
Abstract
The clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system has been seized upon with a fervor enjoyed previously by small interfering RNA (siRNA) and short hairpin RNA (shRNA) technologies and has enormous potential for high-throughput functional genomics studies. The decision to use this approach must be balanced with respect to adoption of existing platforms versus awaiting the development of more “mature” next-generation systems. Here, experience from siRNA and shRNA screening plays an important role, as issues such as targeting efficiency, pooling strategies, and off-target effects with those technologies are already framing debates in the CRISPR field. CRISPR/Cas can be exploited not only to knockout genes but also to up- or down-regulate gene transcription—in some cases in a multiplex fashion. This provides a powerful tool for studying the interaction among multiple signaling cascades in the same genetic background. Furthermore, the documented success of CRISPR/Cas-mediated gene correction (or the corollary, introduction of disease-specific mutations) provides proof of concept for the rapid generation of isogenic cell lines for high-throughput screening. In this review, the advantages and limitations of CRISPR/Cas are discussed and current and future applications are highlighted. It is envisaged that complementarities between CRISPR, siRNA, and shRNA will ensure that all three technologies remain critical to the success of future functional genomics projects.
Background and Introduction
Bacteria and archaea defend themselves from mobile genetic elements (such as phages and plasmids) by recognizing and then degrading their DNA. Entering foreign DNA is bound by constitutively expressed host RNAs that are complementary to the incoming sequence; these RNAs concomitantly recruit a host endonuclease that cleaves the DNA. 1 In Streptococcus pyogenes, the Cas9 endonuclease is responsible for processing of the “surveillance” RNAs and for performing DNA cleavage (see Fig. 1 ). The system constantly evolves, as the cleaved fragments of foreign DNA are incorporated into the clustered regularly interspaced short palindromic repeats (CRISPR) locus. In turn, transcription of these fragments yields new surveillance RNAs (called CRISPR-RNAs, or crRNAs) in a type of bacterial adaptive immune response, so that previously encountered pathogens of bacteria are eliminated. There are many variants of the generic theme presented above. For example, a minimum of 75 Cas9 potentially functional orthologs were found among ~200 bacterial species and many more individual crRNAs. 2 This is just the tip of the iceberg, as CRISPR-Cas9 is just one of several class II subtypes; class I and III systems each contain many other types of endonuclease/RNA permutations.3,4 Although studied extensively in the virology and bacteriology fields for many years, this RNA-guided DNA cleavage system has now been widely adopted by the entire life science community, and novel applications of the CRISPR-Cas systems are appearing at a breakneck pace. In this review, the major focus will be the use of CRISPR-Cas9, although other endonucleases are being used for specific applications.5–7
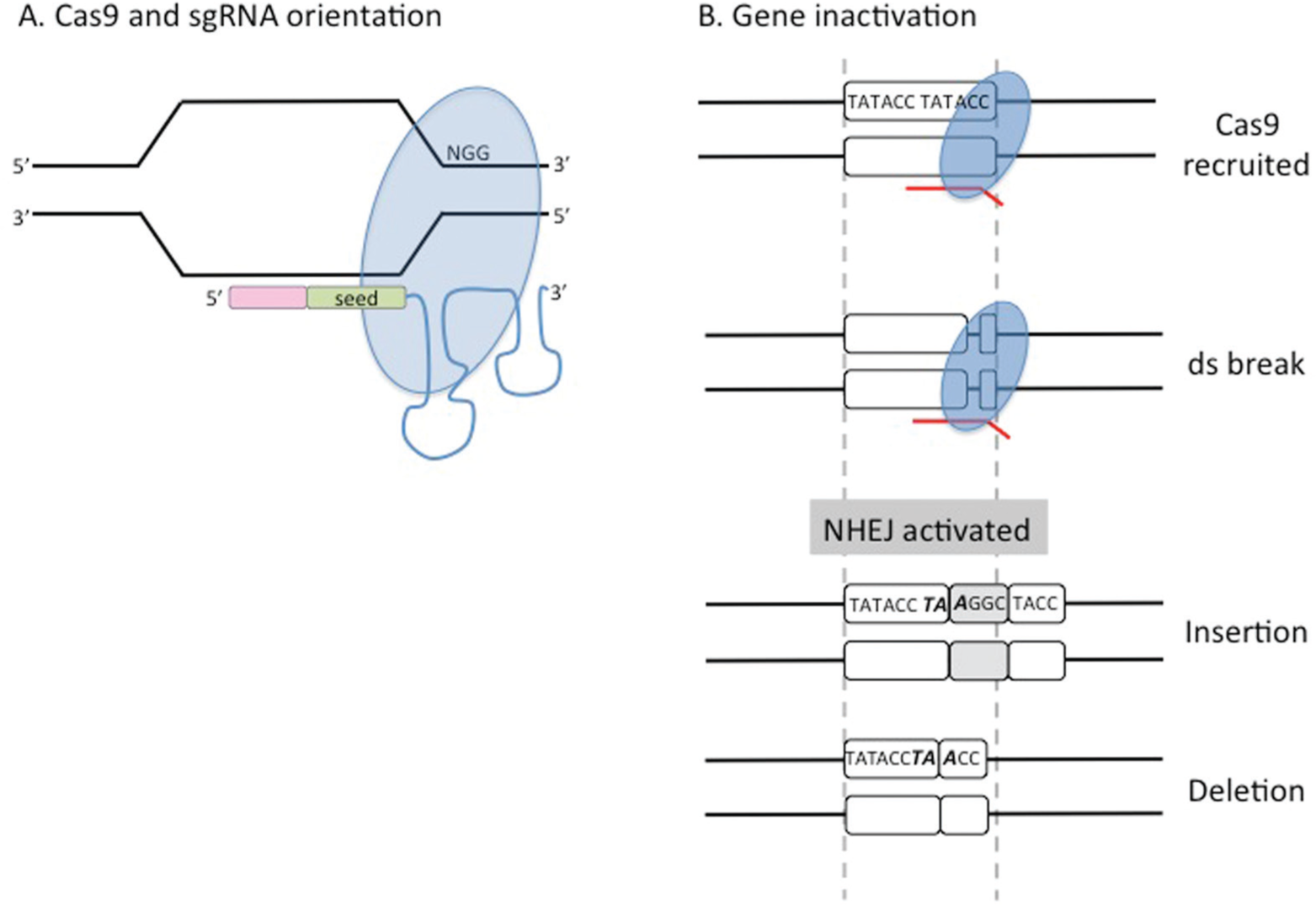
Schematic of Cas9-dependent DNA cleavage. (
From a screening perspective, the selection of the appropriate CRISPR/Cas technology (and the subsequent design and execution of the project) depends on the type of question being asked. Broadly speaking, most research groups considering using genome editing in a screening context will have one of two different goals. The first is for high-throughput screening to identify genes involved in a particular biological process. The second is to use the technology to construct cell lines that harbor disease-specific genetic alterations or that contain cell signaling network–specific reporters.
