
Abstract
Recurrent genetic mutations in isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) have been identified in multiple tumor types. The most frequent mutation, IDH1 R132H, is a gain-of-function mutation resulting in an enzyme-catalyzing conversion of α-ketoglutarate (α-KG) to 2-hydroxyglutarate (2-HG). A high-throughput assay quantifying consumption of NADPH by IDH1 R132H has been optimized and implemented to screen 3 million compounds in 1536-well formats. The primary high-throughput screening hits were further characterized by RapidFire–mass spectrometry measuring 2-HG directly. Multiple distinct chemotypes were identified with nanomolar potencies (6–300 nM). All inhibitors were found to be inactive against the wild-type IDH1 homodimers. An IDH1 heterodimer between wild-type and R132H mutant is capable of catalyzing conversion of α-KG to 2-HG and isocitrate to α-KG. Interestingly, one of the inhibitors, EXEL-9324, was found to inhibit both conversions by the IDH1 heterodimer. This indicates the R132H/WT heterodimer may adopt conformations distinct from that of the R132H/R132H homodimer. Further enzymatic studies support this conclusion as the heterodimer exhibited a significantly lower apparent Michaelis-Menten constant for α-KG (Km =110 µM) compared with the R132H homodimer (Km = 1200 µM). The enhanced apparent affinity for α-KG suggests R132H/WT heterodimeric IDH1 can produce 2-HG more efficiently at normal intracellular levels of α-KG (approximately 100 µM).
Introduction
Mutations in isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) have been identified using high-throughput gene sequencing in both gliomas and leukemias.1–3 The most prevalent mutation in IDH1 is a point mutation that results in a change from arginine to histidine (R132H). The active wild-type IDH1 enzyme is a homodimer that converts isocitrate to α-ketoglutarate (α-KG) while reducing NADP+ to NADPH. The mutant R132H IDH1 protein, however, has lost this function. It was originally proposed that mutant IDH1 acts as a dominant negative inhibitor of the wild-type enzyme. 4 However, it was discovered that this mutant protein actually gained a novel catalytic activity, converting α-KG to 2-hydroxyglutarate (2-HG) while oxidizing NADPH to NADP+. 5
The gain-of-function mutation has attracted considerable attention as this altered metabolism may be critical to the survival and proliferation of certain cancer cells. In glioma and leukemia patients with IDH1 or IDH2 mutations, the level of 2-HG is increased by as much as 100-fold.5–7 It is believed that 2-HG interferes with the activity of enzymes that require α-KG as a substrate, possibly via competitive binding, as indicated by the overlapping binding mode in the crystal structures of both α-KG and 2-HG bound to the JmjC domain of histone demethylase CeKDM7A. 8 One class of enzymes that is thought to be inhibited by 2-HG is the methylation enzymes. In both AML and glioma tissue, cells harboring a mutant IDH1 showed a different pattern of DNA hypermethylation compared with its wild-type cohort. 9 In vitro, it has also been demonstrated that TET2, a 5-methylcytosine hydroxylation enzyme, could be inhibited by overexpression of R132H IDH1. 9 It has also been shown that ectopic expression of R132H could inhibit the histone demethylases involved in the demethylation of H3K4, H3K9, H3K27, and H3K79. 10 Finally, prolylhydroxylase domain–containing protein 2 (PHD2) also requires α-KG to function and can be inhibited by 2-HG. 11 PHD2 hydroxylates hypoxia inducible factor 1α (HIF-1α) and allows it to interact with Von Hippel–Lindau disease tumor suppressor protein, which triggers ubiquination and proteosomal degradation pathways. HIF-1α regulates a host of tumor growth and survival mechanisms. Therefore, IDH1 mutation can indirectly affect the expression of multiple genes in a cancer cell. Consistent with this, recent publications indicate that inhibition of IDH1 and IDH2 mutants suppresses tumor initiation and maintenance in AML models and promotes cellular differentiation. 12 Rohle et al. 13 also demonstrated that inhibition of mutant IDH1 delays the growth and promotes differentiation of glioma cells.
