
Abstract
The clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 system is a widely used genome-editing tool with great clinical potential. However, its application is limited because of low editing efficiency of some target sequences and off-target effects. As this system contains only the Cas9 protein and a single-guide RNA (sgRNA; engineered from crRNA and tracrRNA), the structure and function of these components should be studied in detail to address the current clinical needs. Consequently, we investigated the structural and sequence features of the core hairpin (the first stem loop of sgRNA) of SpCas9 sgRNA. We showed that the core hairpin structure of sgRNA is essential for SpCas9/sgRNA-mediated DNA cleavage and that the internal loop structure in the core hairpin plays a vital role in target DNA cleavage. We observed that the root stem structure within the core hairpin preferentially forms Watson-Crick base pairs and should be of a specific length to maintain an appropriate spatial conformation for Cas9 binding. However, the length of the leaf stem structure of the core hairpin is flexible, having a variable nucleotide composition. Furthermore, extension of the leaf stem structure enhances the DNA cleavage activity of the Cas9/sgRNA complex, and this could be used to enhance the efficiency of gene editing. These observations provide insight into the sgRNA/Cas9 interaction, indicating that sgRNA modification could be a strategy for improved DNA editing efficiency, and optimized sgRNA can be further used for genome-wide functional screening and clinical application.
Introduction
The clustered regularly interspaced short palindromic repeats (CRISPR) system is an adaptive immunity system of bacteria and archaea that defends the host against invading foreign plasmids or viruses.1,2 Based on the number of effector proteins participating in the nucleic acid interference stage of CRISPR editing, two CRISPR classes are recognized: class 1 systems containing multiple effector proteins and class 2 systems, which include only one effector protein.3–5
The class 2 type II CRISPR/Cas9 system is the most extensively studied CRISPR system and the first-ever CRISPR system to be applied to genome editing in mammalian cells. 6 Its functional complex includes the Cas9 protein, a CRISPR-related RNA (crRNA), and a transactivating crRNA (tracrRNA). 7 Cas9 is a large multidomain protein that facilitates RNA binding and guides target DNA binding and cleavage. Further, crRNA hybridizes with tracrRNA to form double-stranded RNA. For the sake of convenience of CRISPR/Cas9 use, crRNA and tracrRNA are linked by an artificial GAAA loop to form a single-guide RNA (sgRNA). 7 The RNA duplex binds to the Cas9 protein, which results in a conformational change of the protein. Then, the ribonucleoprotein (RNP) complex searches the genome for the cleavage site. This requires the presence of a protospacer-adjacent motif close to the target sequence, to differentiate the invading DNA from host DNA. After interaction with the protospacer adjacent motif, the RNP complex undergoes a structural change, the sgRNA target region pairs with the complementary DNA sequence, and a double-strand break is induced at the DNA in the target locus. DNA double-strand breaks are then majorly repaired via nonhomologous end-joining or homology-directed repair, leading to DNA modification.8,9
High editing efficiency is essential for the application of CRISPR/Cas9, and it is extensively used in many research efforts.10,11 Previous studies mainly focused on the structure of the Cas9 protein, Cas9/sgRNA binary complex, and Cas9/sgRNA/DNA ternary complex, to elucidate the key functional sites and interactional mechanisms of the CRISPR/Cas9 system.10–12 Nevertheless, additional features of the sgRNA sequence and structure associated with the activity of Cas9/sgRNA complex are yet to be explored.
sgRNA consists of two regions: a 5′-target sequence and a 3′-scaffold sequence. The conserved modules of sgRNAs have been identified and characterized, and the orthogonality between sgRNAs and Cas9 proteins has been explored. 13 Studies of sgRNA target sequence have identified target features for high editing efficiency and have established tools for target sequence selection.14–18 Furthermore, the sgRNA target sequence was truncated, sgRNA basic scaffold was artificially reformed, and sgRNA sequence was chemically modified to improve the genome-editing efficiency. In addition to the improvements of efficacy, studies of the sgRNA secondary structure helped to reduce the off-target effects of this editing system.19–26
Regardless of the above efforts, however, little is known about the features of the sgRNA scaffold sequence. Consequently, in the current study, we designed a screening system in Escherichia coli to investigate the sequence composition of the sgRNA core hairpin structure, which is the first stem loop in sgRNA and is the complementary region between crRNA and tracrRNA. We also explored the nucleotides of the core hairpin structure that influence the cleavage activity of SpCas9/sgRNA complex in mammalian cells. We subsequently demonstrated the feasibility of promoting the cleavage efficiency via sgRNA sequence optimization and presented modified sgRNAs that improve CRISPR/Cas9 genome-editing efficiency in mammalian cells. We anticipate that the presented findings will provide a strategy for improving the editing efficiency for therapeutic applications.