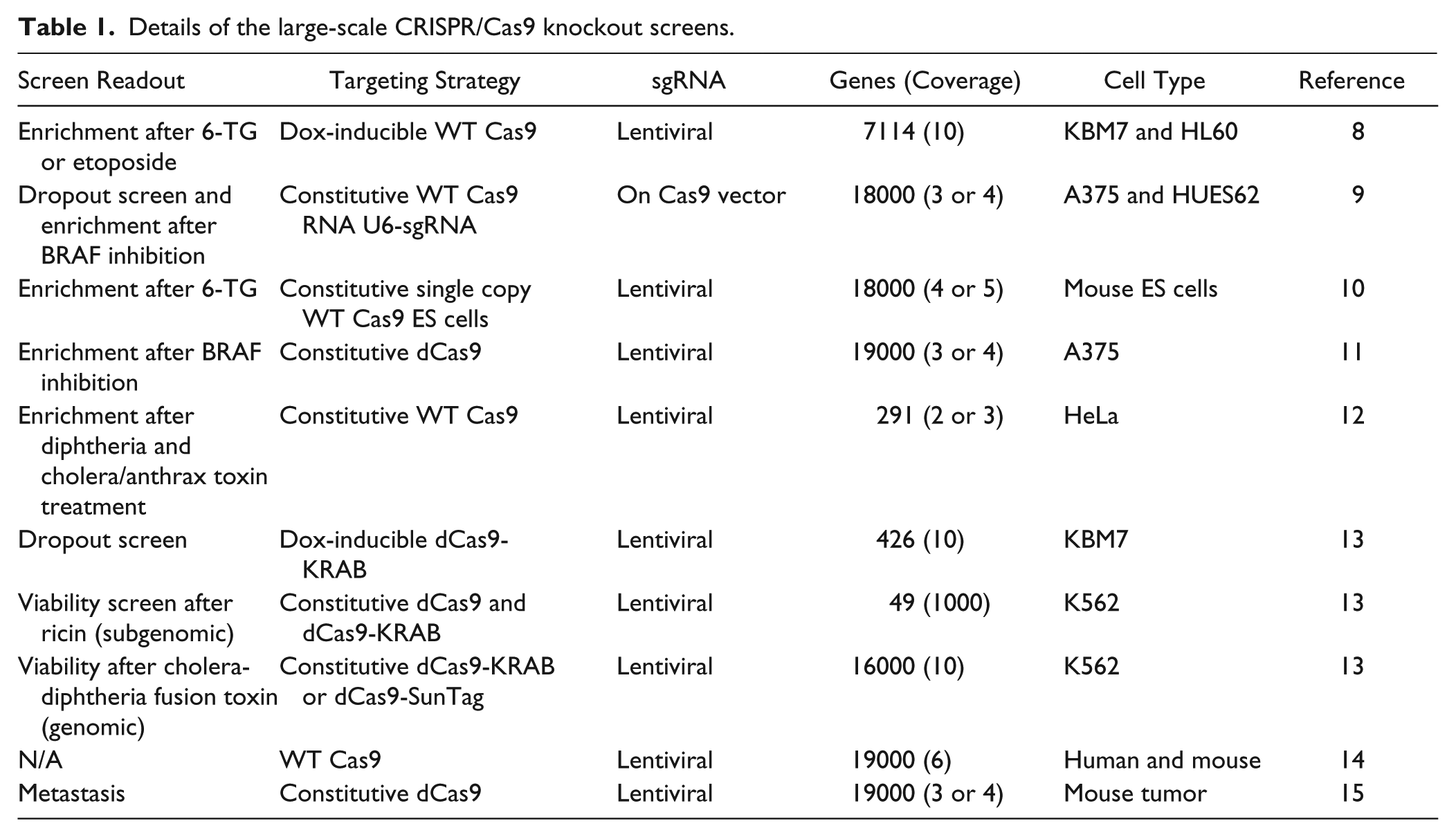
The use of CRISPR/Cas9 in a high-throughput screening context has, to date, been generally restricted to a pooled approach (see Table 1 for a summary). This in some regards follows the well-beaten path used previously for high-throughput lentiviral short hairpin RNA (shRNA) screens. The power of the pooled approach is clear: hundreds or thousands of genes can be interrogated in a single experiment at relatively low cost. However, the very nature of pooled screens requires some type of enrichment procedure to identify novel targets for follow-up studies. Thus, the pooled strategy is particularly suitable for synthetic lethality screens; here, the aim is to identify genes that modulate cell viability in response to cytotoxic molecules or in the context of oncogene activation and/or tumor suppression. By contrast, the pooled approach is not suitable for any type of image-based phenotypic screen that requires individual cell resolution. This is because the identification of genes in pooled screens requires expansion and then lysis of surviving cells prior to decoding molecular barcodes. Initially, this deconvolution stage was performed using microarray technology, but this is likely to be wholly replaced by next-generation sequencing-based methods in the future. Therefore, image-based screening using CRISPR/Cas9 will require an arrayed format, in which only one gene is targeted in each individual well of an assay plate. A particularly powerful alternative use of CRISPR/Cas9 in a high-throughput context is to use the technology to generate isogenic cell lines. These can then be used in combination with other technologies to perform functional genomic studies. This will be discussed in more detail in the “CRISPR/Cas9 and Other Editing Technologies” section.
Details of the large-scale CRISPR/Cas9 knockout screens.
System Choices
For loss-of-function studies, there are several CRISPR/Cas9 systems available. In each case, Cas9 is directed to genomic loci by Watson-Crick base pairing mediated via an RNA moiety known as the single-guide RNA (sgRNA). For the purposes of this review, the term sgRNA refers to a chimeric RNA molecule, which is generally ~102 bases in length. The first ~20 bp are used for targeting; these are followed by ~42 bases that are required for Cas9 binding and a further ~40 nucleotide sequence that provides additional structural requirements for the Cas9 complex and termination of RNA pol III–dependent transcription.