Interestingly, cells harboring IDH1 mutations such as R132H are heterozygous in that one copy of IDH1 protein is wild-type whereas the other one is mutated. 2 The existence of the heterodimeric IDH1 protein may suggest the importance of the wild-type reaction in facilitating the mutant reaction. Genetic knock-in experiments introducing an IDH1 R132H encoding mutation into the genome of HCT116 cells demonstrate that the heterozygous IDH1 R132H/WT protein induces genome-wide alterations in DNA methylation. 14 The heterodimeric IDH1 protein may behave differently biochemically as compared with the mutant homodimer.15,16
Therefore, R132H IDH1 has become an intriguing target for cancer therapeutic intervention. To identify novel inhibitors of mutated IDH1, we developed a high-throughput assay by measuring the utilization of NADPH by the R132H IDH1 homodimer via the NADPH-dependent conversion of resazurin to resorufin in the presence of diaphorase. The resazurin-resorufin system has been used frequently in screening enzymes using NAD(P)H-NAD(P)+ as co-factor(s), including IDH1 mutant protein.17–22 Various inhibitors have been reported previously, including one that is currently in human trials against cancers.12,21,22 Here we report a high-throughput screen of more than 3 million compounds and the subsequent finding of novel chemical entities that were specific inhibitors of the R132H IDH1 mutant. As a secondary screen, we developed a biochemical assay that quantifies the formation of 2-HG using the RapidFire–mass spectrometry (RF-MS) system, which provides an intermediate throughput of approximately 9 s per sample. 23 Using RF-MS, we have demonstrated that IDH1 R132H/WT heterodimer is capable of catalyzing both the wild-type reaction and the R132H mutant reaction. It is interesting that the R132H/WT heterodimer possesses a higher affinity toward its substrate α-KG as compared with the R132H homodimer. Remarkably, the R132H/WT heterodimer becomes sensitive to an inhibitor in converting isocitrate to α-KG while the same inhibitor is completely inactive toward the same reaction that is catalyzed by the wild-type homodimer. Our result suggests some intriguing considerations in targeting IDH1 mutants in human cancers in which all mutants known so far exist as heterodimers.
Materials and Methods
Reagents
Unless specified otherwise, all reagents were purchased from Sigma-Aldrich Co. (St. Louis, MO). All solutions used were freshly prepared.
IDH1 Expression and Purification
BL21 (DE3) cells (Invitrogen, Carlsbad, CA), transformed with plasmid containing the full-length IDH1 R132H variant gene with a C-terminal (His)6 tag, were grown to an OD600 of 0.7 to 0.9 at 28 °C and induced with 0.1 mM IPTG. The temperature was shifted to 18 °C, and the culture was allowed to grow for 15 h before harvest. Cells were pelleted by centrifugation at 500g for 30 min. The pellet was frozen in dry ice and ethanol and stored at −80 °C for purification.
Cell pellets were resuspended in ice-cold lysis buffer (50 mM HEPES pH 8.0, 500 mM NaCl, 5% v/v glycerol, 1 mM TCEP, protease inhibitor cocktail) and lysed using a microfluidizer (Microfluidics, Inc., Westwood, MA). Cellular debris was removed by centrifugation at 22,000g for 30 min. Imidazole stock solution (adjusted to pH 7.5) was added to the supernate to a final concentration of 20 mM. IDH1 was purified by batch mode isolation using Ni-NTA (Qiagen, Venlo, the Netherlands) with gentle mixing for 1 h at 4 °C. The resin was collected in a gravity-flow column (Bio-Rad, Hercules, CA) and washed with 20 column volumes (CV) of wash buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 20 mM imidazole). IDH1 was eluted with 5 CV of wash buffer containing 150 mM imidazole. The resulting eluate was further purified in a Superdex 200 column (GE Life Sciences, Pittsburgh, PA) equilibrated with 50 mM HEPES pH 7.5, 300 mM NaCl, 5 mM DTT. Fractions containing IDH1 were pooled and concentrated using an Amicon Ultra concentrator (10 kDa cutoff; Millipore, Billerica, MA). The protein was buffer exchanged to storage buffer (50 mM HEPES pH 7.5, 200 mM NaCl, 10% glycerol, 3 mM DTT) using a PD-10 column (GE Life Sciences) and stored at −80 °C. The IDH1 R132H mutant was confirmed by gene sequencing and peptide mapping.
Purification of heterodimers (wild-type variant tagged with a C-terminal [His]6 and R132H variant tagged with a C-terminal FLAG tag) followed the same initial Ni-NTA capture as above. The heterodimer was subsequently buffer exchanged to binding buffer (50 mM HEPES, pH 7.5, and 300 mM NaCl) and isolated by batch purification using anti-FLAG agarose (Sigma-Aldrich) with gentle mixing for 3 h at 4 °C. The resin was collected in a gravity flow column and washed with 20 CV of binding buffer. Elution was achieved by 5 CV of binding buffer containing 0.1 mg/mL FLAG peptide (Sigma-Aldrich). The protein was then further processed in the same manner as above. Dimer formation was confirmed by static light-scattering analysis through an SEC-250 column (Bio-Rad) connected to a Mini-Dawn (Wyatt, Santa Barbara, CA).
Characterization of IDH1 Reactions by RF-MS
RapidFire system setup
The RF-MS system consists of a RapidFire RF200 system (Agilent, Santa Clara, CA) interfaced with an API4000 mass spectrometer (AB Sciex, Foster City, CA), as described before. 17 A Zymark Twister robotic arm is present to handle standard microtiter plates. The entire system is run with RapidFire software and Analyst software for the RF200 system and the mass spectrometer, respectively.
The mobile phase consisted of 1 mM tribytylamine and 1.5 mM acetate in water (solvent A) and 100% methanol (solvent B). Samples were aspirated directly from 384-well plates into a 10 µL sample loop and passed through an in-line purification SPE system with C4 cartridges (Agilent) with solvent A at a flow rate of 1.5 mL/min for 3 s. After the desalting step, the analyte retained on the cartridge was eluted to the mass spectrometer with solvent B at a flow rate of 0.4 mL/min for 5 s. The cartridge was reequilibrated with solvent A at a flow rate of 1.5 mL/min for 0.5 s. In total, the entire sampling cycle was 9 s per well.