Materials and Methods
Plasmid Construction
SpCas9-D10A protein and mutant sgRNA library used for screening in E. coli were individually expressed under the control of the T7 promoter from the pACYCDuet-1 vector (Novagen, Darmstadt, Germany). SpCas9-D10A sequence was inserted into the plasmid using the restriction enzymes Pst I and Not I to generate pACYCDuet1-SpCas9-D10A. The original second T7 promoter on pACYCDuet-1 plasmid was deleted using Not I and Avr II to remove the unnecessary ribosome-binding site. Constructs containing T7 promoter and mutant sgRNA sequences were synthesized by using random nucleotides according to different designs of the experiments (Biomed, Beijing, China) and inserted into the pACYCDuet1-SpCas9-D10A plasmid using Not I and Avr II, generating pACYCDuet1-SpCas9-D10A-sgRNA libraries using Gibson Assembly Master Mix (New England BioLabs, Ipswich, MA). Single-strand annealing (SSA) red fluorescent protein (RFP) reporter plasmid was constructed based on vector pETDuet-1 (Novagen). The incomplete RFP (396 bp) sequence was placed in front of two TAA stop codons and the target sequence. Next to the target sequence, an out-of-frame complete RFP sequence was inserted. The sequence encoding for wild-type SpCas9 protein was codon-optimized for mammalian cells by Lasergene. It was inserted in the expression vector pcDNA3.1 (Invitrogen, Carlsbad, CA) using restriction enzymes Kpn I and Xba I, and nuclear localization sequences were inserted at both the C- and N-termini of the protein. sgRNA was expressed under the U6 promoter from plasmid pGPU6/GFP/Neo (GenePharma, Shanghai, China). sgRNA was inserted in the vector using restriction enzymes Bbs I and BamH I. All sequences are provided in the
Core Hairpin Structure Screening in E. coli
E. coli BL21 strain was transformed with Cas9-D10A/sgRNA plasmid pool and the SSA RFP reporter plasmid by electroporation. Briefly, 150 µL of electrocompetent E. coli BL21 cells (Biomed) were mixed with 300 ng Cas9/sgRNA plasmid pool and 100 ng SSA RFP reporter plasmid and electrotransformed using Lonza Amaxa Nucleofector 2b (Program 7). subsequently, 500 µL of room-temperature SOC medium was added and the sample agitated vigorously (220 rpm) at 37 °C for 1 h. Next, 1 mM isopropyl β-d-1-thiogalactopyranoside was added, and the cells were cultured for another 1 h. The transformants were spread on LB agar plates supplemented with ampicillin and chloramphenicol and incubated at 37 °C; 18 h later, red clones were picked using a fluorescence microscope. The total plasmids were extracted (Biomed), and the RFP plasmids were degraded by Xho I digestion for a second round of screening. We then checked for complete elimination of RFP plasmids after transformation into BL21 cells. The process of the second screen was the same as the primary screen. The red clones were then picked and sequenced using Sanger sequencing (Biomed).
Mammalian Cell Culture and Transfection
HEK293T cells were cultured in high-glucose Dulbecco’s modified Eagle medium (Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum in an incubator set at 37 °C and under 5% CO2 atmosphere. The cells (at approximately 70% confluence) were transfected in 48-well plates using Lipofectamine 2000 (Thermo Fisher, Waltham, MA) with 300 ng Cas9 expression plasmid and 100 ng sgRNA plasmid. The cells were harvested 72 h after transfection for T7 endonuclease I (T7EI) assay.
T7EI Assay
For the experiment, 72 h posttransfection, genomic DNA was extracted from HEK293T cells using TIANamp Genomic DNA kit (TIANGEN, Beijing, China). The T7EI experiment was performed as previously described with some modification.
27
Briefly, Q5 high-fidelity DNA polymerase (New England BioLabs) was used to amplify genomic sequence containing the target sequence using 100 ng genomic DNA as a template. HBB gene primers are shown in the
Statistical Analysis
Data are represented as mean ± SD of two or three independent experiments. An unpaired two-tailed t test was applied to analyze the p value between the two groups (*P < 0.05, **P < 0.01).
