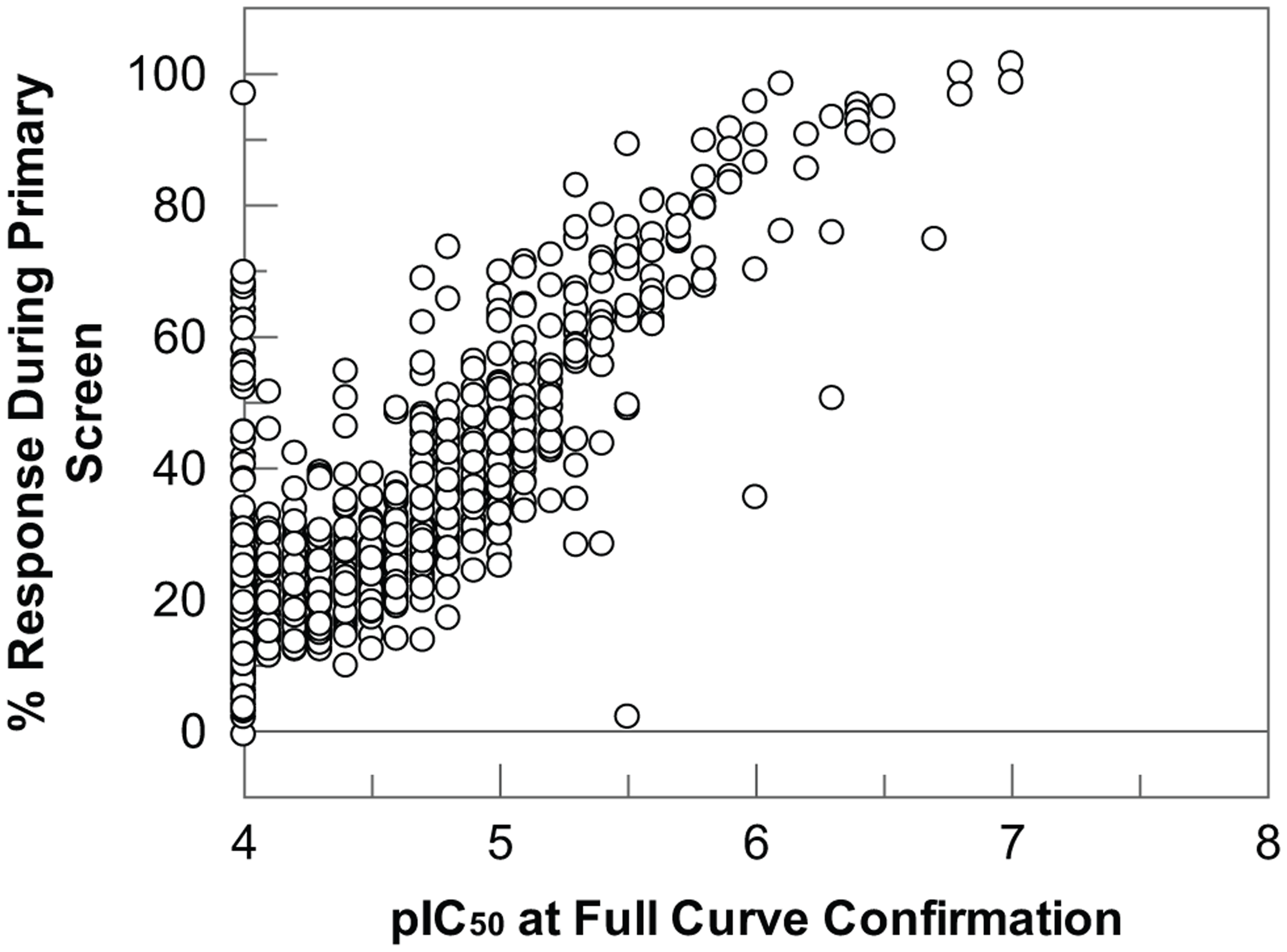
Correlation of percent inhibition in the single-concentration screen vs. pIC50 value determined at full-curve confirmation for all compounds progressed. Compounds that sit on the y-axis at pIC50 4 actually reported <4—that is, they did not reach 50% inhibition at the highest concentration in the full-curve testing and were therefore recorded as inactive.
Compounds of further interest were taken forward for confirmatory assays, resynthesis, and evaluation of predicted binding mode and developability criteria. Some examples of the compounds identified by this process are shown in Table 1 , with their respective IC50 plots from the LRRK2 RapidFire assay shown in Figure 5 . They include many well-known LRRK2 hinge-binding motifs such as 7-azaindole (compound 1), 32 adenine (compound 2), cinnoline (compound 3), 33 indazole (compound 4), 34 and triazolopyridazine (compound 6). 35 Particularly noteworthy was the identification of compound 5, a close analogue of Fasudil, a marketed Rho-kinase inhibitor with LRRK2 activity. 13 In all cases, the ligand efficiencies (LEs, defined as 1.37 × pIC50/HAC) 36 were greater than 0.40, representing attractive starting points for further hit expansion.