The IDH1 wild-type reaction (isocitrate to α-KG)
The typical reaction mixture contained 1 mM isocitrate incubated with 5nM R132H/WT heterodimer, 2.5 nM WT/WT homodimer, or 2.5 nM R123H/R132H homodimer in the presence of 1 mM NADP+ in the assay buffer (buffer A) containing 50 mM Tris (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 0.3% BSA, and 0.01% Brij-35 for 1 h at room temperature. The assay was conducted in 384-well microtiter plates with a final reaction volume of 20 µL unless specified otherwise. The reaction was stopped by 100% methanol with vigorous mixing. The reaction mixture was then injected into an API 4000 mass spectrometer by the RapidFire sampling system. The product, α-KG, was detected by RF-MS using multiple reaction monitoring (MRM) mode at a transition from 144.7 to 100.84 in mass-to-charge ratio. The formation of 2-HG was also monitored using MRM (transition from 146.661 to 102.676) to investigate enzyme activities to convert isocitrate to 2-HG.
To determine the apparent Km and kcat of isocitrate, concentrations of isocitrate (up to 1 mM) were varied in the IDH1 wild-type reaction. Isocitrate was incubated with 1 mM NADP+ in the above buffer for 1 h. The reaction was stopped and product (α-KG) was detected by RF-MS as described above.
IDH1 mutant reaction (α-KG to 2-HG)
The typical reaction mixture contained 1 mM α-KG incubated with 1 mM NADPH and 5 nM R132H/WT heterodimer, 2.5 nM WT/WT homodimer, or 2.5 nM R123H/R132H homodimer in buffer A for 1 h. The reaction mixture was processed as described above. The product (2-HG) was detected using RF-MS.
To determine the apparent Km and Kcat of α-ketoglutarate, concentrations of α-KG (up to 2 mM) were varied in the IDH1 R132H mutant reaction, as described above. The reaction was stopped at various times with 100% methanol, and product was detected using RF-MS.
α-KG Apparent Km and Kcat determination
Concentrations of α-KG (up to 2 mM) were varied in the IDH1 R132H mutant reaction, as described above. The product (2-HG) was detected using RF-MS as described above.
NADP+ apparent Km and Kcat determination
NADP+ at various concentrations was incubated with 1 mM isocitrate and either 5 nM R132H/WT heterodimer or 2.5 nM WT/WT homodimer in the above buffer for 1 h. The reaction was stopped, and product (α-KG) was detected using RF-MS.
NADPH Km and Kcat determination
NADPH at various concentrations was incubated with 2 mM α-KG and either 5 nM R132H/WT heterodimer or 2.5 nM R132H/R132H homodimer in the above buffer. The reaction was stopped at various times with 100% methanol, and product was detected using RF-MS.
IDH1R132H Homodimer High-Throughput Assay
The high-throughput screening (HTS) campaign was performed using an integrated automated assay platform that has been described previously. 18 The assays were conducted in a 1536-well microtiter plate format with a final compound concentration of 5 µM in a total reaction volume of 6 µL. Compound library samples for HTS application were orthogonally pooled as mixtures of 10 compounds per well, with duplicate representation for each compound in columns 3 to 22 of 384-well plates. Compound plates were diluted with the assay buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 0.3% BSA, 0.01% Brij-35, 20 µM NADPH, and 400 µM α-KG using the Flexdrop liquid dispenser to a compound concentration of 10 µM. Aliquots of 3 µL from four 384-well compound plates were transferred to a 1536-well white medium binding polystyrene plate (Greiner Bio-One, Monroe, NC) using a PlateMate Plus. A 3 µL volume of 5 nM IDH1 R132H homodimer was added to columns 5 to 48 using a Flexdrop dispenser. Columns 1 to 4 received an equal volume of buffer in the absence of the enzyme. The final assay wells contained 2.3 nM IDH1 R132H mutant protein, 200 µM α-KG, 10 µM NADPH, and 5 µM compounds. Each assay plate was mixed gently and subsequently centrifuged in the VSpin for 10 s before it was placed in a Cytomat incubator at 25 °C for 60 min. After the incubation, a 3 µL volume of detection solution containing 10 µM resazurin, 10 µg/mL diaphorase, and 0.1% Tween 20 was added, and the fluorescence was read on EnVision with an excitation wavelength filter of 530 nm (30 nm band width) and emission wavelength filter of 590 nm (30 nm band width).
HTS Data Analysis
A total of 3.0 million compounds in approximately two thousand 1536-well plates were evaluated in the IDH1 R132H HTS campaign. Assay Analyzer, data management software from Genedata Screener (Basel, Switzerland), was used to facilitate the quality control and analysis of the screening data, as described previously. 24 In general, raw plate data was imported into Assay Analyzer at the end of each daily run and inspected for any unexpected systematic patterns due to liquid handling. Sample wells exhibiting 30% inhibition or higher (more than three times the standard deviation of sample wells) were identified and deconvoluted for primary hits.