Results and Discussions
Screening of sgRNA Core Hairpin Sequence Features in E. coli
According to previous studies, the unique structure of sgRNA allows it to bind Cas9 protein.10,13 However, little is known about the specific features of sgRNA. Based on its RNA secondary structure, we divided the sgRNA structure into three regions: the spacer region, core hairpin structure, and other hairpin structures. The core hairpin structure refers to the complementary region between crRNA and tracrRNA, and it contains three modules: root stem, internal loop, and leaf stem. The secondary structure of the sgRNA core hairpin was predicted by using RNAfold Server (
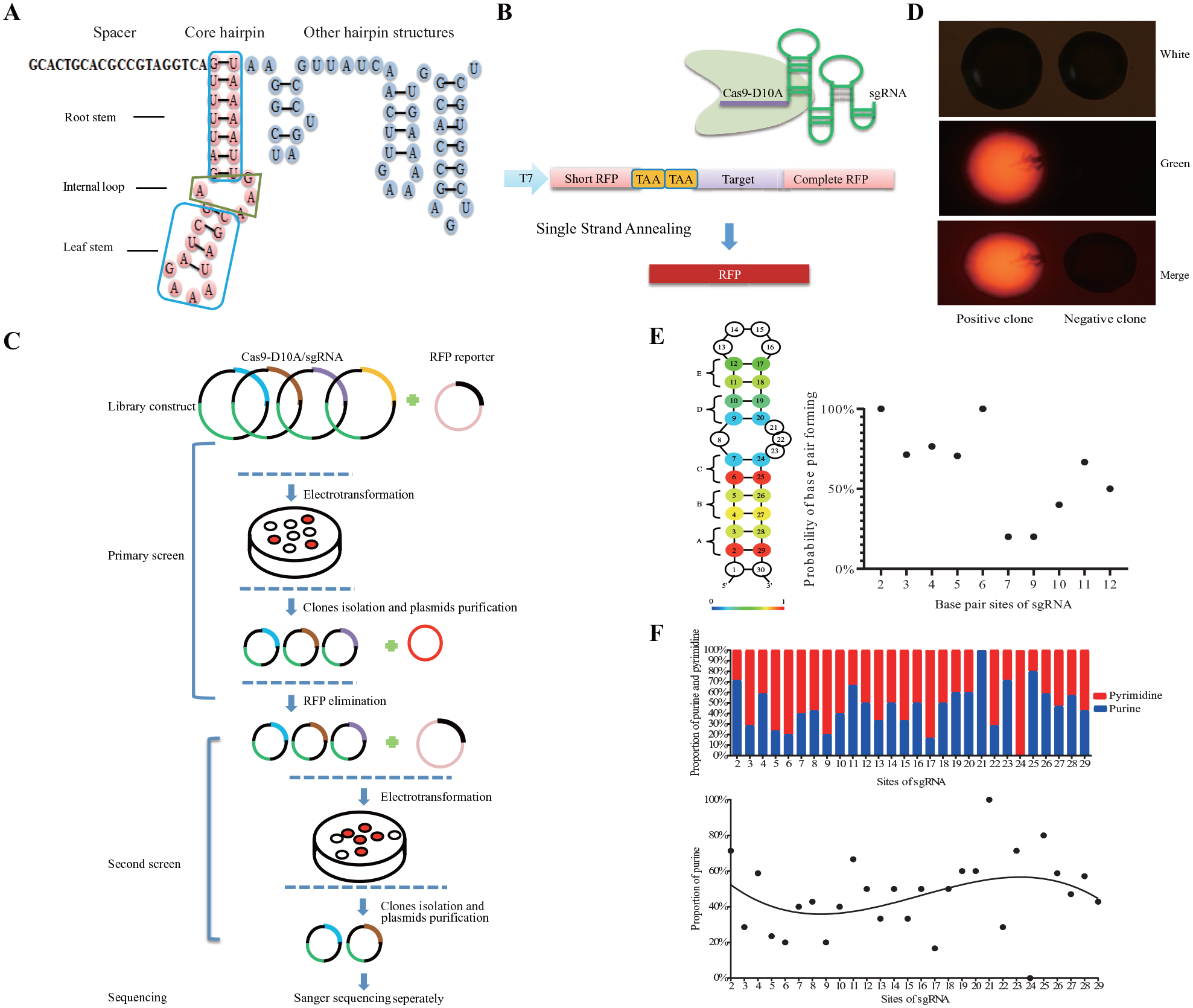
Screening of single-guide RNA (sgRNA) core hairpin sequence features in Escherichia coli. (
Accordingly, we performed a large-scale screening of the features of the sgRNA core hairpin sequence by using an SSA-based RFP reporter (
The screening process involved four steps: sgRNA library construction, primary screen, secondary screen, and sequencing (
We then used statistics to analyze the base pairing status of all identified positive hits. A heat map was constructed to show the base-paring pattern in Design A–E. Most of the bases preferentially formed pairs in the root stem (Design A–C), indicating that the root stem structure is necessary for the sgRNA function (
Base-Pairing Probability of Each Site of sgRNA Core Hairpin.
