
Abstract
Targeted genome editing using CRISPR/Cas9 is a relatively new, revolutionary technology allowing for efficient and directed alterations of the genome. It has been widely used for loss-of-function studies in animals and cell lines but has not yet been used to study circadian rhythms. Here, we describe the application of CRISPR/Cas9 genome editing for the generation of an F-box and leucine-rich repeat protein 3 (Fbxl3) knockout in a human cell line. Genomic alterations at the Fbxl3 locus occurred with very high efficiency (70%-100%) and specificity at both alleles, resulting in insertions and deletions that led to premature stop codons and hence FBXL3 knockout. Fbxl3 knockout cells displayed low amplitude and long period oscillations of Bmal1-luciferase reporter activity as well as increased CRY1 protein stability in line with previously published phenotypes for Fbxl3 knockout in mice. Thus, CRISPR/Cas9 genome editing should be highly valuable for studying circadian rhythms not only in human cells but also in classic model systems as well as nonmodel organisms.
Genetic loss-of-function studies have been proven to be indispensable for a mechanistic analysis of almost any cellular process. Until recently, the generation of cells or organisms with a targeted deletion or alteration of specific genes mostly exploited homologous recombination (HR)—for example, in germline competent stem cells, integrating exogenous DNA with sequence homology to the target region. However, HR is a very rare event, and thus genome editing was tedious, time-consuming, and expensive. The recent years have witnessed enormous progress in the development of new, much more efficient technologies that not only are revolutionizing genetics but also have enormous potential for biotechnology and medicine. While all these approaches exploit endonucleases that perform double-strand breaks (DSBs) at specific genomic positions, they differ in the ways of guiding the endonucleases to the desired genomic locus. Several excellent reviews provide overviews about similarities and differences between these new technologies (Gaj et al., 2013; Wijshake et al., 2014; Maggio and Gonçalves, 2015). Briefly, both zinc-finger nucleases (ZFNs) and transcription activator–like effector nucleases (TALENs) use a tethering strategy, where genomic targeting is accomplished by fusing the catalytic domain of an endonuclease (mostly FokI) to assemblies of target sequence-specific DNA-binding domains (zinc fingers or DNA-binding domains from type III effector proteins from Xanthomonas spp., respectively). In contrast, in the most recently described CRISPR/Cas9 nuclease (Clustered Regularly Interspaced Short Palindromic Repeats/Cas9 nuclease) genome editing technology, the endonuclease Cas9 is recruited by small RNAs through base pairing to the target DNA. This is much easier compared with the ZFN and TALEN strategies because it does not require multiple cloning steps to assemble the target sequence-specific DNA binding domain array; CRISPR/Cas9 vector generation is a one-step procedure for cloning a single oligonucleotide (representing the guide RNA) into the Cas9 expression vector (e.g., Ran et al., 2013). Common to all three approaches is the exploitation of one of the two major endogenous DNA repair pathways upon endonuclease-mediated DSBs for genome editing: Nonhomologous end-joining (NHEJ) is error-prone and mostly leads to short insertions or deletions (indels) at the target site, often resulting in frame-shift mutations and premature STOP codons when targeted to open reading frames; thus, NHEJ can be used when a gene knockout is desired. In contrast, to create more defined genetic alterations (a knock-in), one should take advantage of HR-mediated repair, where an exogenously introduced repair template is required. In addition to the ease, efficiency, and cost-effectiveness of these new technologies to modify the genome, one further major advantage is the applicability in nonmodel organisms, which traditionally have been not tractable by genetic tools.