Characterization of IDH1 Inhibitors
Some modifications to the HTS protocol were used for biochemical characterization of the primary hits. All experiments were performed in the 384-well format with proportionally larger volumes (20 µL in most cases) or as indicated in the figure legends. The deconvoluted HTS hits were cherry-picked from the original stock solutions and tested at 10 µM concentration in triplicate by the RF-MS method. The confirmed cherry-pick hits were characterized in a 10-point dose-response study to generate the compound concentration required to yield 50% IDH1 inhibition (IC50). Generally, 0.5 µL DMSO containing varying concentrations of the test compound (ranging from 0 to 160 µM) was mixed with 20 µL of 2.3 nM IDH1 R132H mutant protein, 200 µM α-KG, and 10 µM NADPH. The nonlinear fits to the dose-response curves and to the Michaels-Menton enzyme kinetic curves were performed using Prism 4.0 (GraphPad, San Diego, CA).
Mechanism-of-Action Studies of Inhibitors
EXEL-9324 inhibition of the α-KG to 2-HG reaction
EXEL-9324 at various concentrations was mixed with buffer containing 10 µM NADPH, 1 mM α-KG, and either 5 nM R132H/WT heterodimer or 2.5 nM R132H/R132H mutant homodimer and incubated for 1 h. Reaction was stopped with 100% methanol, and product was measured using RF-MS. Substrate competition studies were performed by varying the concentrations of NADPH or α-KG. Apparent inhibition constant (Ki) against α-KG was determined from the values of IC50 at different concentrations of α-KG.
EXEL-9324 inhibition of the conversion of isocitrate to α-KG and 2-HG
EXEL-9324 at various concentrations was mixed with buffer containing 0.5 mM NADP+, 0.2 mM isocitrate, and either 5 nM R132H/WT heterodimer or 2.5 nM WT/WT homodimer for 1 h. Reaction was stopped with 100% methanol, and product (α-KG and 2-HG) was measured using RF-MS. Substrate competition studies were performed by varying the concentrations of isocitrate. Apparent Ki against isocitrate was determined from the values of IC50 at different concentrations of isocitrate.
Results and Discussion
Characterization of R132H IDH1 Mutant Proteins Using RF-MS
Purified R132H mutant IDH1 homodimer catalyzes the formation of 2-HG from α-KG in the presence of NADPH, assessed by directly measuring 2-HG using RF-MS ( Fig. 1A ), whereas the wild-type IDH1 exhibits negligible activity. Interestingly, the R132H/WT heterodimer can catalyze this conversion as well ( Fig. 1A ). In addition, the heterodimer can catalyze the wild-type reaction, converting isocitrate to α-KG in the presence of NADP+ ( Fig. 1B ); subsequently, 2-HG is produced ( Fig. 1C ).