For complete and irreversible loss of function, three systems are most commonly used, each of which engenders DNA cleavage at the target genomic site. The first uses wild-type Cas9 to irreversibly knock out gene function. Wild-type Cas9 contains two nuclease domains (RuvC I and HNH) that cut sense and antisense DNA strands to create a blunt end double-strand break, 16 which is repaired by nonhomologous end joining (NHEJ). As NHEJ is imprecise, it leads to deletions or insertions (indels); some of these events will be in-frame and may have little effect on gene function, whereas others will disrupt the original coding sequence, thus creating a functionally inactivated gene/protein. Generally speaking, 66% of indels will generate frameshift mutations while the rest will be in-frame. However, the precise frequency at a given locus will vary depending on several factors; this is also case for CRISPR/Cas9. Examples range from ~50% in the adenoviral genome, 17 more than 80% in human KBM7 cells and in CHO-K1 cells,8,18 and almost 90% in a murine in vivo model. 19 Although the underlying reasons for this variability are unclear, the choice of sgRNA/target site does seem to play a role.9,10
The second system employs the so-called “nickase” Cas9 variant, which harbors a D10A amino acid mutation in the RuvC I nuclease domain. This variant generates a single-strand break only on the DNA strand to which the sgRNA binds. Thus, using paired sgRNAs (spaced by ~30–150 bp, one complementary to the sense strand and the other complementary to the antisense strand), Cas9-D10A generates two “nicks” on opposite strands that are treated as a double-strand break and repaired by NHEJ. This reduces the chance of off-target repair, because the probability that two off-target binding events occur with sufficient proximity to trigger NHEJ is low.
The third module uses a Cas9 variant containing mutations in both the RuvC I and HNH nuclease domains. This catalytically inactive Cas9 (dCas9) is then fused to a Fok1 nuclease domain.20,21 Because Fok1 activity is absolutely dependent on dimerization to generate a double-strand break, two Fok1-dCas9 proteins must be brought into proximity at the target site. As with the nickase Cas9, this is achieved using paired sgRNAs. However, it is proposed that the off-target effects (OTE) of Fok1-dCas9 are lower than the nickase, because the latter enzyme does not require dimerization for activity.20,21 Figure 2 summarizes the various activities of nickase and Fok1-Cas9.
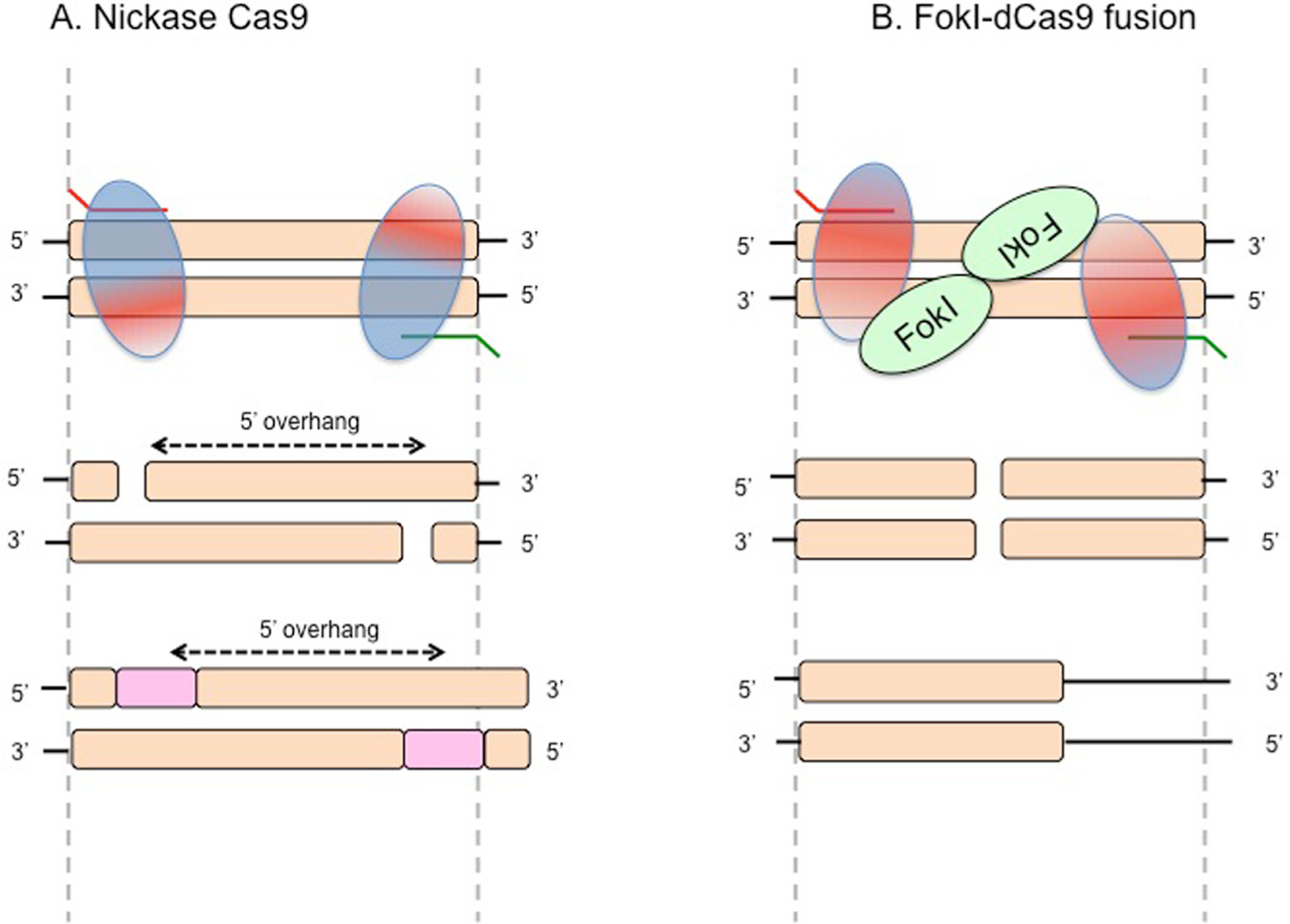
Improving specificity of Cas9-mediated cleavage. (
In the absence of fusion to other nuclease domains, the dCas9 protein can still reduce gene expression via direct transcriptional interference or by recruitment of transcriptional repressors. This iteration, termed CRISPRi (and not to be confused with iCRISPR, which employs an inducible wild-type Cas9 for genome editing 22 ), has undergone a rapid technological evolution that has opened up many possible lines of investigation. 23 Initially, targeting of dCas9 alone to promoter regions was shown to inhibit transcription in a passive mode, presumably by blocking RNA polymerase loading or elongation. 