Examples of LRRK2 Inhibitors Identified by the RapidFire Screening Platform.
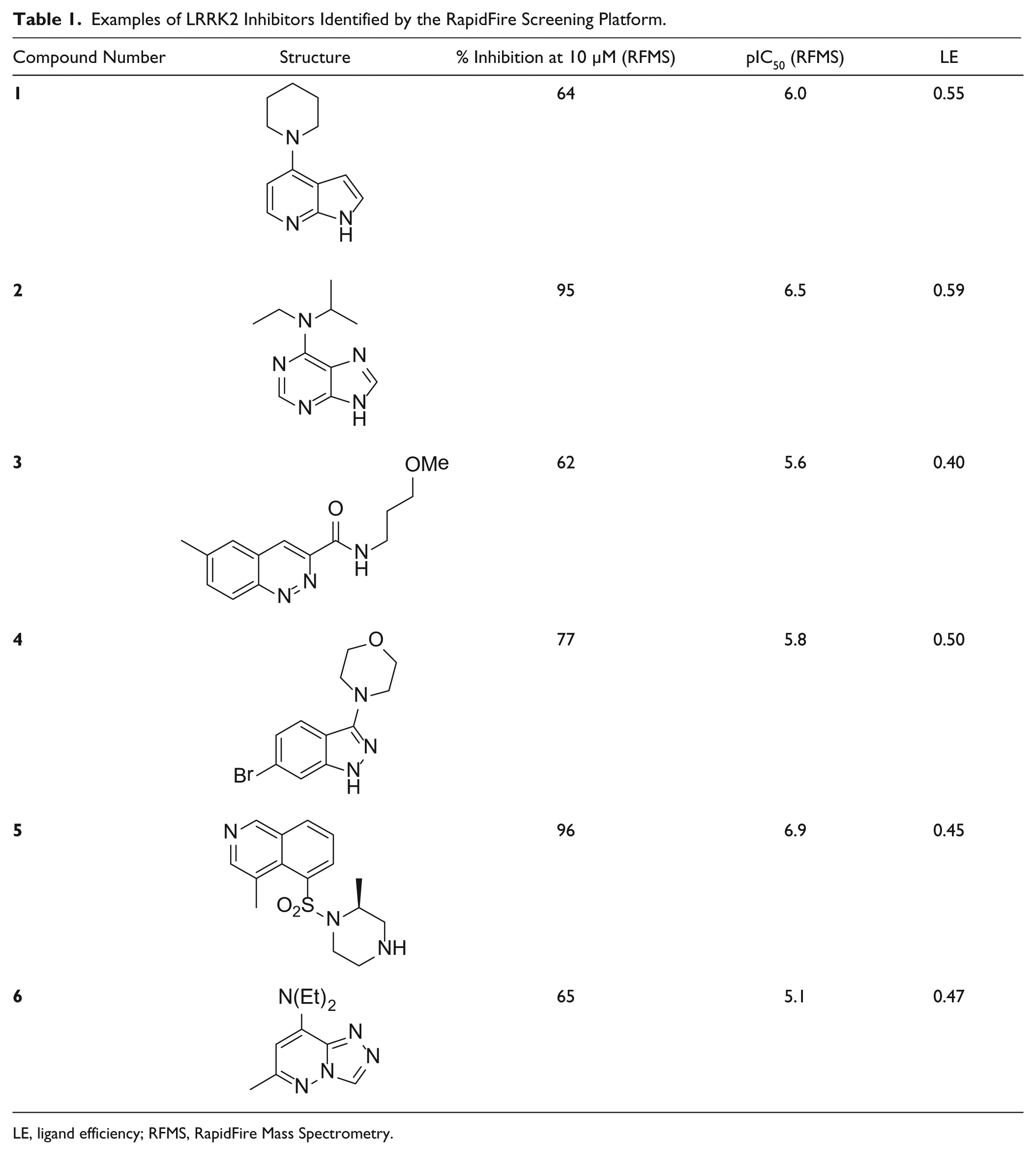
LE, ligand efficiency; RFMS, RapidFire Mass Spectrometry.