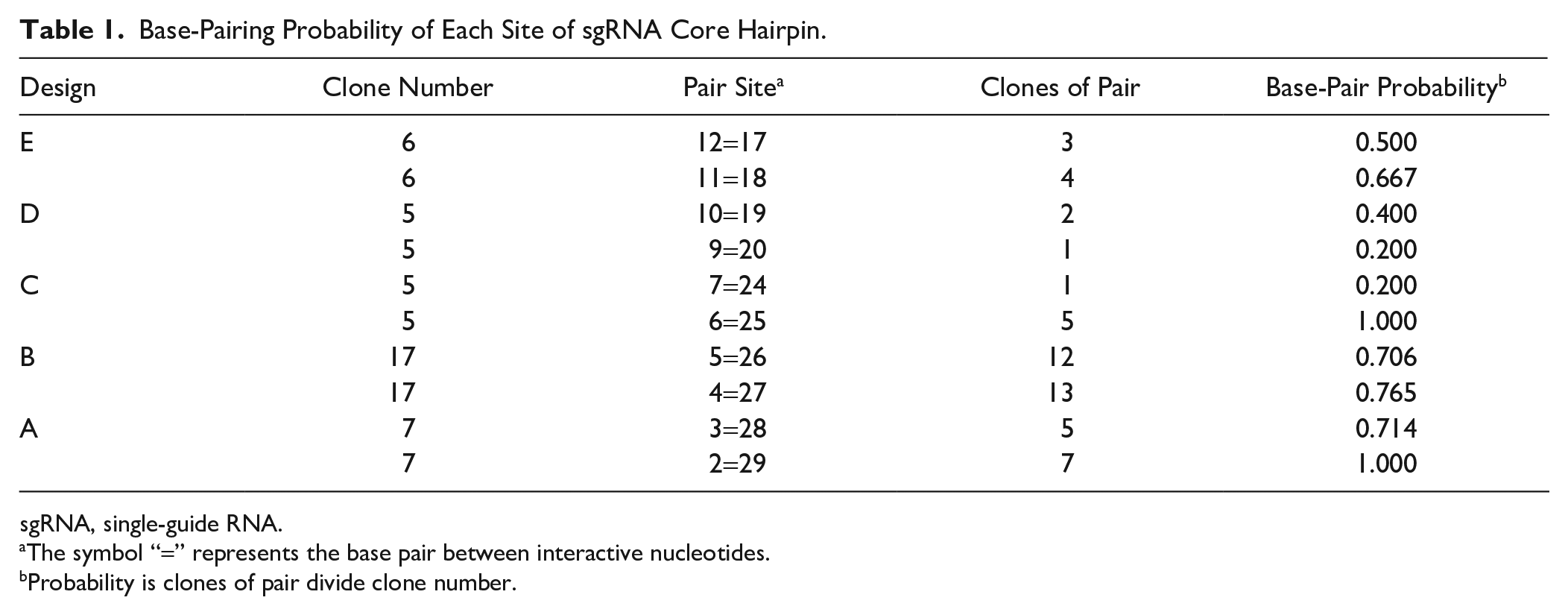
sgRNA, single-guide RNA.
The symbol “=” represents the base pair between interactive nucleotides.
Probability is clones of pair divide clone number.
The distribution of paired bases in the sgRNA core hairpin also depends on the structure of the root stem and leaf stem. We observed a relatively low proportion of purines in the 5′-end of the sgRNA core hairpin and a relatively high proportion of purines in the 3′-end of the sgRNA core hairpin (
We also investigated the loop size and composition of the internal loop and the leaf stem. We inserted additional nucleotides into the internal loop and the leaf stem loop, as part of the screening results of Design H to Design R (
Collectively, the core hairpin structure of sgRNA tended to form stable Watson-Crick base pairs in the root stem but had a loose conformation in the leaf stem. The bases adjacent to the internal loop had tendency to be unpaired. Although it is not accurate to evaluate the editing efficiency of different sgRNA scaffolds in E. coli as fluorescence intensity is slightly affected by clone size or bacterial growth, our screening method in E. coli can also be used to screen for mutant Cas9 protein with PAM sequences other than NGG, target sequence preferences for gene editing, and so on.
Internal Loop of the Core Hairpin Structure Is Critical for the DNA-Editing Activity of Cas9/sgRNA Complex
Because the editing efficiency of different sgRNAs could not be compared using screening results in E. coli, we investigated the editing efficiency of different sgRNAs by using the T7EI assay in HEK293T cells. We chose a target on the HBB gene for Cas9/sgRNA cleavage in mammalian cells (
First, we replaced the internal loop 8A:21AAG with 8AC=21AG and 7G with 7A to destabilize the internal loop structure. We used the T7EI assay to evaluate DNA cleavage activity of the Cas9/sgRNA complex in HEK293T cells. The results revealed that the complex failed to cut the relevant genomic target in this scenario, indicating that the internal loop was important for the activity of Cas9/sgRNA complex (