23 Subsequently, however, the transcriptional repressor, KRAB, was directly fused to dCas9 to engender active repression. When targeted to promoter regions, or to the nontemplate strand elsewhere in the coding sequence, the dCas9-KRAB fusion exhibited a wide range of repression potencies (between 3- and 100-fold, depending on the target). Very recently, a third version of CRISPRi has been developed, in which KRAB is not fused directly to dCas9 but rather is encoded separately as a fusion partner with the Com RNA-binding protein. 24 In this instance, the Com-KRAB module is recruited to the genomic target via a dCas9/scaffold RNA complex. The scaffold RNA (scRNA) is a variant of the conventional sgRNA that includes the RNA sequence to which Com binds. One clear advantage over the dCas9-KRAB fusion approach is that scRNA can be programmed with different RNA modules that permit recruitment of a variety of epigenetic modifiers. In this way, scRNA allows dCas9 to direct either repression or activation, depending on the requirements of the research group. Although this review is focused on loss-of-function genomic studies, it should be mentioned that dCas9 can also be used to recruit activators of transcription to promoters. There are two main systems currently used at the genome scale. The CRISPRa system 25 can be multimerized to enhance transcriptional activation. This is achieved by fusing dCas9 to a so-called “SunTag,” which is composed of up to 24 copies of the GCN4 peptide epitope. This facilitates the binding of up to 24 copies of sc-Fv fragments, each of which is fused to the transcriptional activator, VP-64. 26 CRISPRa has also been combined with scRNAs 27 ; it therefore provides a robust and high-throughput means to selectively up-regulate gene expression in eukaryotic cells (see also Fig. 3 for a summary). The CRISPRi/a combination is likely to find many applications in rewiring of select genetic networks and may be particularly suited to biotechnological environments. Indeed, proof of concept for this is already available, because the system has been used to rewire metabolism in yeast. 24 Similar to the scRNA/CRISPRa approach, the SAM genome editing suite 11 employs a modified sgRNA that can recruit multiple transcriptional activators. Using this technology at the genome-wide scale has uncovered novel candidate genes involved in the resistance to a small molecular inhibitor of the BRAFV600E variant.
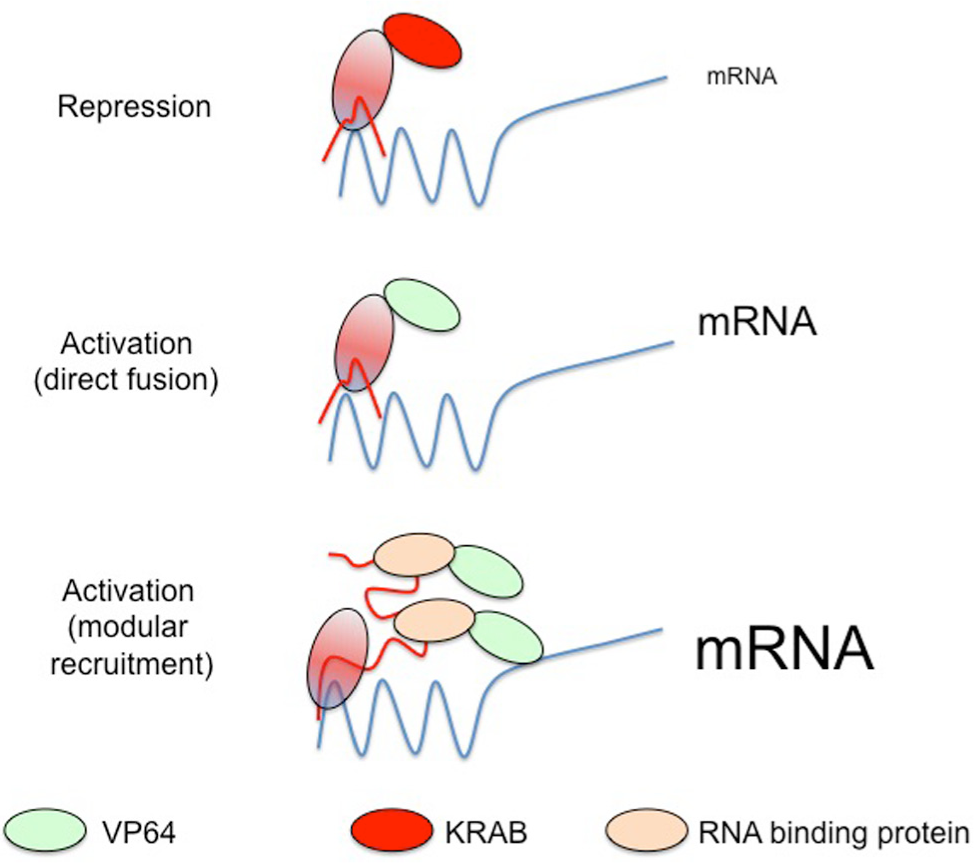
Additional applications of Cas9. In addition to acting at the DNA level, Cas9 can be reprogrammed to silence or activate transcription. When targeted to DNA alone by a single guide RNAs, dCas9 can attenuate mRNA expression passively by blocking polymerase passage or by inhibiting recruitment of elongation factors; it can also actively repress genes if fused to the KRAB repressor. Conversely, dCas9 can be fused to the VP64 transcriptional activation domain, leading to higher levels of mRNA expression (center). The latest generation of activators relies on a modified sgRNA that contains multiple RNA structural motifs. These motifs are bound by specific RNA-binding proteins, which are in turn fused to transcriptional activators or repressors (only the former is shown for clarity). This multimerization dramatically enhances the level of activation or repression.