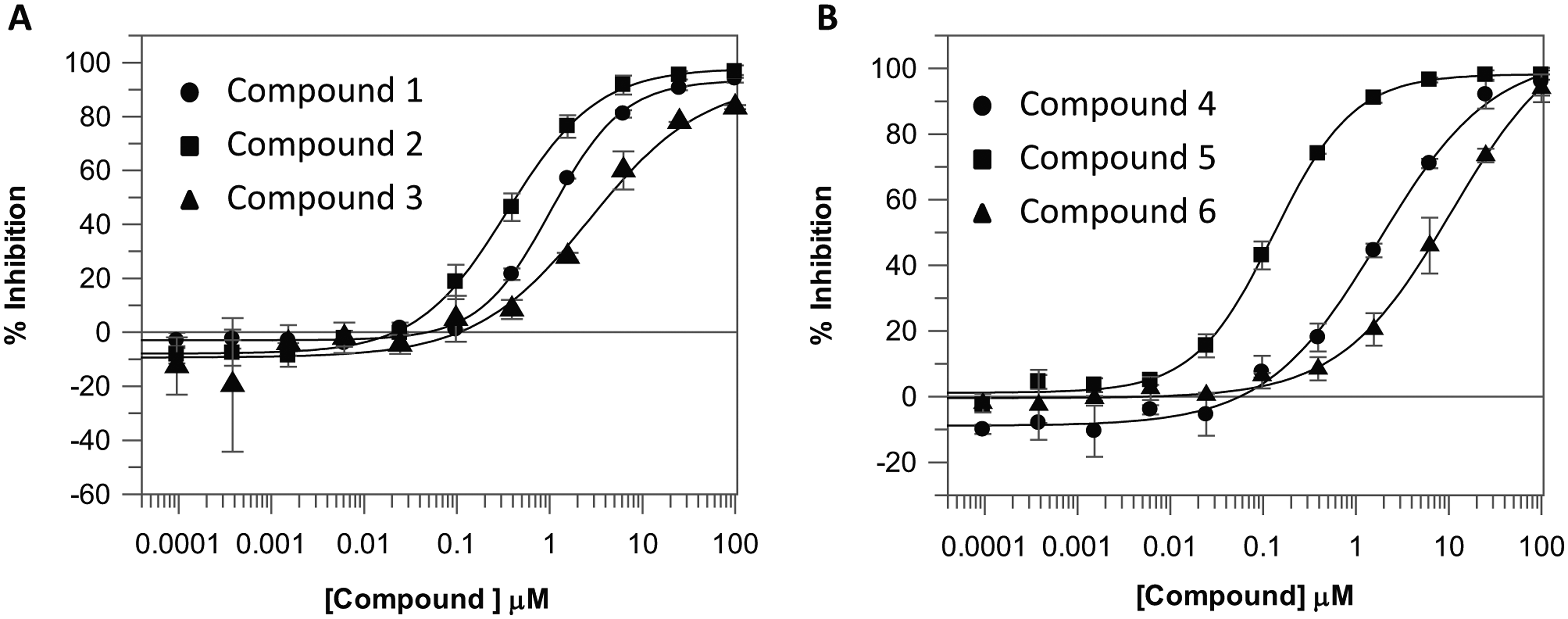
IC50 curves for (
The ability of the RapidFire assay in its current form to detect known LRRK2 binding motifs gives confidence in the output of the screening campaign in this format. The RapidFire platform, with a sampling rate of 10 s per well, is not of sufficient throughput to enable full diversity screening of the entire GSK Compound Collection (~2 million molecules) with single substrates. However, it is possible to use multiplexed peptide assays to increase throughput, 37 which could enable a broader screening campaign against LRRK2. Although the focus of the screening effort described here is related to LRRK2 kinase activity, this protein also contains a GTPase domain, and recently, compounds that bind to this site have been described. 38 One could envisage a multiplexed mass spectrometry–based assay that enables simultaneous monitoring of both kinase and GTPase activities of LRRK2. There is also precedent for using more complex matrices such as cell lysates, blood, or plasma on the RapidFire system, 24 and it may therefore be possible to establish a direct kinase activity assay for LRRK2 on this platform using a more native reagent such as a whole-cell lysate. Screens in a cellular context could overall be more relevant for LRRK2 if membrane association, complex formation, and dimerization are key components of its activity within a cell. 23 Although others have reported cellular readouts for LRRK2, these are generally indirect, monitoring phosphorylation of, for example, Ser935. 16 One would, however, have to carefully control such an assay to ensure specificity for LRRK2. In general, the direct nature of mass spectrometry–based detection lends itself well to setting up multiple assays for a large, complex protein target such as LRRK2, without having to employ a plethora of different detection reagents or antibodies.
In conclusion, we have developed a direct, label-free substrate phosphorylation assay for LRRK2 on the RapidFire platform, using the well-known, moesin-derived LRRKtide peptide as a substrate. The assay is sensitive at nM concentrations of LRRK2 protein and gives robust screening data, while kinetic parameters match well to previously reported assay formats with LRRKtide. We have successfully applied this assay to screen a diverse set of 100,000 compounds in a short timeframe. Compounds were carefully selected with optimal physicochemical properties for a CNS target, and known LRRK2 inhibitors and binding motifs were identified, which gives confidence in the data generated. The RapidFire system offers a number of advantages over conventional fluorescent and indirect detection technologies previously employed for LRRK2, and in the future, the assay described here could be adapted to probe other aspects of LRRK2 function and biology.
Footnotes
Acknowledgements
The authors thank Kelly Locke for compound profiling work following the screen, Qian Liu for input into modeling, Yang Qiu and Robin Carr for useful discussions on strategy, Emma Jones and Stuart Baddeley for review of the manuscript, and many colleagues in GSK’s Platform Technology and Science Unit and Neurosciences Therapy Area Unit who contribute to GSK’s work on LRRK2 drug discovery and development.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.