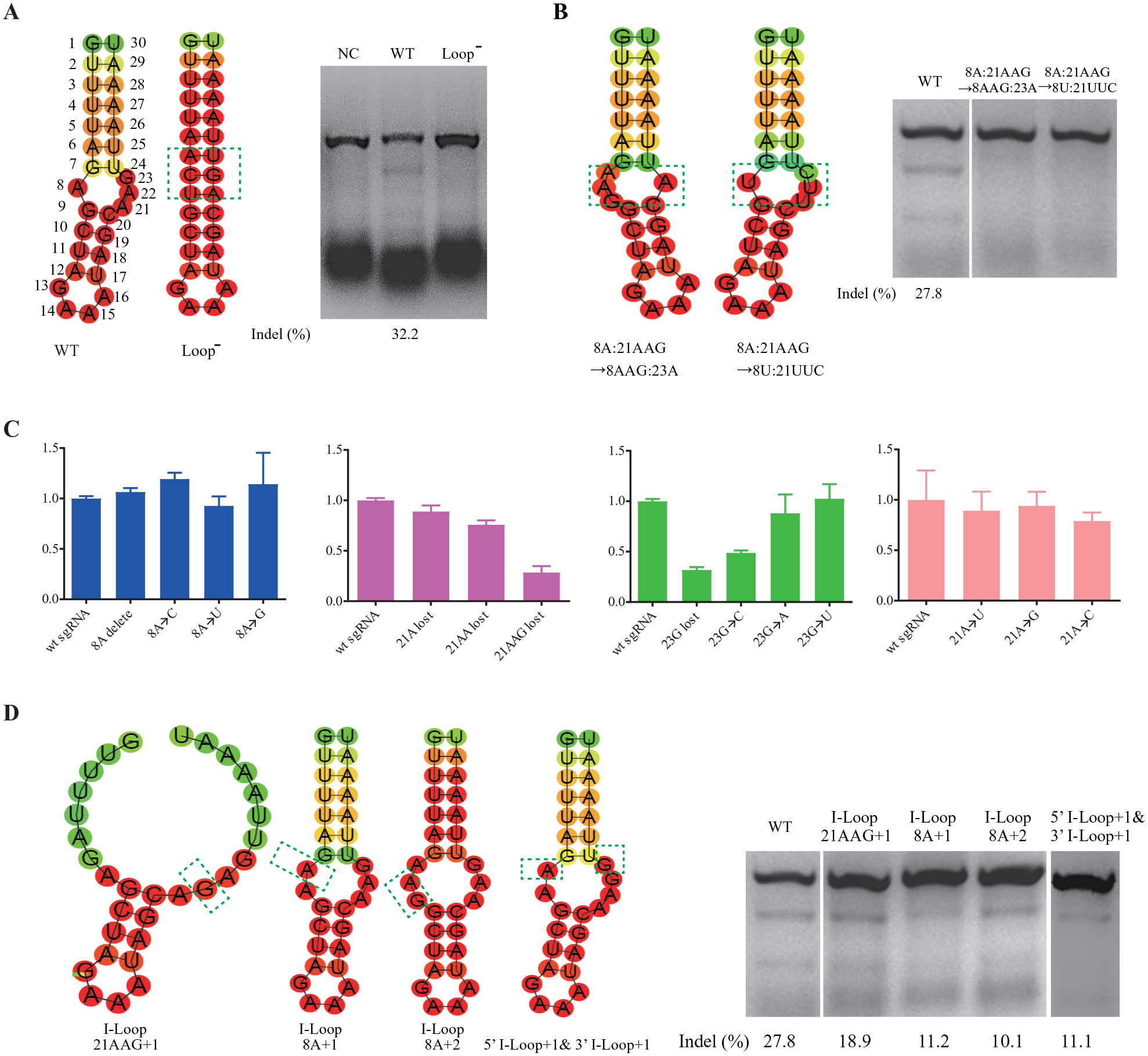
Features of the internal loop structure of single-guide RNA (sgRNA) core hairpin. (
We then altered the spatial conformation of the internal loop. We reversed the orientation of the internal loop by swapping nucleotides 8A and 21AAG. T7EI assay results suggested that the Cas9/sgRNA complex lost its DNA cleavage activity when sgRNA conformation was altered, emphasizing that the conformation of the groove formed by the internal loop was crucial for the interaction between Cas9 and sgRNA (
We also investigated the sequence of the internal loop. We replaced nucleotides 8A and 21AAG with 8T and 21TTC, respectively. This eliminated the DNA cleavage activity of the Cas9/sgRNA complex (
Finally, we enlarged the size of the internal loop structure by adding one or more nucleotides in the loop region. We inserted one G in 21AAG, one A before 8A, and two As before 8A. Consequently, the DNA-editing efficiency was reduced to 18.9%, 11.2%, and 10.1%, respectively. By comparison, the editing efficiency of wild-type sgRNA was 27.8% (
Collectively, the presence and appropriate conformation of the internal loop in the sgRNA core hairpin was necessary for the DNA-editing activity of the Cas9/sgRNA complex. The functioning of the Cas9/sgRNA complex also depended on the internal loop sequence, and nucleotide in position 23 appeared to bind certain amino acids of the Cas9 protein directly. Indeed, recent studies of the structure of the Cas9/DNA/sgRNA ternary complex revealed that the guanine nucleotide at position 23 forms hydrogen bonds with Phe351 and Asp364 of Cas9 in a base-specific manner, 10 confirming the above findings.
Cas9 Recognition of sgRNA Depends on the Appropriate Spatial Conformation and Specific Sequence of the Root Stem
The root stem of sgRNA core hairpin is a U-A base-pair repeat region close to the sgRNA target sequence. The root stem region is not required for Cas9 DNA cleavage activity in vitro.