Another choice is whether to adopt an “all-in-one” vector that contains both Cas9 and the sgRNA or to create a dual system that separates the two. The dual system allows the creation of a parental cell line that expresses Cas9 (either constitutively or under the control of an inducible promoter). With this approach, clones with different NHEJ capacities may be selected 12 ; this should be evaluated during system setup. Various methods have been used to generate the Cas9-expressing cell line; these include lentiviral infection 8 and Flp-mediated recombination. 28 An advantage of creating a master cell line is that data from multiple repeated screens can be performed in the same cell line and cross-compared with relative ease. In contrast, the all-in-one approach may be more suited to screens in which the cell number is limiting, such as in vivo screening, or to those that require primary cells and in which generation of a master Cas9 stable line is not possible. However, use of all-in-one vectors may be susceptible to two kinds of false-negative artifacts. First, some of the constructs may happen to enter individual cells with inherently low NHEJ efficiency. Second, the efficiency of Cas9-mediated cleavage may also vary at the individual cell level. Despite such caveats, very recent data indicate that inducible all-in-one vectors can efficiently knock out genes in vivo, with acceptable levels of OTE.29,30
Selection of Cell Type
Ideally, the primary driver for cell type selection should be the biology in question. However, the practical challenges associated with using some cell types means this is not always possible. Also, recent studies indicate that, at least in mouse, genes are expressed from both alleles in the majority of the individual cells of a population. 31 Furthermore, there can be compensatory up-regulation of expression from wild-type alleles in a heterozygous state. This should be taken into consideration when performing genome editing-based screens, as removal of only one allele could contribute to a false-negative result in the screen. Although haploid strains are readily available for yeast, researchers are more restricted when it comes to higher eukaryotic cell types. However, the human haploid KBM7 cell line (which carries only one copy of each chromosome except for chromosome 8 and part of chromosome 15) has successfully been used for functional genomic screens, including those mediated by CRISPR/Cas9.8,32 Interestingly, reprogramming of KBM7 to a pluripotent state gave rise to an adherent cell line derivative, HAP1, which has lost the second copy of chromosome 8. 33 In a marvelous convergence of technologies, CRISPR/Cas was used to excise the remaining chromosome 15 fragment, purportedly giving rise to the first fully haploid human cell line. 34 Haploid murine cells are also available and have been used in genetic screens. For example, the Penninger lab generated haploid mouse ES cells (mES) and used them to identify mediators of ricin toxicity. 35 In addition, the Wutz lab isolated haploid mES cells and performed a forward genetic screen for resistance to 6-thioguanine. 36 These cell lines have now been used in other laboratories to perform genomic screens (for examples, see refs. 37–39).
sgRNA Design
As a larger number of sgRNAs are tested, the properties that define the optimal composition are evolving. Several high-throughput sequencing reports have confirmed that the 20 nt sgRNA should be immediately upstream of an NGG (rather than an NAG) triplet.40,41 Given equal targeting efficiency, sgRNAs targeting the 5′ region of a gene are reportedly more effective than those that target the 3′ end 8 ; this may be because knocking out the last exons of some genes may be less deleterious than an early disruption to the coding sequence. Particular care should be taken when targeting genes with multiple splicing isoforms. In such cases, either the common 5′ exon or an isoform-specific exon or splice site should be targeted by the sgRNA, depending on the goal of the editing.
Logistical Considerations
Once the appropriate system has been selected, users must consider the screen in terms of scale, cost, and management of the associated reagents. At the time of writing, three separate laboratories have published their results of genome editing–based knockout screens at the genome-wide scale (see Table 1 ). In all screens, the assays were designed to identify genes that altered general cellular viability or those that modulated sensitivity to cytotoxic agents. Aside from the biological relevance of this type of screen, it carries the practical advantage that it can be carried out in a pooled format. This is because the score for each sgRNA in the screen is based its relative enrichment or depletion from homogenized test samples (determined by high-throughput sequencing) when compared with controls. In two of the genome-wide screens, the scale in terms of cell culture and harvesting was also made more manageable by the use of suspension cells (K562 and KBM7 leukemia cell lines). However, a range of both human and murine adherent cells was used in the other screens, although of course in this case, additional preparatory work such as washing and trypsinization for cell harvesting is required. A screen performed by Wang and colleagues 8 involved 10x sgRNA coverage for each of ~7000 genes. Although not genome-wide, the scale can be considered similar to the three studies mentioned above, which used on average 4x sgRNA coverage for ~18,000 genes. Other screens at smaller scale have been performed, and this may be the model adopted by many academic research labs with no access to a core facility that can manage a larger sgRNA library collection. For example, Wei et al. 12 used HeLa cells in a viability screen of a focused 291-gene set with 2 or 3x sgRNA coverage. CRISPRi has also been for genome wide-scale screens 13 and was able to identify genes that determine sensitivity to a cholera-diphtheria fusion toxin. The authors went on to demonstrate that knockdown efficiency using sgRNAs that yielded hits was at least 80% at the mRNA level for each gene. Thus, although CRISPRi does not generate genetic knockouts like wild-type or paired nickase Cas9 systems, it is still suitable for loss-of-function screening.
Cost and Reagent Management
In the case of the pooled genome-wide screens, lentiviral vectors were used to deliver the sgRNAs. This format and method of delivery are more cost-effective than arrayed screens using chemical small interfering RNAs (siRNAs) that require expensive transfection reagents and other screening consumables. Although the greatest cost savings can be made if the library is built, produced, curated, and packaged in house, this requires a significant investment of time, liquid-handling equipment, and resources. Such a strategy is warranted if many large-scale screens are to be repeatedly run. Alternatively, several companies are beginning to offer commercial lentiviral CRISPR/Cas editing tools, although in contrast to lentiviral-shRNA libraries, these are not yet available at the genome-wide scale. As with the siRNA and shRNA predecessors, these are likely to be updated as the scientific data inform best practice. For example, the original versions of both Dharmacon and Ambion siRNA libraries (in Europe now sold by GE Healthcare and ThermoFisher/Life Technologies, respectively) were virtually completely redesigned to reduce both off-target and cytotoxic effects based on continued discoveries in the field.42 –46
Dealing with OTE
Following the first wave of functional genomics screens using chemically synthesized siRNA, there was the realization that OTE played a considerable role in the poor hit overlap found by groups performing very similar screens. OTE are also major factors in the low validation rate of hits from primary screens. Primarily, these effects are due to binding of siRNAs to nontarget mRNAs via the seed region of both the sense and antisense strands. 47 This recapitulates the activity of cellular microRNAs, as the seed (nucleotides 2–8) is presented to the 3′UTR of nontarget mRNAs by the cytoplasmic RISC complex. OTE in shRNA-based screens are also a concern. This is because shRNAs against multiple unintended targets can be generated by alternate transcriptional start sites within lentiviral shRNA constructs. 48 This phenomenon may have a significant contribution to the low confirmation rates in shRNA screens and the relatively low cross-validation rate between shRNA and siRNA screens.49 –51 Given these issues, researchers in the CRISPR/Cas9 field have invested significant efforts in the determination of OTE and their mitigation.
