
Abstract
Phenotypic screening had successfully been used for hit generation, especially in the field of neglected diseases, in which feeding the drug pipeline with new chemotypes remains a constant challenge. Here, we catalyze drug discovery research using a publicly available screening tool to boost drug discovery. The Malaria Box, assembled by the Medicines for Malaria Venture, is a structurally diverse set of 200 druglike and 200 probelike compounds distilled from more than 20,000 antimalarial hits from corporate and academic libraries. Repurposing such compounds has already identified new scaffolds against cryptosporidiosis and schistosomiasis. In addition to initiating new hit-to-lead activities, screening the Malaria Box against a plethora of other parasites would enable the community to better understand the similarities and differences between them. We describe the screening of the Malaria Box and triaging of the identified hits against kinetoplastids responsible for human African trypanosomiasis (Trypanosoma brucei), Chagas disease (Trypanosoma cruzi), and visceral leishmaniasis (Leishmania donovani and Leishmania infantum). The in vitro and in vivo profiling of the most promising active compounds with respect to efficacy, toxicity, pharmacokinetics, and complementary druggable properties are presented and a collaborative model used as a way to accelerate the discovery process discussed.
Introduction
Kinetoplastids are a group of flagellated protozoa responsible for potentially fatal diseases in humans as well as in other mammals. Human African trypanosomiasis (HAT; better known as sleeping sickness), human American trypanosomiasis (Chagas disease), and visceral leishmaniasis (VL; also known as kala-azar) are caused by Trypanosoma brucei (with rhodesiense and gambiense subspecies), Trypanosoma cruzi, and Leishmania donovani, respectively, and account for the major kinetoplastid diseases. Neglected diseases affect populations mainly located in low-income tropical countries and are closely associated with high burdens of mortality and morbidity as well as social stigmatization.
Current existing drugs used in the treatment of kinetoplastid diseases have several drawbacks and limitations in terms of efficacy, as exemplified by the low drug susceptibility profile of specific T. cruzi lineages to benznidazole or nifurtimox for Chagas disease, 1 the resistance of VL to antimonials in India or Nepal, 2 and the lack of efficacy of miltefosine and AmBisome for VL in East Africa.3,4 The limited safety of some of the drugs used to treat kinetoplastid diseases is also a major concern, leading in some instances to the discontinuation of treatment, as in the case of benznidazole and nifurtimox for Chagas disease,5,6 or causing a non-negligible drug-induced mortality in HAT patients when treated with melarsoprol. 7 The lack of pediatric formulations for some of those drugs together with contraindications for pregnant women and those of childbearing age further limit the use of existing treatments. 8 In addition, the various requirements of a cold chain for transport and storage (e.g., AmBisome), parenteral administration by clinical staff, and sometimes lengthy hospitalization (e.g., eflornithine) make them inadequate treatment options for field use and are clear obstacles in terms of drug access for the patients. Finally, some of those drugs have suffered from a lack of availability due to production as well as distribution shortages, as recently documented in the case of benznidazole for the treatment of Chagas disease. 9 The next generation of drugs therefore aim to be effective and safe orally administrated drugs, with a long shelf life in tropical field conditions and simple, short-course drug administration regimens (maximum 10 d, ideally 1–3 d).
Several discovery approaches have been investigated to identify new active starting points for drug development against kinetoplastid pathogens, based on the selection of compound collections and evaluation in either target-based or whole-cell assays. These include, on one hand, the screening of drugs and preclinical candidates developed for other indications against the parasite in question, which may lead to drug repurposing, and, on the other hand, the screening of libraries made up of compounds or chemical classes that are known to affect mammalian targets/biochemical pathways that may also be present in parasites. These compounds/drugs can originate from data mining the literature. More recently, the development of technological platforms for high-throughput screening (HTS) has opened new areas of investigation, allowing the evaluation of significantly larger compound collections (10,000 to several million) representative of a much broader chemical diversity against T. b. brucei, T. cruzi, and L. donovani whole-cell assays. The merits and drawbacks of these various discovery strategies with respect to compound selection and in vitro activity have been detailed and critically discussed elsewhere. 10
The Medicines for Malaria Venture (MMV) has compiled the “Open Access Malaria Box,” a collection of 400 compounds selected from more than 19,000 structurally unique molecules with confirmed activity against the blood stage of Plasmodium falciparum in three large phenotypic (HTS) campaigns undertaken by GlaxoSmithKline (GSK), 11 Novartis-GNF, 12 and St. Jude Children’s Research Hospital.13,14 The Malaria Box, launched at the end of 2009, aims to provide the research community with a collection of starting points for hit-to-lead drug discovery campaigns and a tool to facilitate the understanding of the underlying parasite and host biology. All compounds are commercially available, allowing sourcing of compounds and their near neighbors for efficient confirmation of activity and exploration of structure-activity relationships. Repurposing of the Malaria Box has successfully identified new scaffolds in the field of cryptosporidiosis 15 and schistosomiasis. 16 In addition to initiating new hit-to-lead activities, screening the Malaria Box against other parasites should enable the community to better understand the similarities and differences between them.