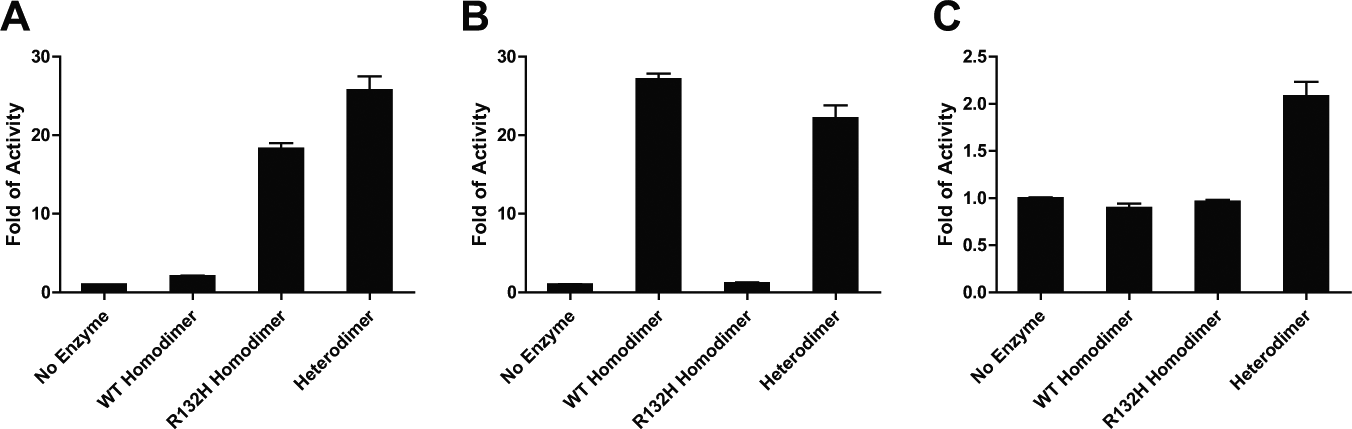
Enzymatic activities of wild-type isocitrate dehydrogenase 1 (IDH1) homodimer, R132H IDH1 homodimer, and R132H/WT heterodimer as measured by fold increase in product formation quantified using RapidFire–mass spectrometry. (
Further enzyme kinetic measurement reveals that the R132H/WT heterodimer has similar apparent Km values for NADPH as compared with the R132H mutant homodimer but significantly different Km values for α-KG ( Fig. 2A ). As listed in Table 1 , the Km for α-KG is 110 ± 20 µM for the R132H/WT heterodimer, approximately 11-fold lower than that for the R132H/R132H homodimer. The apparent rate constant (kcat) appears to be the same. Table 1 summarizes the kinetic parameters of the 2 reactions for different dimers. The enhanced apparent affinity for α-KG suggests R132H/WT heterodimeric IDH1 can produce 2-HG, an onco-metabolite, more efficiently at normal intracellular levels of α-KG (approximately 100 µM). 5 Thus, the heterodimer IDH1 may potentially be more potent in tumor initiation, consistent with the clinical observation that all IDH1 mutations in tumors are heterozygous. 2 When isocitrate is used as the substrate, the R132H/WT IDH1 heterodimer can catalyze the production of α-KG, with similar Km values for isocitrate as compared with the wild-type IDH1 homodimer enzyme ( Fig. 2B ; Table 1 ). The relative scatter in the rate determination may be due to interference in RF-MS from high concentrations of isocitrate (data not shown). The Km values for wild-type IDH1 and R132H/R132H mutant homodimer generally agree with the reported literature values that appeared to vary significantly among various laboratories, sometimes by one order of magnitude. For example, the reported Km values of NADP+ for the wild-type IDH1 ranged from 4.7 to 49 µM.4,5,15 For the R132H/R132H mutant homodimer, the reported Km values of NADPH ranged from 0.044 to 0.44 µM.5,21 The reported Km values of α-KG for the R132H/R132H mutant homodimer have been more consistent, ranging from 820 to 965 µM.5,21 Various assay conditions, including detection methods, protein preparation, and buffer conditions, may have contributed to the large variation in Km values.
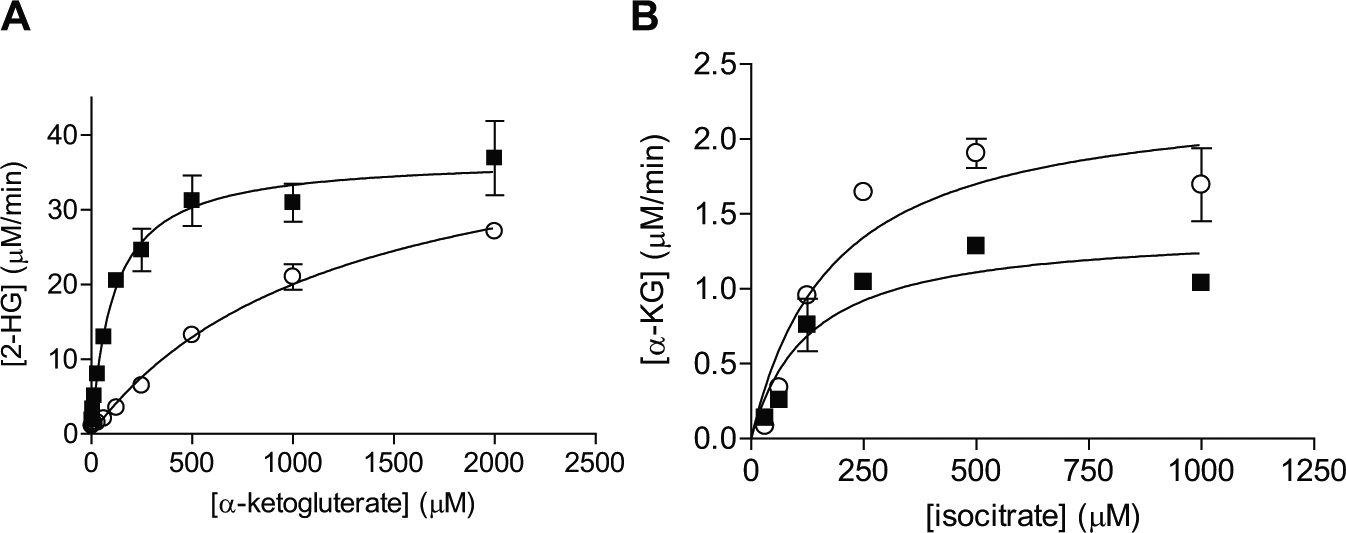
(
Apparent Michaelis-Menten Constants for the R132H/R132H Homodimer and R132H/WT Heterodimer with Various Substrates (µM).

n.a. = no observed activity; IDH1 = isocitrate dehydrogenase 1.