Because of the diversity in experimental systems and analysis pipelines within the published literature, it is challenging to derive a common value for the rate of OTE caused by genome editing. Indeed, the rates appear to depend on multiple factors, including cell background, sgRNA length and sequence, type of nuclease used, and the way in which off-target sites are defined. However, a general survey of the first-generation larger-scale cell-based screens indicates that OTE are considerable. Despite this caveat, there was reassuring confirmation of on-target hits in all of these screens. Below, an overview of OTE and confirmation rates for several screens is presented.
Using an enhanced green fluorescent protein reporter assay in U2OS cells, Fu et al. 52 reported that the frequency of confirmed OTE can range from ~3% to 60%, depending on the sgRNA used. This frequency has also been confirmed at some endogenous sgRNA target sites. 41 Koike-Yusa et al. 10 performed a genome-wide screen in murine cells and successfully identified all the major components of the GPI biosynthetic pathway, a key process that determines sensitivity to alpha-toxin. They went on to show that 18 genes were identified as hits by at least two of four sgRNAs in a screen for mediators of sensitivity. Of the six that were followed up, two were designated as false-positive and were likely off-target. In the same report, 10 genes were identified as determinants of sensitivity to 6-TG treatment; this included several mismatch repair (MMR) proteins, which are known players in the pathway. However, none of three novel candidate genes of this 10-gene subset were confirmed in follow-up, suggesting that they were also off target. An independent screen for 6-TG resistance also identified the expected MMR proteins 8 ; however, although off-target frequency was estimated to be low in this case, it was not experimentally determined. In another positive selection screen for determinants of sensitivity to the oncogenic RAF inhibitor, PLX, one-third (4/12) of the sgRNAs tested gave off-target modifications with a frequency of ~2% to 100% compared with the intended on-target site. 9
In chemical siRNA screens, it has been shown that reducing the concentration of siRNA can to some extent reduce the rate of OTE. However, this strategy does not appear particularly effective with CRISPR/Cas9, because reducing the level of either Cas9 or gRNA decreased both the rate of OTE and that of the desired editing event. 52 As mentioned above, another strategy to reduce OTE is to use the nickase Cas9 with paired sgRNAs, as this reduces the possibility of off-target NHEJ compared with wild-type Cas9.30,53 However, to date, there are no large-scale published screening data that compare the frequency of OTE using this approach to methods using wild-type Cas9. Another approach to reduce OTE is by shortening the actual guide sequence from 20 to 17 or 18 nucleotides. At first glance, this is paradoxical, because a shorter sequence has potentially more genomic sites with which it can hybridize. However, it is postulated that these so-called truncated guide RNAs (tru-gRNAs) reduce the affinity of the Cas9 complex for its potential binding sites. 54 Thus, binding at off-target sites that might otherwise occur due to base pair mismatches would be reduced still further, thereby preventing strand cleavage. Using a new technique for characterization of CRISPR/Cas-dependent OTE, Tsai et al. 41 have recently quantified double-strand breaks on a genome-wide scale. This GUIDE-seq approach revealed that, depending on the sgRNA used for targeting, between 0 and 150 OTE were generated. In a more recent study, Kim et al. 55 reported a very low frequency of OTE in human cells and provided guidelines for sgRNA modification to reduce their promiscuity. In addition to modifying sgRNAs, whether the Cas9 orthologs from other organisms 2 have inherently lower propensities to generate OTE or whether directed evolution may reduce the OTE properties of Cas9-like nucleases remains to be determined.
The studies mentioned above all used cell lines that are either immortalized or transformed, which might contribute to some of the OTE observed. Thus, it is possible that the use of more “normal” cells may reduce OTE. In support of this, it does appear that the frequency of OTE is lower where CRISPR/Cas has been used for in vivo genome editing of normal cells such as murine zygotes and somatic cells and human induced pluripotent stem cells. Although the reasons for this are not fully understood, this indicates that allelic cell lines generated from normal cells by precision editing will be a valuable addition to the functional genomics toolbox.
CRISPR/Cas and Other Editing Technologies
Considerable efforts in both academic and commercial environments have been invested in the development of other genome modification suites. Whether CRISPR/Cas9 proves to be the modern-day asteroid that lays all these other technologies to rest remains to be seen. However, it is likely that, in many instances, there will be applications for each technology, and perhaps sometimes a combination of platforms will prove yet more powerful. In this section, a few examples are provided; for more details on each of the other technologies mentioned, the reader is referred to several review articles.56 –59 One of the main advantages of CRISPR/Cas9 compared with the earlier genome-editing nucleases such as zinc finger nucleases (ZFNs) and transcription activator–like effector nucleases (TALENs) is the increased flexibility in terms of number of target sites per genomic locus. This provides more choice in terms of precisely where functional knockouts are generated. In addition, multiplexing is much more feasible with CRISPR/Cas9 because a single nuclease construct can be targeted to multiple loci simply by co-introducing several sgRNAs 60 ; in contrast, multiplexing with ZFNs and TALENs is far more demanding in terms of design and construction time. In some cases, a greater on-target specificity of TALENs when compared with ZFNs has been demonstrated. 