7
Considering the conserved nature of crRNA and tracrRNA sequences, we speculated that the U-A repeat region in the core hairpin binds to the Cas9 protein directly. To investigate the role of this region in DNA target cleavage, we shortened it. Initially, we removed one base pair at a time from the U-A repeat region (
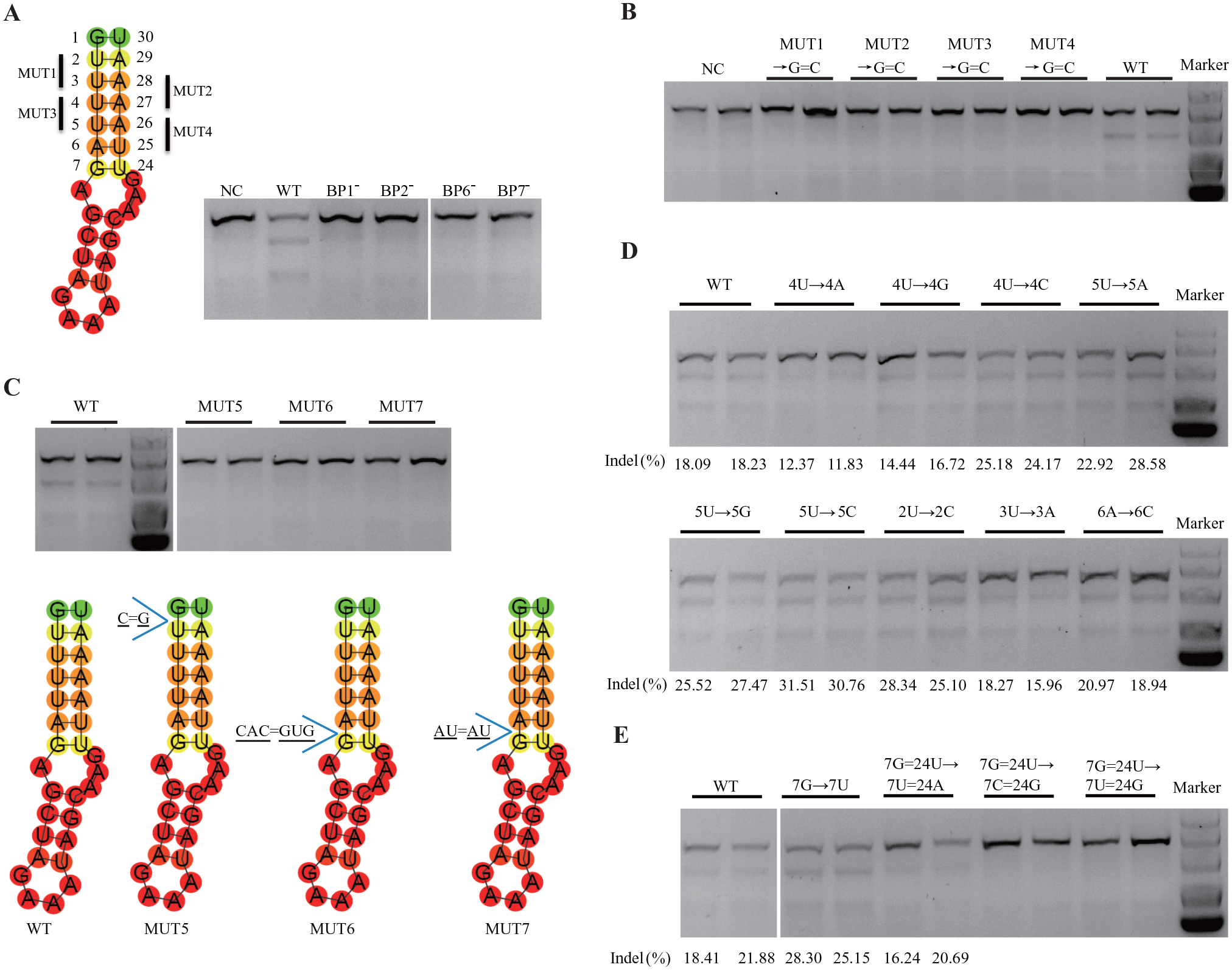
Features of the root stem structure of single-guide RNA (sgRNA) core hairpin. (
Because the screening analysis in E. coli suggested a preference for perfect Watson-Crick base pairing in the sgRNA root stem, we next explored the sequence features of the root stem. We replaced nucleotides at position 4U or 5U with A, G, or C; each replacement resulted in an unpaired nucleotide loop at that site. Changing 4U to A or G reduced the DNA-editing activity slightly, whereas changing 4U to C and 5U to A, G, or C increased the DNA-editing efficiency, based on the results of the T7EI assay (
In the RNA secondary structure, G and U may be paired to maintain stability.34,35 Initially, we speculated that nucleotides G7 and U24 (at the edge of the root stem near the internal loop) could adopt wobble pairing or Watson-Crick pair and that replacing nucleotides at these locations would enhance the stability of sgRNA, thus increasing the editing activity of the Cas9/sgRNA complex. However, we observed that the replacement of both G7 and U24 with Watson-Crick base pairs (C7=G24) and opposite wobble pair (U7:G24) impaired the activity of the complex (
To sum up the presented findings thus far: (1) the root stem of the sgRNA core hairpin could tolerate one base-pair mismatch and remain functional, (2) the root stem had to be exactly six base pairs in length to embed itself in the Cas9 protein groove, (3) nucleotides G7 and U24 did not form a wobble pair or a Watson-Crick pair, and (4) the base of G7 faced outward of the RNA helix axis to contact amino acids of the Cas9 protein.