Genetic loss-of-function studies have also been indispensable for understanding the molecular basis of circadian clocks—endogenous oscillators that drive daily rhythms in physiology, metabolism, and behavior of almost all light-sensitive species ranging from bacteria to humans (for a review, see Dibner et al., 2010). For example, in mammals, numerous clock gene knockout studies in mice (e.g., van der Horst et al., 1999; Bunger et al., 2000; Zheng et al., 2001; Debruyne et al., 2006) and also large-scale RNA interference experiments in cell culture (Maier et al., 2009; Zhang et al., 2009) helped to shape our current view on the molecular makeup of the circadian oscillator: The fundamental mechanism of rhythm generation is a delayed negative feedback loop of gene regulation, in which Period and Cryptochrome genes are activated by the transcription factor heterodimer CLOCK/BMAL1 and repressed by their own products, the PER and CRY proteins. The delay in negative feedback is probably accomplished by multiple mechanisms including posttranslational modifications of CRY and PER proteins that regulate essential events such as complex formation, subcellular localization, and stability. CRY protein stability is regulated by AMP kinase-mediated phosphorylation (Lamia et al., 2009) and the balanced and localization-specific activity of the E3-ligases FBXL21 and FBXL3, the latter being responsible for promoting the ubiquitination and proteasomal degradation of CRY proteins (Hirano et al., 2013; Yoo et al., 2013). Hypomorphic Fbxl3 mouse mutants (Siepka et al., 2007; Busino et al., 2007; Godinho et al., 2007), complete Fbxl3 knockouts (Hirano et al., 2013; Shi et al., 2013), and RNAi-mediated Fbxl3 downregulation (Maier et al., 2009) lead to increased CRY protein levels and—probably as a result—to very long circadian periods.
In chronobiology, the new genome editing technologies have rarely been applied. To our knowledge, the only published examples are the use of ZFN-mediated genome editing by the Reppert laboratory to successfully target the type 2 vertebrate-like cryptochrome gene of the monarch butterfly (Merlin et al., 2013). In addition, the Wang laboratory used TALENs to create Per2 knockouts in zebrafish (Wang et al., 2015). Notably, so far the use of CRISPR/Cas9 for genome editing—although the most easy and efficient method—has not been reported in chronobiology. As a proof-of-concept, we have applied this technology to generate knockouts of the clock gene Fbxl3 in human U2-OS cells—a highly used model cell line to study the molecular mechanism of the human circadian clock. This worked extremely well (with 70%-100% efficiency at both alleles) and is very simple and robust to apply even for nonspecialized laboratories. As expected, Fbxl3 knockout cells show long-period, low-amplitude rhythms on the population level and also for all single clones we investigated. We analyzed CRISPR/Cas9-induced genome modifications with several assays including sequencing both Fbxl3 alleles, demonstrating that the modifications in the genome (insertions and deletions at the target site) cause the knockout of the protein by introducing premature STOP codons in the open reading frame. We also analyzed potential nonspecific off-target effects and could not detect any.
Materials and Methods
Plasmids, Oligonucleotides, and Sequencing
Oligonucleotides specific for the target site of Fbxl3 were designed using the Optimized CRISPR Design tool (http://crispr.mit.edu/) and ligated into the lentiCRISPR v2 plasmid (Addgene #52961) (Sanjana et al., 2014) using a BsmBI restriction site. PCR-products for sequencing were phosphorylated and ligated into a pUC19 vector. Single clones were sequenced using the M13 forward primer.
Lentivirus Production and Transduction
Lentiviruses were produced in HEK293T cells as previously described (Maier et al., 2009), and virus-containing supernatants were filtered. U2-OS Bmal1-luciferase reporter cells were transduced with 1 mL of virus filtrate supplemented with 8 ng/µL protamine sulfate. After 24 h, the cells were selected for transduced cells using 10 µg/mL puromycin.
T7 Endonuclease Assay
Genomic DNA was harvested using the Direct PRC cell lysis kit (Peqlab, Erlangen, Germany) and Proteinase K (Roth, Karlsruhe, Germany). The Cas9 target region in the Fbxl3 locus was PCR-amplified using forward primer 5′-CACACATCTCAGTGACAGAAG-3′ and reverse primer 3′-CACAATAATTCAATGTAATTCACAG-5′ with Platinum Pfx DNA polymerase according to the manufacturer’s protocol (Life Technologies, Darmstadt, Germany). The PCR-product was gel-purified using the QIAEX II Gel extraction kit (Qiagen, Hilden, Germany), denatured at 95 °C for 5 min, and slowly reannealed via a temperature ramp from 85 to 35 °C (20 sec per −0.5 K). Subsequently, the annealed DNA was digested using T7 endonuclease 1 (New England Biolabs, Frankfurt am Main, Germany) for 90 min at 37 °C. Fragmentation of heteroduplex DNA with single-stranded regions was visualized in an ethidium bromide–stained 10% polyacrylamide gel. The rate of indel mutation was calculated as the ratio of the digested band intensities to the total intensities of all 3 bands.