Lowest concentration tested in the experiment
HTS against IDH1 R132H Homodimer
To identify small-molecule inhibitors of the R132H IDH1 mutant protein, an HTS against the Exelixis compound collection was performed in March 2010. A coupled assay measuring the utilization of NADPH was developed by monitoring the NADPH-dependent conversion of resozurin to resorufin by diaphorase. This same format has been used for IDH1 R132H mutant by other groups, most recently by the National Institutes of Health and Broad Institute. Although the IDH1 R132H/WT heterodimer might be a more biologically relevant target, the R132H/R132H homodimer was selected as the target in the HTS campaign based on the fact that the homogeneous assay measuring the level of NADPH is not suitable when both wild-type and mutant reactions occur concurrently. Moreover, any potential hits against the R132H/R132H homodimer can be further characterized against the wild-type/R132H heterodimer.
The HTS assay was initially developed in 384-well microtiter plates.
Figure 3
shows the fluorescent signal decreases over time (labels on the left y axis), indicating the depletion of NADPH, for the reaction with the mixture of 200 µM α-KG, 10 µM NADPH, and 5 nM R132H IDH1 homodimer. The decrease in NADPH is correlated well with the production of 2-HG, as monitored by RF-MS (
Fig. 3
, labels on the right y axis). An enzyme titration at 1 h performed in 1536-well microtiter plates demonstrated a linear range up to 3 nM R132H IDH1 homodimer (
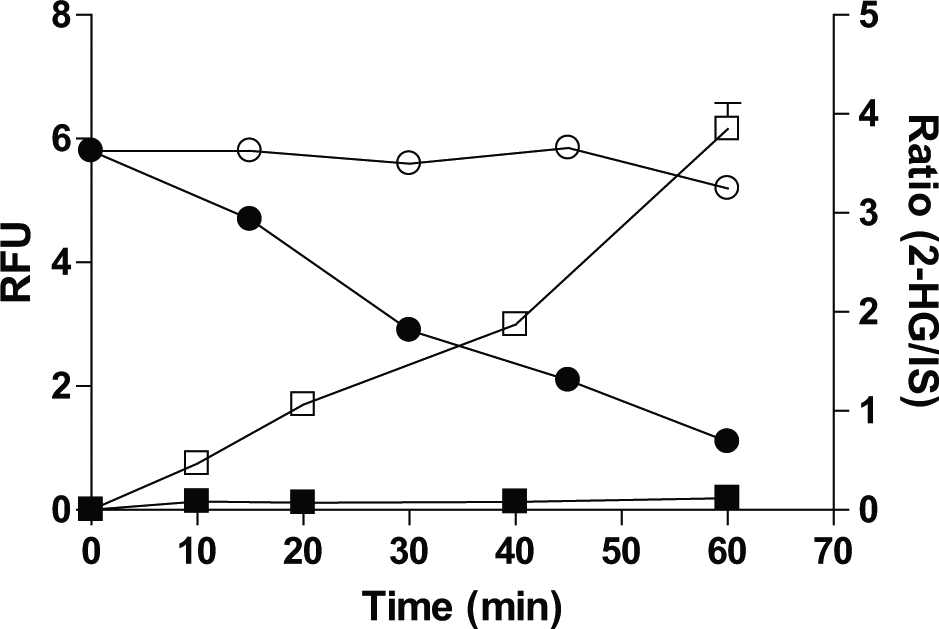
Time course of NADPH depletion as monitored in the coupled assay in the absence (O) and presence (●) of isocitrate dehydrogenase 1 (IDH1) R132H homodimer, as shown with the left x axis in a 384-well format. Time course of 2-hydroxyglutarate formation as monitored in RapidFire–mass spectrometry in the absence (■) and presence (□) of IDH1 R132H homodimer as shown with the right x axis in 384-well format.
The HTS assay flow chart is shown in
Summary of High-Throughput Screening Statistics.