61 This is largely due to the higher degree of specificity at the level of DNA binding for the TALENs. How CRISPR/Cas9 compares with other nucleases in terms of OTE is an active area of research. In a direct comparison of targeting a single locus, Veres and colleagues 62 reported that the incidence of OTE was very low and comparable for both CRISPR/Cas9 and TALENs. However, another report indicated that the frequency is likely to vary depending on the genomic target, sgRNA sequence, and the amount of nuclease used. 63
Another exciting application for CRISPR/Cas is the generation of an allelic series, in which a parental cell line is engineered to create progeny that express specific genetic alterations. The range of questions that could be addressed by this approach is vast, and not all possibilities can be discussed here. For example, one could generate a panel of cell lines based on mutations found in patient-derived sequencing data; this panel could be interrogated to identify combinations that modulate the response to therapeutic agents. Technically, this is achieved by recruiting Cas9 nickase to the target site with a single sgRNA while concurrently supplying an oligonucleotide donor (ODN) with homology to the target site but that contains the desired nucleotide change(s). Nickase Cas9 then generates a single-stranded break, which is a substrate for homology-directed repair (HDR 64 ); this leads to incorporation of the ODN and subsequent gene correction/mutation. This has now been demonstrated both in cell lines and in murine models.65,66 Again, CRISPR/Cas provides an advantage compared with ZFNs and TALENs here, because the flexibility of sgRNA-dependent targeting increases the likelihood that any particular mutation can be introduced. One of the most established methods for generating an allelic series in mammalian cells via HDR is that which uses adeno-associated virus (AAV). rAAV is a single-stranded DNA virus, and this property renders it a more effective mediator of recombination events when compared with double-stranded DNA vectors. 67 Although much more efficient than using plasmids, the rAAV system still suffers from some drawbacks. For example, AAV-dependent HDR is most efficient when double-strand breaks are induced, 68 yet this type of genomic lesion can also increase the chances of random integration. Next-generation rAAVs that employ a negative selection strategy hold great promise for reducing off-target recombination.69,70 It is now clear that single-strand DNA breaks are sufficient for AAV-driven HDR to occur. 71 Therefore, further improvements may also come from strategies that specifically target AAVs and the ODNs to target loci and that concomitantly induce single-strand breaks. Here, one might envisage the use of nickase Cas9 in combination with a rAAV that delivers an ODN. Proof of principle for the wild-type Cas9/rAAV combination is already available, as it has been used to enhance rAAV-dependent targeting of the CDK2 locus by up to 10-fold. 72 Conversely, several reports indicate that AAV vectors are improving the cellular target range and efficacy of CRISPR/Cas9 genome editing. 73 For example, AAV vectors have been used to codeliver Cas9 and sgRNA to the mouse brain for loss-of-function studies. 74 This dual system is required because conventional AAV vectors are limited in the size of the DNA inserts they can carry (~4.8 kb), and Streptomyces pyrogenes Cas9 alone accounts for ~4 kb of this space. To circumvent these size limitations, while still exploiting the high titer and broad tissue target range of AAV, Platt et al. 19 generated mice that either constitutively or conditionally express Cas9. In this way, space was freed in the AAV vector in order to package multiple sgRNAs as well as ODNs. The authors demonstrated simultaneous knockout of the Trp53 and Lkb1 tumor suppressors and knock-in of the oncogenic G12D Kras mutant. All-in-one Cas9/sgRNA AAV vectors have also been reported 75 ; these constructs were successfully used to edit suspension cultures of Jurkat T cells as well as murine hepatocytes in vivo.
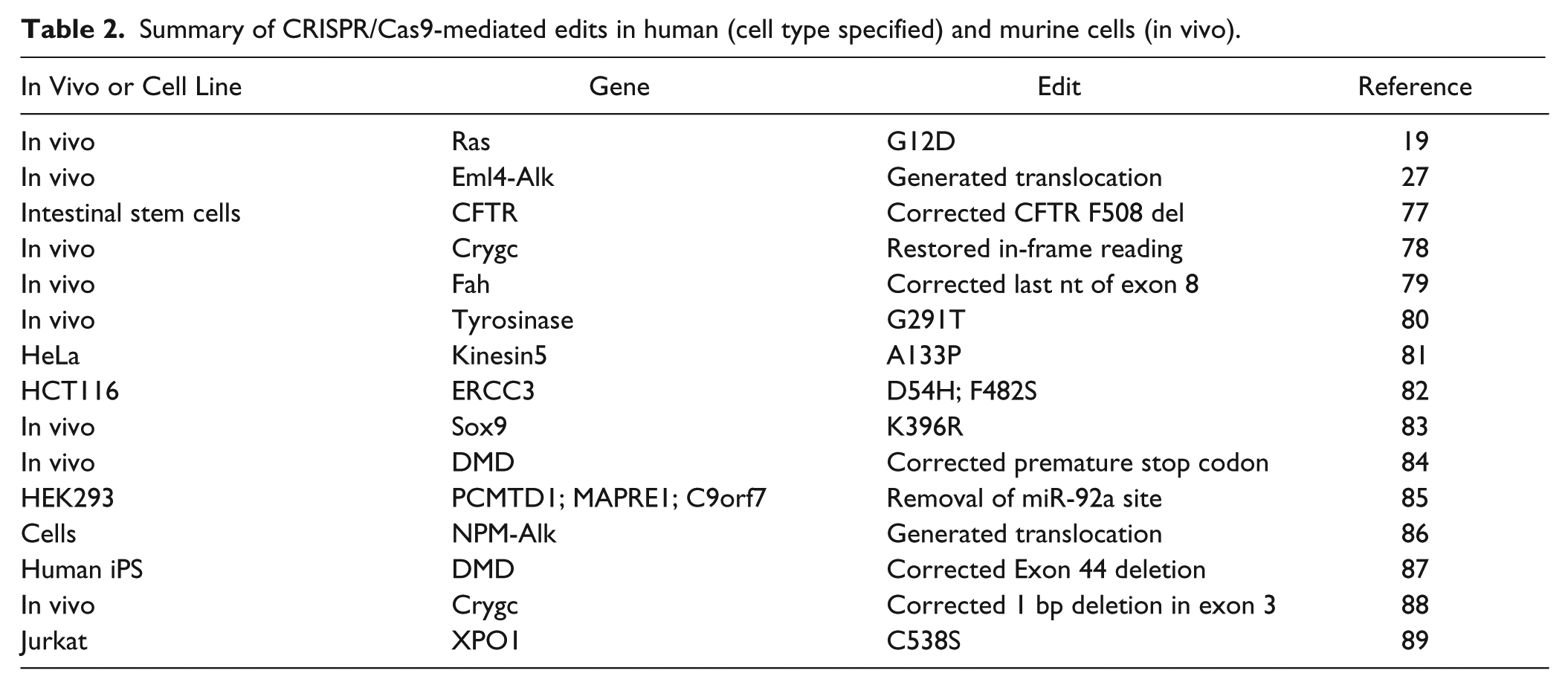
In contrast to AAV, lentiviral vectors are not as limited in terms of insert size and therefore have been used extensively both in cell-based screens (see the “System Choices” section) and in vivo for genome editing. One particularly flexible in vivo system is pSECC, 76 which is an all-in-one lentiviral vector expressing Cas9, sgRNA, and the Cre recombinase. This permits the conditional activation or inactivation of genes that harbor LoxP sequences, which are the targets of Cre. Proof of principle was shown by simultaneous knockout of tumor suppressor genes and activation of Kras. 76 Given that Cre can induce single- and double-strand breaks throughout the genome, one concern is that the combination of Cas9 and Cre might lead to increased off-target editing events. Although this did not appear to be the case for a limited number of predicted off-target loci, more sophisticated genome-wide methods will be required for full validation. Table 2 provides more examples of how CRISPR/Cas-mediated HDR is revolutionizing the field of genetic modification.