Antibodies
Four different anti-hFBXL3 antibodies were purchased from Abcam (Cambridge, UK; ab77200 and ab189794), GeneTex (Hsinchu City, Taiwan; GTX110755), and Novus (Littleton, USA; NB100-53784). These primary antibodies were tested for sensitivity and specificity by Western blotting total cell lysate (80 µg per lane) from either wild-type or FBXL3 RNAi-downregulated U2-OS cells (Maier et al., 2009). For all 4 antibodies, endogenous hFBXL3 could not be detected; bands at the predicted size were either not visible or not reduced in intensity upon knockdown, although knockdown cells showed damped and long period reporter rhythms (not shown). Antibodies from GeneTex and Novus at least recognized ectopically overexpressed hFBXL3. The antibody against CRY1 was generated as follows: A His-tagged C-terminal fragment of mCRY1 (about 150 amino acids) cloned into pET15b (kind gift of J. Ripperger) was overexpressed in Escherichia coli BL21 DE, purified by a Ni-NTA column, and used for rabbit immunization (Eurogentec, Seraing, Belgium).
Bioluminescence Recordings
U2-OS Bmal1-luciferase reporter cells were cultured as described (Maier et al., 2009). Briefly, reporter cells were synchronized using 1 µM dexamethasone for 30 min; washed and cultured in phenol-red–free medium; supplemented with 10% fetal calf serum, antibiotics, and 250 µM D-luciferin (PJK, Kleinblittersdorf, Germany); and placed in either a 96-well plate luminometer (Topcount, Perkin Elmer, Rodgau, Germany) or LumiCycle (Actimetrics, Düsseldorf, Germany). Bioluminescence recordings were continuously monitored for several days. Raw data were detrended by dividing the 24-h running average. Periods were estimated by fitting the cosine wave function using the ChronoStar software (Stefan Lorenzen, Bernhard-Nocht-Institut, Hamburg, Germany) as described by Maier et al. (2009).
Western Blotting
Western blotting was performed essentially as described by Maier et al. (2009). Briefly, cells were harvested in RIPA lysis buffer containing 1:100 protease inhibitor cocktail (Sigma, Munich, Germany). Equal amounts of protein were separated by SDS-PAGE using 4% to 12% Bis-Tris gels (Life Technologies), transferred to nitrocellulose membrane, and incubated with anti-CRY1 antibody (1:250, second bleed) or anti β-ACTIN antibody (1:100,000, A3853, Sigma). Next day, membranes were probed with HRP-conjugated secondary antibodies (donkey anti rabbit, Santa Cruz Technologies, SantaCruz, CA; sc2305, 1:1000 in TBST), and a chemiluminescence assay was performed using Super Signal West Pico substrate (Pierce, Rockford, IL) followed by protein detection.
Quantitative RT-PCR
Total RNA was prepared using Pure Link RNA Mini Kit (Life Technologies) according to the manufacturer’s protocol and then reversely transcribed to cDNA using M-MLV Reverse Transcriptase (Life Technologies). Quantitative PCR was performed with SYBRGreen fluorescence assays and analyzed in a CFX96 machine (Bio-Rad, Munich, Germany). For quantitative PCR, QuantiTect primers (Qiagen) were used (hCry1: QT00025067; hCry2: QT00094920; hDbp: QT00055755) except for Gapdh (hGAPDH_fwd: TGCACCACCAACTGCTTAGC, hGAPDH_rev: ACAGTCTTCTGGGTGGCAGTG). The transcript levels for each clock gene were normalized to Gapdh and evaluated according to the 2-ddCt method.