Length of the Leaf Stem Affects the Activity of Cas9/sgRNA Complex
Jinek et al.
7
demonstrated that the leaf stem of the sgRNA core hairpin is vital for Cas9-mediated DNA cleavage in vitro. Other studies similarly indicated the importance of the sequences near the loop region for the RNP complex.13,36 To investigate the function of the leaf stem in the sgRNA core hairpin in vivo, we designed a set of sgRNAs with leaf stem sequences that were either longer or shorter than the wild-type sgRNA. Notably, we observed that when the leaf stem length was shortened by one base pair, the DNA editing efficiency was higher than that with wild-type sgRNA. Further, instead of losing DNA cleavage activity, sgRNA lacking the entire leaf stem structure also maintained DNA cleavage activity in mammalian cells (
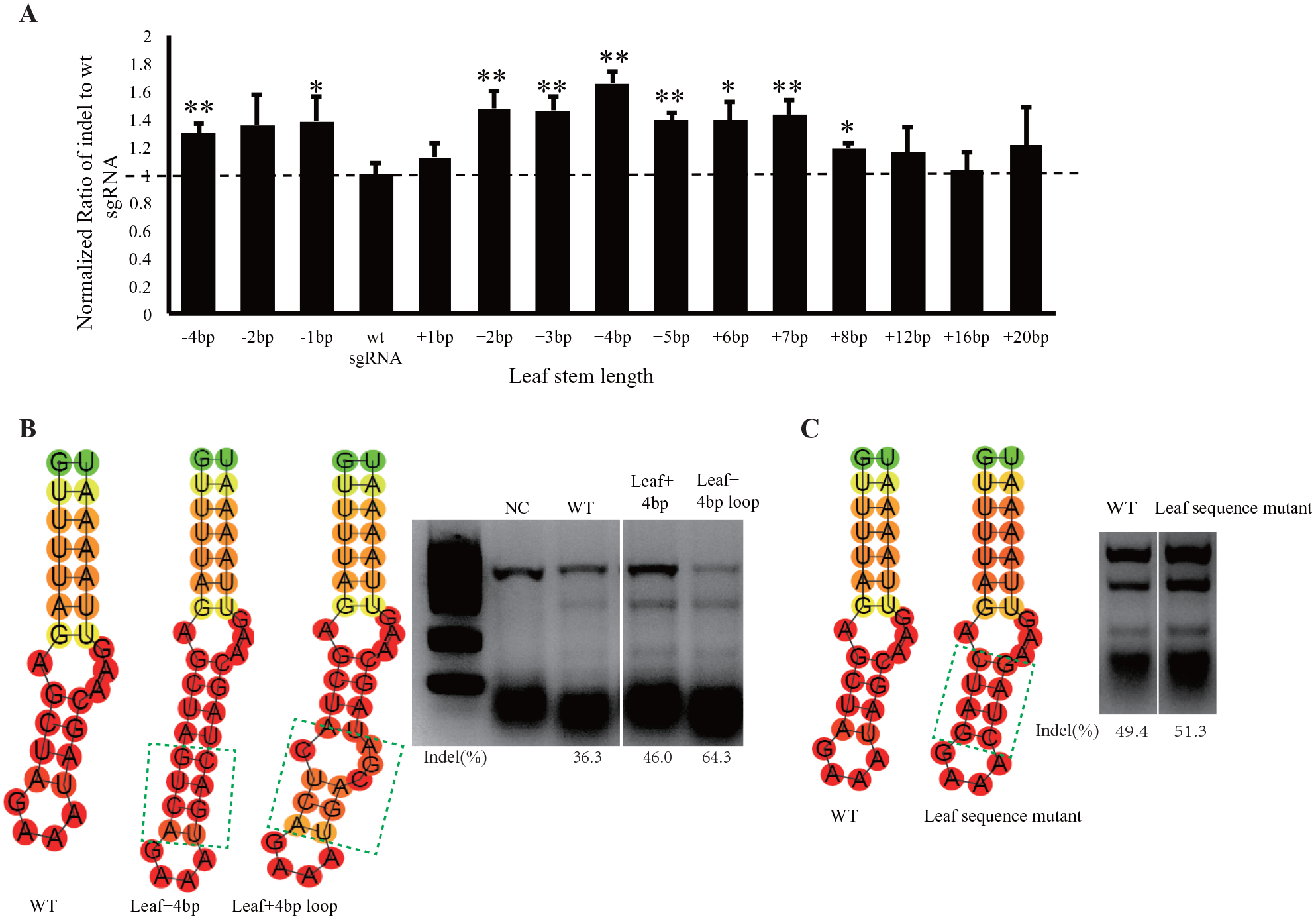
Features of the leaf stem structure of single-guide RNA (sgRNA) core hairpin. (
In our sgRNA sequence screening in E. coli, we observed that the leaf stem did not tend to form Watson-Crick base pairs as compared with the root stem. We next tested the impact of inserting a loop structure into the leaf stem in mammalian cells. We added an additional internal loop structure into the leaf stem region based on the more effective sgRNA structure with a 4 bp leaf stem extension (
We also attempted to alter the sequence of the leaf stem. We exchanged the wild-type sgRNA leaf stem 9GCUA=17UAGC with 9CUGA=17UCAG and found that the mutant sgRNA was still functional in mammalian cells (
Conclusions
The CRISPR/Cas9 system has been successfully implemented to execute sequence-specific cleavage of target DNA in a variety of mammalian cells and model organisms, including Drosophila, zebrafish, frog, mouse, rat, and monkey.38–42 This DNA-editing approach has a great potential for disease treatment, but its clinical applications are limited by safety concerns and low efficiency. In the current study, we aimed to explore the sequence features of the sgRNA core hairpin structure, trying to further study the interaction between Cas9 and sgRNA. We showed that sgRNA could tolerate modification in some regions. These observations may be helpful for improving the editing efficiency of CRISPR/Cas9 system.
The structure of the Cas9/sgRNA complex has been recently analyzed, and the possible mechanism of target DNA cleavage by the RNP complex has been deciphered.12,43–48 In the current study, we systematically analyzed the structural and sequence features of SpCas9 sgRNA core hairpin, in an attempt to identify new approaches for its optimization. The secondary structure of sgRNA contains many stem-loop structures. We focused on the core hairpin, which is the complementary region between the crRNA and tracrRNA. We divided the sgRNA core hairpin structure into three parts, according to its secondary structure: the root stem, the internal loop, and the leaf stem. We first established a screening system in E. coli to explore these sequence features. We selected different parts of the core hairpin and randomly synthesized mutant sequences to construct a mutant sgRNA library. To improve the screening accuracy, we performed two rounds of screening. We analyzed the effective clones and observed that Watson-Crick base pairs were typically present in the root stem region of the core hairpin structure and that there was a potential for sgRNA structure optimization in the upper leaf stem region of the core hairpin structure.