Here we present and critically comment on the results of a novel approach of addressing drug discovery in a collaborative and open research framework in relation to kinetoplastid diseases through the Open Access Malaria Box initiative. This exemplifies the collaborative work between Product Development Partnerships organizations (i.e. MMV and Drugs for Neglected Diseases initiative [DNDi]) and shows coordination efforts in drug discovery activities with international expert centers in parasitology.
Materials and Methods
Open Access Malaria Box
The Malaria Box was supplied in V-shaped 96-well plates in 20 µL of a 10 mM DMSO solution and shipped frozen. The chemical purity based on liquid chromatography–mass spectrometry (LC/MS) was ≥90%. Plate mapping and full data on the Malaria Box with the original GSK/St Jude/Novartis compound number, structure, canonical SMILES, biological data, and select in silico physicochemical parameters is available (in pone.0062906.s001) as well as on the MMV Web site (http://www.mmv.org/research-development/malaria-box-supporting-information), together with a list of vendors and their contact details (in pone.0062906.s002). For those compounds of most interest after a first round of screening, a fresh solid sample was repurchased from vendors (MMV665961 from Otava Ltd with 95% purity, MMV019017 from Pharmeks Ltd with ≥90% purity, MMV000498 and MMV019746 from ChemDiv Inc. with ≥95% purity, MMV000248 as AG-690/ 13706332 and MMV000963 as AS-724/STK154807, MMV-006319 as AS-724/STK839477, and MMV020505 as AN-465/ 43369387 from SPECS, all with >90% purity) prior to retesting for biological activity.
Primary Screening Campaign
Primary screening of the library, consisting of 400 compounds, was undertaken in a dose-response scheme using 96-well plates against T. b. brucei, T. b. rhodesiense, T. cruzi, and L. infantum. Standard screening methodologies were adopted as previously described with slight modifications. 17 All assays were performed in duplicate at the Laboratory of Microbiology, Parasitology and Hygiene at the University of Antwerp (LMPH, Belgium). Compounds were tested at 5 concentrations (32, 8, 2, 0.5, and 0.125 μM) to establish a full dose titration and determination of the IC50 (inhibitory concentration 50%) using Graph Pad Prism software (GraphPad Software, Inc., La Jolla, CA). The IC50 values were calculated using blank noninfected controls as 100% and infected nontreated controls as 0% inhibition. The final test volume was 200 µL, and the in-test concentration of DMSO was below 0.5%, a level that is known not to interfere with the different assays. Selectivity of activity was assessed by simultaneous evaluation of cytotoxicity on a human fibroblast (MRC-5) cell line.
Antileishmanial Activity
L. infantum MHOM/MA(BE)/67 amastigotes were collected from the spleen of an infected donor hamster and used to infect primary peritoneal mouse macrophages. To determine in vitro antileishmanial activity, 3 × 104 macrophages were seeded in each well of a 96-well plate. Culture medium was RPMI-1640 supplemented with L-glutamine (20 mM), 16.5 mM sodium hydrogen carbonate, and 10% fetal calf serum (FCS). After 2 d of outgrowth, 5 × 105 amastigotes/well were added and incubated for 2 h at 37 °C. Prediluted samples to be tested were subsequently added, and the plates were further incubated for 5 d at 37 °C and 5% CO2. Total parasite burdens in the well were microscopically assessed after Giemsa staining and expressed as a percentage of the total burden in the blank controls without compound. In treated wells with high amastigote burdens, an overall estimate of the total burden per well was made without discrimination between the number of infected macrophages and the number of amastigotes per infected cell. In treated wells with low burdens, exact counting was performed. Miltefosine was used as reference and obtained an IC50 value of 7.56 ± 1.25 µM. The data set was deposited in the ChEMBL database under the assay ID CHEMBL2028076.