Summary of CRISPR/Cas9-mediated edits in human (cell type specified) and murine cells (in vivo).
Additional Applications and Improvements
Because multiple gene-targeting events within the same cell are possible using CRISPR/Cas9,60,90 one might engineer cells with combinations of mutations that reflect various stages of neoplastic disease. Such cell lines could then be used in combination with siRNA or shRNA to identify stage-specific synthetic lethal interactions. Such combinatorial screens have already been performed in yeast and Drosophila.91,92 Recently, all pairwise combinations of siRNA against a 323 × 20 mammalian gene subset have been tested in an image-based assay. 93 Adaptation of CRISPR/Cas-engineered cell lines to a similar array-type format will provide a wealth of additional information of mammalian genetic interactions. In terms of feasibility for the average research laboratory, it has been estimated that 12 individual allelic variant cell lines could be generated in as little as 1 month. 22
CRISPR/Cas might also be used to modify mitochondrial genomes. Indeed, the concept and validation of endonuclease-dependent cleavage of mitochondrial DNA (mtDNA) was provided almost 15 years ago using mitochondrially targeted bacterial restriction enzymes, 94 and several other articles have followed (for example, refs. 95 and 96). More recently, the use of TALENs has increased the specificity and range of mtDNA targets. 97 It will be of great interest to determine whether CRISPR/Cas provides additional advantages in this area of research.
Because the conventional CRISPR/Cas9 operates at the level of DNA, it may also provide researchers the opportunity to modulate the expression of some noncoding RNAs that do not get exported to the cytoplasm, such as many of the long-noncoding RNAs (lncRNAs). This is because conventional technologies of siRNA and shRNA act at the level of the cytoplasmic RISC/mRNA complex and therefore may be much less effective against lncRNA. The original genome-wide sgRNA libraries targeted only mRNAs but have recently been upgraded to include miRNA-encoding loci. 14 Thus, extension to targeting loci that encode nuclear RNAs would seem to be a next logical addition to genome-editing–based functional genomics. Indeed, recent data show that the lncRNA, Rian, can be deleted using CRISPR/Cas9. 98 Several lncRNAs were suppressed using CRISPRi, 13 whereas others were transcriptionally up-regulated using SAM. 11 Going beyond this, CRISPR/Cas9 could also be used to probe the functional significance of even nontranscribed regions. Circular RNAs (circRNAs) are also emerging as critical ncRNA biological regulators in many instances.99,100 Many of these are derived from posttranscriptional processing of the normal linear RNA encoded by the respective gene. These circRNAs cannot be selectively modulated by CRISPR/Cas, as interference at the DNA level would block production of both linear mRNA and circRNA. This represents an example of where siRNA-mediated knockdown may still hold some advantages. Although challenging in terms of sequence restrictions, the design of a siRNA that targets the 5′-3′ junction of circRNA could achieve the selective knockdown required. On this subject, selective cleavage of RNA by Cas9 has been demonstrated, 101 offering an additional strategy for targeting circRNAs.
Great efforts are already being made in the ability to predict off-target sites and to improve analysis of CRISPR/Cas9 screening data. These advances are coming from both wet-lab and computational experiments. For example, it is now known that a Cas9/sgRNA complex may bind many more genomic sites than its intended target. 102 However, many (if not most) of these off-target sites are either not edited or are edited at lower frequency than the on-target sequence.54,102 Although the mechanisms for this remain unclear, perhaps (as is the case for other DNA-binding proteins, such as transcription factors) this suggests that Cas9 “samples” the DNA until a productive cleavage event occurs. Whether epigenetic modifications and/or higher-order chromatin structure also contribute to selection of Cas9 sites remains to be determined. In the context of site selection, the finding that Cas9 can cleave at noncanonical PAMs41,103,104 also deserves further investigation. Although this can be viewed as a liability in terms of potential OTE, the ability to control this behavior would open up a large number of additional sites for editing. New computational approaches to identify off-target sites are being updated in response to new experimental data sets. This is likely to be a challenging endeavor, because deep-sequencing results indicate that predicting off-target sites based solely on sequence similarities to the intended site has limited utility.62,105
Finally, there is a bewildering array of analytical tools available for data processing and analysis. Many of these either use or are based on the algorithms designed to analyze other types of high-throughput sequencing data, such as RNA-seq. Although challenging because of the diversity of experiments and CRISPR/Cas9 systems currently in use, some efforts to standardize reporting methods and analytical tools (including statistical tests) should be made. This would certainly enable interested researchers to rapidly compare data sets and will provide accessible benchmarking data, which are invaluable for groups to gauge their own progress as they establish their screening procedures. In this regard, approaches such as MAGeCK, 106 which is specifically designed to analyze data from positive and negative selection CRISPR/Cas screens, may fit the bill.
Summary
CRISPR/Cas is undoubtedly a game changer in the field of functional genomics. In the space of less than 5 years, it has enabled laboratories of all sizes and capabilities to edit genomes at will. Although the initial indications are that the technology is less plagued by off-target effects than siRNA or shRNA, direct comparisons of the three technologies are still required. However, there are some encouraging findings. For example, several common hits that mediate resistance to the BRAF inhibitor, PLX-4720, were identified in CRISPR and shRNA loss-of-function screens.9,107 Furthermore, there are overlapping hits between a CRISPR transcriptional activation screen conducted by Zhang and colleagues 11 and a cDNA overexpression screen carried out by the Garraway lab. 108 Specifically, each group identified genes that, when overexpressed, conferred resistance to inhibitors of the RAS/RAF/MAPK pathway. Theoretically, CRISPR/Cas loss-of-function screens could reveal hits that were not found in shRNA screens. This is because shRNA-mediated knockdown acts at the level of RNA, rather than by deleting the coding sequence; thus, partial mRNA knockdown may lead to false-negatives in shRNA screens. Of course, CRISPR/Cas9 screens will also have a certain false-negative rate because of variations in sgRNA targeting efficiency, Cas9 activity, and the efficacy of NHEJ in each cellular background. Future benchmarking studies should be prospective, rather than rely on reanalysis of existing data sets. The availability of relatively inexpensive methods to analyze genome-wide modifications, combined with the wealth of computational expertise in genomic data analysis, will also allow the field to rapidly optimize targeting strategies and reagents. In the interests of rapid and continual scientific progress, it is hoped that such information is shared with, and remains in, the larger academic community.
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.