Results and Discussion
To test whether CRISPR/Cas9-mediated genome editing of clock genes is feasible and efficient in circadian reporter cells, we chose to target Fbxl3, since in mice knockout of this gene results in such a severe lengthening of the circadian period (Hirano et al., 2013; Shi et al., 2013) that genome editing might lead to detectable phenotypes even if the efficiency was moderate or low. To generate circadian reporter cells with targeted deletion in the Fbxl3 gene, we equipped U2-OS Bmal1-luciferase reporter cells (Maier et al., 2009) with two different CRISPR/Cas9 constructs (Fbxl3_#1 and Fbxl3_#2). They both target exon 2 of the Fbxl3 gene shortly downstream (100 bp or 175 bp) of the START-codon within the F-box region (Figure 1A). Single-guide RNAs were designed using the Optimized CRISP Design Tool developed by the Zhang laboratory, which suggests using guides with specific target cleavage sites while minimizing possible off-target effects (Ran et al., 2013). The corresponding oligonucleotides were ligated into the lentiCRISPR v2 plasmid (Sanjana et al., 2014), and lentiviruses were produced and used to transduce U2-OS Bmal1-luciferase reporter cells. To analyze the efficiency of genome modification in our cell population, we performed a T7 endonuclease assay (Cho et al., 2013) that analyzes the degree of insertions and deletions (indels) upon CRISPR/Cas9 application. To this end, the genomic target region was PCR-amplified, double-strand DNA melted, and slowly reannealed. Due to the arbitrary nature of the NHEJ repair resulting in many different indels at the target site, reannealed DNA heteroduplexes contain single-strand regions that can be digested with T7 endonuclease. Homoduplexes without single-strand regions occur when Cas9 does not generate genomic double-strand breaks. For both of our CRISPR/Cas9 constructs, we found 67% indels for Fbxl3_#1 and 66% indels for Fbxl3_#2 (Figure 1B). To get a more detailed view on the Cas9-mediated genomic modification, we cloned the amplified target region from Fbxl3_#1 cells into bacterial vectors and sequenced 14 clones. None of them were identified as Fbxl3 wild-type sequence; that is, all showed indels at the targeted location. We found 11 sequences with deletions (ranging from 2 to 46 nucleotides) and 3 sequences with insertions (1-2 nucleotides). Nine of these alterations caused shifts in the open reading frame leading to STOP-codons shortly after the genomic modification; in 5 other cases, in-frame deletions resulted in the loss of 1 to 3 amino acids. Unfortunately, our attempts to quantify FBXL3 protein levels