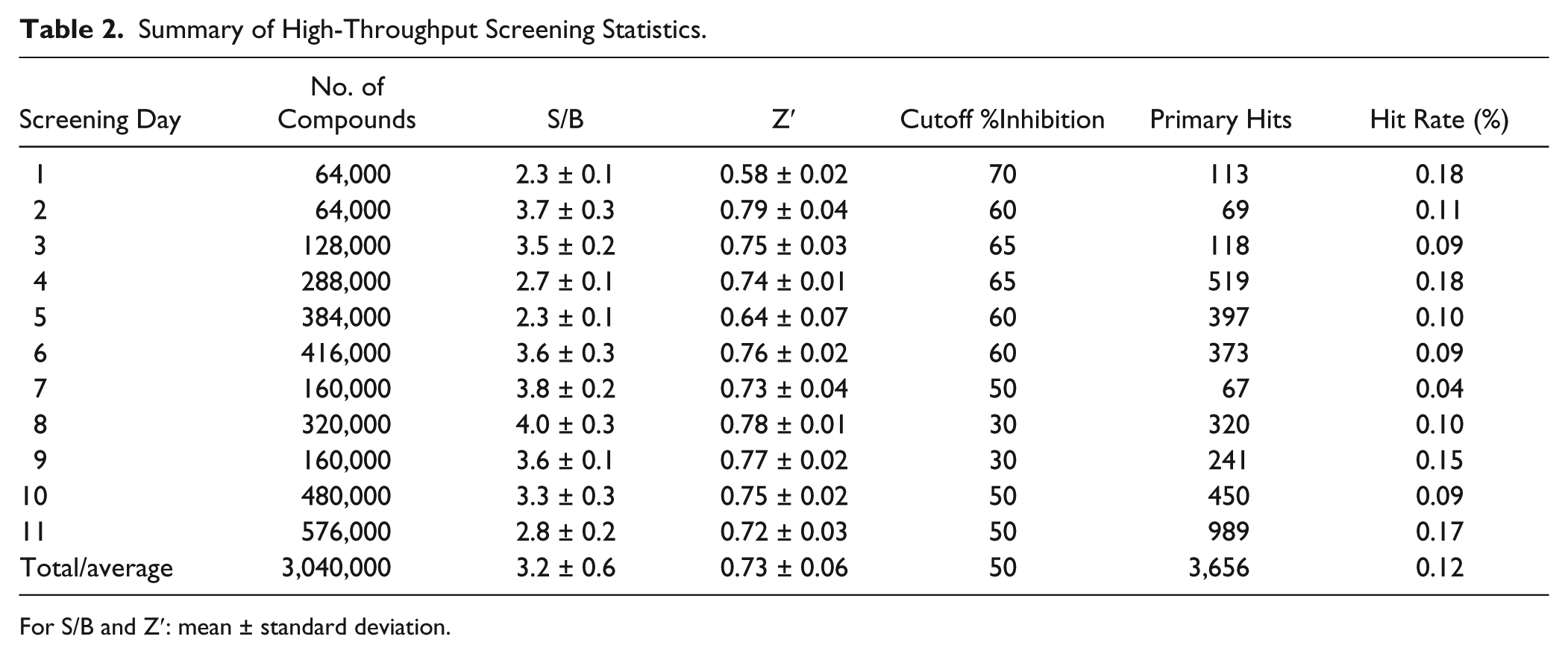
For S/B and Z′: mean ± standard deviation.
The primary hits were individually cherry-picked from the DMSO stock solutions and tested at 10 µM in the RF-MS format. Among 680 compounds with 50% of more inhibition, 160 unique compounds were selected for 10-point dose-response determinations, based on molecular weight (between 200 and 600 daltons) and structural diversity, judged by a simplified cyclic assembly approach.
25
The IC50 values correlated well with single-point inhibition (
Mechanism of Inhibition of Selected Inhibitors
At least one chemical series was investigated further through focused SAR efforts. Here we present one lead compound from this series, EXEL-9324. When tested with varying concentrations of the substrates, EXEL-9324 was found to be competitive against α-KG but not NADPH (
The value of apparent Ki for EXEL-9324 against the R132H homodimer while varying α-KG is 298 nM as determined from the slope of the linear curve between IC50 and α-KG concentration ( Fig. 4A ). Since EXEL-9324 was identified in an HTS campaign against the R132H homodimer, its activities against the R132H/WT heterodimer and WT IDH1 were subsequently investigated. As expected, EXEL-9324 was found to inhibit the formation of 2-HG catalyzed by the R132H/WT heterodimer with an apparent Ki value of 72 nM against α-KG ( Fig. 4B ). This tighter binding of EXEL-9324 to the R132H/WT heterodimer is consistent with the tighter binding of α-KG to the heterodimer. EXEL-9324 was inactive against the formation of α-KG catalyzed by the WT IDH1 protein ( Fig. 4C ). Surprisingly, EXEL-9324 showed dose-dependent inhibition against the same reaction catalyzed by the R132H/WT heterodimer IDH1 protein with an IC50 value of 800 ± 50 nM ( Fig. 4C ). This observation would also be consistent with little to no wild-type IDH1 homodimer present in the purified heterodimer protein because EXEL-9324 has no inhibitory effect on the wild-type IDH1 enzyme ( Fig. 4C ). The Ki for EXEL-9324 against the R132H/WT IDH1 heterodimer while varying isocitrate is 298 nM ( Fig. 4D ), similar to the Ki value against the R132H homodimer while varying α-KG.