As it may be difficult to evaluate the editing efficiency of mutant sgRNAs in E. coli, we used the T7EI assay to test the cleavage efficiency of different sgRNAs in mammalian cells. We found that the internal loop contributes to sgRNA conformation and identified an important interaction position: the site 23G. In mammalian cells, the number of base pairs in the root stem region is fixed, only six base pairs in length, to maintain the interaction with Cas9 protein. Increasing or decreasing the number of base pairs will affect the cleavage activity of the complex. By contrast with the E. coli screening data, which indicated that the root stem tends to contain completely complementary base pairs, mismatches were tolerated in the root stem region in mammalian cells. According to the presented investigation of the leaf stem in the core hairpin structure, the matched base pairs are dispensable for the interaction with Cas9 protein, and although altering the number of base pairs affected the Cas9 cleavage activity, insertion of an unmatched bulge structure did not inactivate the functional RNP complex. We also found that the leaf stem could be potentially modified, which suggests that the region tolerates artificial reforming. According to other studies, MS2 RNA sequence can be inserted into this region, to allow the recruitment of MS2 coating protein without affecting the Cas9/sgRNA complex assembly,49,50 which is consistent with the data from the current study.
We were unable to analyze the complete sgRNA structure because of the time- and cost-consuming nature of the experiments, but it remains a viable direction for further investigations. Further, we believe that aside from the target sequence, the sgRNA structure also affects the DNA-editing efficiency, and it is of great importance to develop screening systems to analyze other important sites of sgRNA. In recent years, an increasing number of CRISPR systems have been identified and exploited, such as the CRISPR/Cpf1 and CRISPR/C2C1 systems.51–53 We anticipate that future studies will focus on engineering the CRISPR system by altering the wild-type CRISPR crRNA to further improve the efficiency of editing and facilitate clinical applications.
Supplemental Material
Supplementary_Data – Supplemental material for Core Hairpin Structure of SpCas9 sgRNA Functions in a Sequence- and Spatial Conformation–Dependent Manner
Supplemental material, Supplementary_Data for Core Hairpin Structure of SpCas9 sgRNA Functions in a Sequence- and Spatial Conformation–Dependent Manner by Mingjun Jiang, Yanzhen Ye and Juan Li in SLAS Technology
Footnotes
Acknowledgements
We thank Hanshuo Zhang, Yuyang Dong, and Huinan Lu for their suggestions on the experiments.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.