in the cellular populations failed, since all four commercially available anti-hFBXL3 antibodies we tested did not detect endogenous FBXL3 in human U2-OS cells (see Materials and Methods). To analyze the specificity of the Cas9-mediated cleavage, we PCR-amplified the genomic region of the most likely Fbxl3_#2 off-target site (according to the Optimized CRISP Design Tool), which is located on chromosome 11 and has 4 mismatches compared with the target site. Again, we cloned the amplified putative off-target region into bacterial vectors and sequenced 4 clones, all of which showed wild-type sequence indicating absent or very low off-target cleavage (not shown). Together, our genomic analyses demonstrate a highly efficient and specific genome editing in U2-OS cells at this locus with a very likely effect on FBXL3 protein.
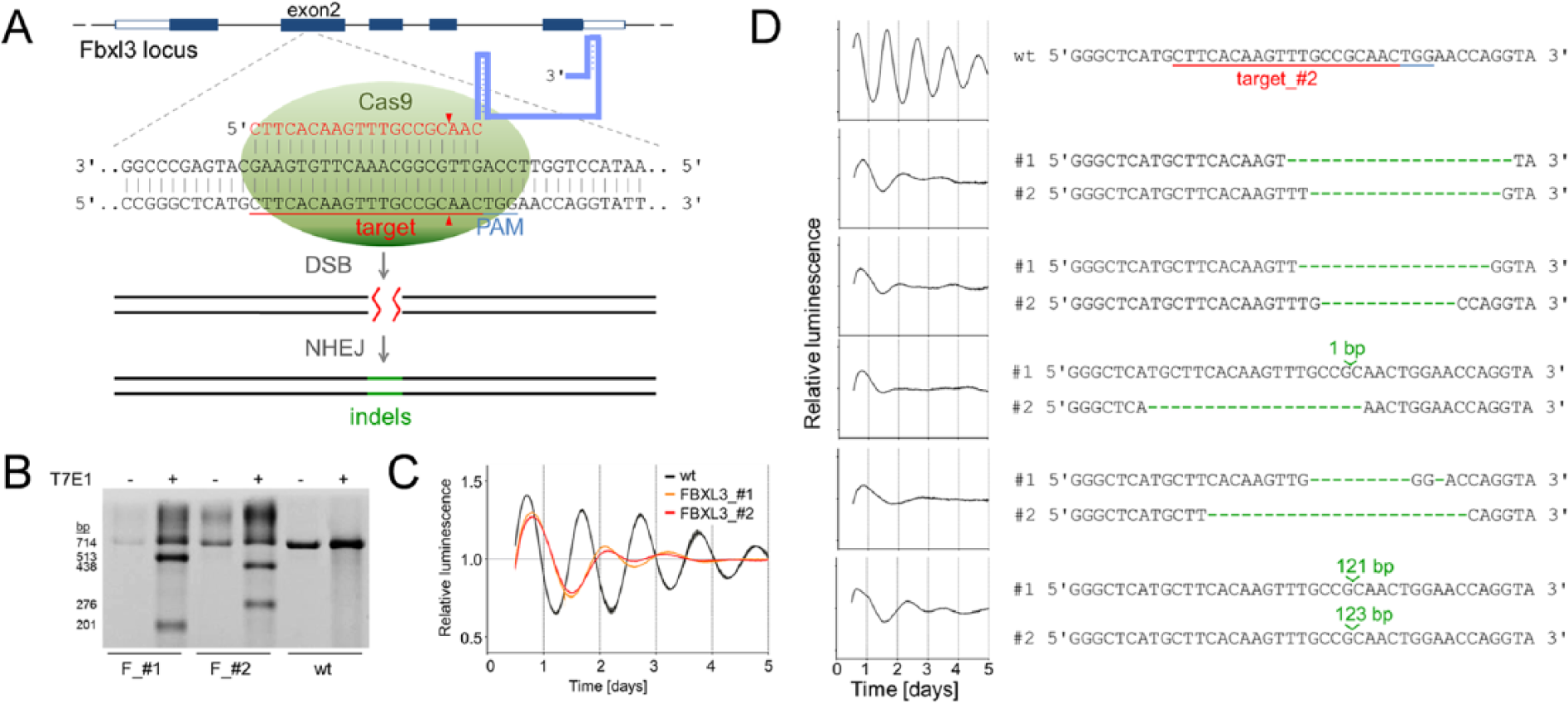
Generation of Fbxl3 knockout cells for circadian phenotyping. (A) Schematic representation of the CRISPR/Cas9 genome editing strategy. A single guide RNA (sgRNA) targeting exon 2 of the human Fbxl3 locus consists of a 20-nt guide sequence (red) and a scaffold (blue). Upon base-pairing of the guide sequence with the genomic target region 5′ to a required so-called PAM motif (5′-NGG), Cas9 performs a double strand break (DSB) about 3 bp upstream of the PAM (red triangle). This DSB is religated through nonhomologous end joining (NHEJ), which is an error-prone repair mechanism leading to insertion/deletion (indel) mutations. (B) T7 endonuclease (T7E1) assay to quantify indel mutations. Genomic DNA of 2 different Fbxl3 Cas9 targets (F_#1 and F_#2) was PCR-amplified, heat-denatured, and slowly reannealed resulting in single-strand regions at indel sites. T7E1 digestion (+) cleaves amplicons from Cas9-edited cells but not from wild-type (wt) U2-OS reporter cells. (C) U2-OS cells harboring a Bmal1-luciferase reporter were lentivirally transduced with 2 different CRISPR/Cas9 constructs (Fbxl3_#1 and Fbxl3_#2) and synchronized with dexamethasone, and luciferase activity was continuously monitored. Detrended average time series (± standard deviation; n = 3) for the 2 CRISPR/Cas9 constructs and an untreated control are given. (D) Circadian phenotypes are correlated with indel mutations at both alleles of the Fbxl3 locus. Single cells from the Fbxl3_#2 population were isolated, and circadian bioluminescence rhythms were measured as described in (C). In contrast to wt reporter cells (shown is a representative single clone), strongly damped, long period rhythms were associated with indel mutations (green) in both (#1 and #2) Fbxl3 alleles.