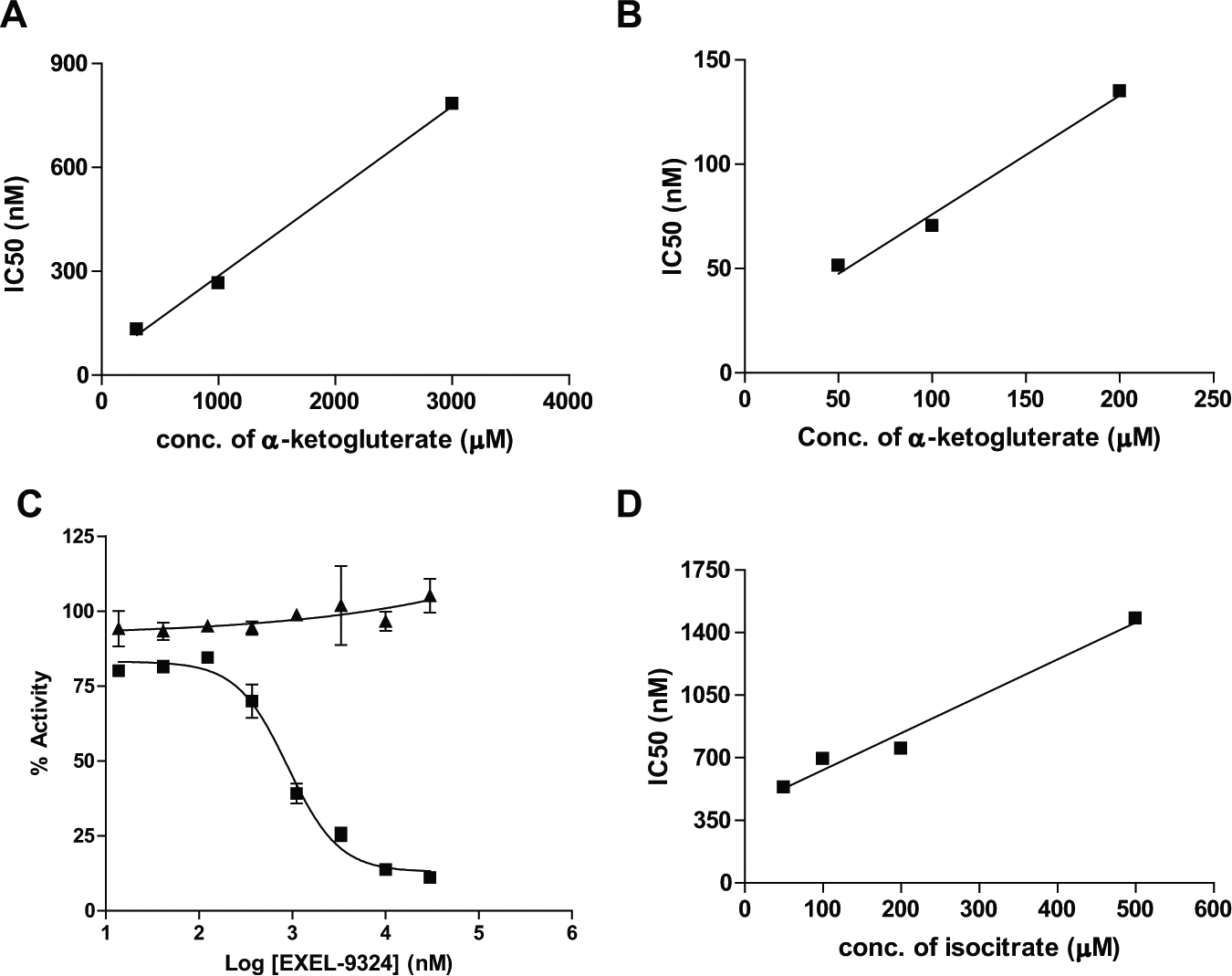
Characterization of the mechanism of inhibition for EXEL-9324. (
In conclusion, HTS of 3 million compounds yielded multiple chemical scaffolds that inhibit the IDH1 R132H mutant enzyme in its conversion of α-KG to 2-HG. When tested, these inhibitors exhibited no inhibition against the wild-type IDH1 in its conversion of isocitrate to α-KG. Among the inhibitors that were tested against the substrates, NADPH or α-KG, it is interesting to note that they are all competitive inhibitors against α-KG, not NADPH. This may point to the possibility that inhibitors of NADPH are relatively rare in the Exelixis compound collection. Yet it is also consistent with the original design of the HTS assay to be performed at the NADPH concentration that is higher than its Km value. Other inhibitors have also been reported to be competitive against α-KG, not NADPH,21,22 indicating a bias in developing a small-molecule inhibitor of IDH1 mutant targeting the α-KG binding site. One of the inhibitors, EXEL-9324, was shown to inhibit the conversion of isocitrate to α-KG catalyzed by the heterodimeric R132H/WT IDH1 but not by the homodimeric WT/WT IDH1, indicating the conformation of the active sites may be different among various dimeric arrangements. The difference in enzyme kinetics between the R132H/WT heterodimer and the R132H mutant homodimer further supports this notation, indicating a higher affinity toward α-KG for the physiologically relevant heterodimer. This lower Michaelis-Menten constant for α-KG may provide a metabolic advantage, namely, an enhanced ability to use cellular α-KG, which has been reported to be in the 100 µM range, 5 to produce 2-HG, for cells harboring one copy of the R132H mutation. The identification of inhibitors selective for the IDH1 R132H mutant protein provides useful tool compounds for further assessing the role of IDH1 R132H in tumor development.
Footnotes
Acknowledgements
We wish to thank all members of Exelixis Lead Discovery for assistance in the HTS, Exelixis Medicinal Chemistry for compound resynthesis, Dr. Peter Lamb for comments on the manuscript and discussions, and the reviewers for their insightful comments.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.