If the CRISPR/Cas9-induced genomic alterations at the Fbxl3 locus cause a functional knockout of FBXL3 in U2-OS Bmal1-luciferase reporter cells, circadian dynamics of these cells should be similarly disrupted as seen in conventional loss-of-function studies. Mice with either hypomorphic Fbxl3 alleles (Siepka et al., 2007; Godinho et al., 2007) or complete gene knockouts (Hirano et al., 2013; Shi et al., 2013) as well as circadian reporter cells upon RNAi-mediated Fbxl3 silencing show a substantial lengthening of the circadian period by up to 4 h (Maier et al., 2009; Zhang et al., 2009). To study the circadian dynamics of our CRISPR/Cas9 edited U2-OS reporter cells, we synchronized them with dexamethasone and continuously measured Bmal1-luciferase derived bioluminescence over several days. Indeed, both targeted (Fbxl3_#1 and Fbxl3_#2) cell populations showed a strongly damped rhythm with a severe lengthening of the circadian period by several hours (due to the low amplitude and rapid damping of the oscillation of these cells, the extent of period lengthening is difficult to quantify, Figure 1C). These cellular phenotypes are indeed very similar to or even more severe than those from classic knockout mice or RNAi experiments (see Brown et al., 2005), which is indicative of an effective FBXL3 knockout in a very large fraction of the cell populations.
Are both alleles of Fbxl3 equally hit in our CRISPR/Cas9 targeted U2-OS cells? To study the genomic modifications on a single cell level, we isolated 11 cells from the Fbxl3_#2 population by limited dilution, PCR-amplified the genomic target region from single cells, and subcloned the products from 7 randomly chosen cells for sequencing. Although we did not identify a single Fbxl3 wild-type sequence, we found insertions or deletions in both alleles of 5 cells (for 2 cells, sequence information for only 1 allele could be obtained) (Figure 1D). Since almost all these indels led to STOP codons, no functional FBXL3 protein is being made. As expected from the circadian phenotypes of the cell populations (Figure 1B), each of these cell clones (as well as all 6 other isolated clones; not shown) displayed substantially damped, long period Bmal1-luciferase rhythms, whereas 30 single clones from wild-type cells oscillated with high amplitude and an average period of 24.7 ± 0.6 h (Figure 1D). Together, these data show that CRISPR/Cas9-mediated genome editing in U2-OS cells is largely targeting both alleles, simultaneously making it an efficient and rapid tool for creating clock gene knockout cells in a single editing step.
If no functional FBXL3 protein is being made in our putative knockout cells, the molecular circadian oscillator should be similarly affected as in hypomorphic Fbxl3 mouse mutants (Siepka et al., 2007; Busino et al., 2007; Godinho et al., 2007) or complete Fbxl3 knockouts (Hirano et al., 2013; Shi et al., 2013): CRY protein levels should be relatively high due to increased protein stability, while corresponding mRNA levels should be low due to increased repressor function of CRYs. Indeed, we found that transcript levels of Cry1 and Cry2 as well as the E-box-controlled gene Dbp were reduced 3- to 5-fold in single clones from the Fbxl3_#2 population compared with the wild-type population (Figure 2, upper part)—very similar to what has been described in the literature for Fbxl3 mutants (e.g., Siepka et al., 2007). In addition, although Cry1 transcript levels were only about 20% of wild-type levels, CRY1 protein abundance was not reduced but even slightly increased in the CRISPR/Cas9 targeted cell clones (Figure 2, lower part). Together, this strongly indicates an increased CRY1 protein stability due to the CRISPR/Cas9-mediated lack of the E3-ligases FBXL3 required for efficient proteasomal degradation of CRYs.
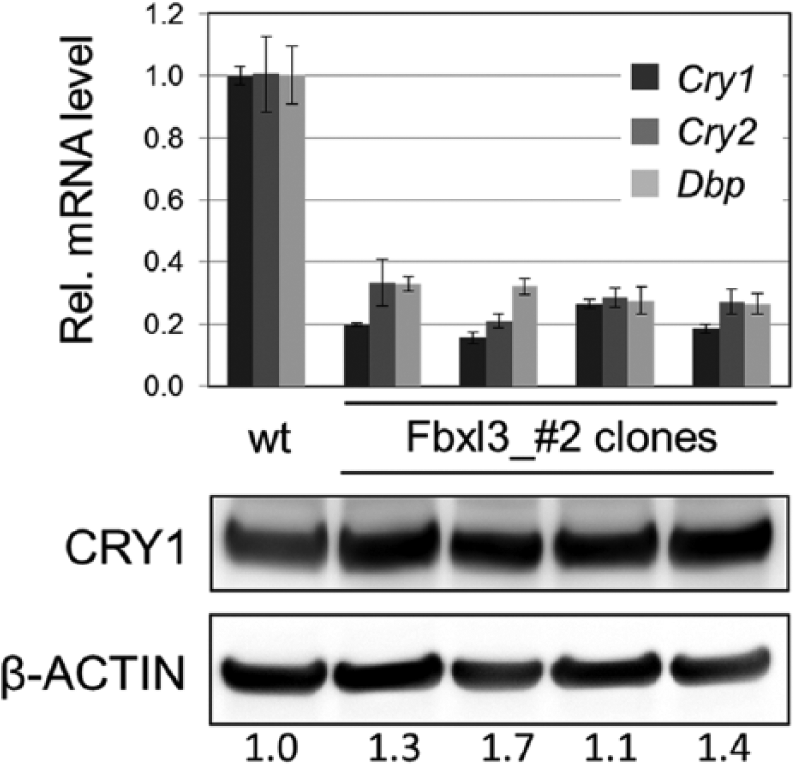
CRY1 protein stability is increased in Fbxl3 knockout cells. Upper part: Transcript levels of Cry1, Cry2, and Dbp either from the wild-type U2-OS cell population or from single clones of the Fbxl3_#2 population were determined by quantitative RT-PCR. Expression levels are given relative to Gapdh and normalized to the average expression level of the wild-type cells (mean ± SD, n = 3). Note that although Cry1 mRNA abundance in knockout cells is low, corresponding CRY1 protein levels are not, but are even slightly increased as determined by Western blot analysis (lower part). Numbers: protein levels relative to β-ACTIN and normalized to the level in wild-type cells.
In summary, we created the first clock gene knockout in a human cell line using CRISPR/Cas9 genome editing. The procedure is easy and fast (a single cloning step, virus production, and transduction), worked with 70% to 100% efficiency (depending on the assay—T7 endonuclease assay or sequencing), and was specific for the target site. While not used in this study, the double-nicking strategy (Trevino and Zhang, 2014) can further reduce the risk of off-target cleavage. Here, simultaneous single-strand breaks (nicks) at opposite strands of the DNA target site using a mutant version of Cas9 and appropriately spaced guide RNAs create double-stranded breaks while increasing the specificity for cleavage. In addition to creating gene knockouts in human cells or in other nonmodel organisms, CRISPR/Cas9 genome editing allows a variety of further applications. First, it allows the introduction of specific mutations, such as point mutations, loxP sites, or reporter tags using repair DNA templates and HR. Since the efficiency of HR-mediated repair is reported to be much lower than for NHEJ, efficient screening strategies to identify positive cell clones are required (e.g., see Bell et al., 2014). Second, CRISPR/Cas9 technology enables customizable gene regulation. To this end, the endonuclease activity of Cas9 is disabled through the insertion of point mutations, and the mutant Cas9 is fused to transcriptional activator or repressor domains (e.g., Agne et al., 2014). By the use of guide RNAs, Cas9-effector fusions are targeted to one or even multiple genomic loci to activate or repress the transcription of endogenous genes. Regulating clock gene expression from endogenous promoters would be an interesting strategy also for studying the circadian core oscillator. Third, pooled CRISPR/Cas9 libraries have been used for conducting genetic screens in mammalian cells (e.g., Wang et al., 2014; Shalem et al., 2014; Koike-Yusa et al., 2014; Zhou et al., 2014). Using this technology to screen for clock modifying genes would probably require a way to phenotype single cells for clock properties, since—due to the still imperfect efficiency of CRISPR/Cas9—assessing a cell population would not necessarily be more powerful than RNAi screens. Fourth, CRISPR/Cas9 genome editing using adeno-associated viral vectors has been used recently in vivo to knock out genes in mouse brain (Swiech et al., 2015)—again, a very interesting technology for potential genome editing in specific brain areas important for chronobiology, such as the suprachiasmatic nucleus, the dorsomedial hypothalamus, or the paraventricular nucleus of the hypothalamus. In conclusion, we predict that CRISPR/Cas9 genome editing and regulation will become a major genetic tool also in chronobiology, with our study generating Fbxl3 knockout cells only being a starting point.
Footnotes
Acknowledgements
We thank Sebastian Jäschke for technical assistance, Thomas Wallach for critical reading of the manuscript, and Rupert Öllinger for helpful discussions. This work was funded by the German Research Foundation (DFG, grant SFB 740/D2).