Antitrypanosomal Activity
For HAT assays, T. b. brucei (strain Squib-427) and T. b. rhodesiense (strain STIB 900) were cultured at 37 °C and 5% CO2 in Hirumi-9 medium, 18 supplemented with 10% FCS. Assays were performed in 96-well plates, each well containing 10 µL of the compound dilution and 190 µL of the parasite suspension (1.5 × 104 trypomastigotes/well for T. b. brucei and 4 × 103 for T. b. rhodesiense). Parasite growth was assessed after 72 h at 37 °C by adding resazurin. 19 After 6 h (T. b. rhodesiense) or 24 h (T. b. brucei), fluorescence was measured (λex 550 nm, λem 590 nm). Suramin was used as reference and obtained an IC50 value of 0.05 ± 0.01 µM. The data sets were deposited in the ChEMBL database under the assay IDs CHEMBL2028074 and CHEMBL2028075, respectively.
For Chagas disease, T. cruzi Tulahuen CL2 was maintained on MRC-5 cells in minimal essential medium (MEM) supplemented with 20 mM L-glutamine, 16.5 mM sodium hydrogen carbonate, and 5% FCS. In the assay, 4 × 103 MRC-5 cells and 4 × 104 parasites were added to each well, and after incubation at 37 °C for 7 d, parasite growth was assessed by adding the alpha-galactosidase substrate chlorophenol red alpha-D-galactopyranoside. 20 The color reaction was read at 540 nm after 4 h, and absorbance values were expressed as a percentage of the blank controls. Benznidazole was used as reference and obtained an IC50 value of 3.18 ± 0.11 µM. The data set was deposited in the ChEMBL database under the assay ID CHEMBL2028073.
Cytotoxicity against MRC-5 Cells
MRC-5 SV2 cells were cultivated in MEM, supplemented with L-glutamine (20 mM), 16.5 mM sodium hydrogen carbonate, and 5% FCS. For the assay, 104 MRC-5 cells/well were seeded onto the test plates containing the prediluted sample and incubated at 37 °C and 5% CO2 for 72 h. Cell viability was assessed fluorimetrically 4 h after the addition of resazurin. Fluorescence was measured (λex 550 nm, λem 590 nm) and the results expressed as percentage reduction in cell viability compared with control. Tamoxifen was used as reference and obtained an IC50 value of 11.17 ± 0.16 µM. The data set was deposited in the ChEMBL database under the assay ID CHEMBL2028077.
Medicinal Chemistry Analysis of Hits Identified from Primary Screening
Cluster analysis of the entire Open Access Malaria Box (n = 400) was completed using a predefined fingerprinting set (DotFPCF_1024, number of clusters = 50, Tanimoto similarity). A priority hit list was defined based on the following criteria: (1) number of hits within the cluster based on predefined activity and selectivity criteria; (2) absence of toxicophores based on filters developed in house, which include a list of 110 undesirable chemical moieties, permanently charged molecules, and frequent hitters based on PAINS filters 21 ; (3) druglike structural features (based on Lipinski’s rule of 5 scoring) 22 ; (4) scaffold novelty with respect to kinetoplastid activity; and (5) potential for central nervous system (CNS) penetration, in the case of T. b. rhodesiense hits only (TPSA <70 Å2).
Pharmacokinetic Studies for Selected Hits in the Malaria Box
Fasted animals (male CD-1 mouse, 7–9 wk of age, 30 ± 10 g, n = 3) obtained from an approved vendor (SLAC Laboratory Animal Co. Ltd., Shanghai, China; or Sino-British SIPPR/BK Laboratory Animal Co. Ltd, Shanghai, China) were administered with a single oral dose (~50 mg/kg) using DMSO:1% HPMC (5:95) as vehicle. Blood sampling was performed via the submandibular or saphenous vein (t = 0.083, 0.25, 1, 2, 4, 6, and 9 h) and transferred into microcentrifuge tubes containing sodium heparin (1000 IU/µΛ) as anticoagulant and placed on wet ice until processed for plasma by centrifugation at 3000×g for 15 min at 4 °C. Plasma samples were stored in polypropylene tubes, quick frozen over dry ice, and kept at −70 ± 10 °C until liquid chromatography–tandem mass spectrometry (LC/MS/MS) analysis. LC/MS/MS methods for the quantitative determination of compound plasma levels were developed with an internal standard. Finally, concentration in plasma versus time data were analyzed by noncompartmental approaches using the WinNonlin software program (Phoenix WinNonlin, version 6.2.1; Pharsight, Mountain View, CA) and expressed as Cmax, Tmax, T½, and AUC(0-t). Graphs of plasma concentration versus time profile are provided in the supplementary material (
Hit Confirmation against T. b. rhodesiense STIB 900
Compounds prioritized from primary screening were retested at Swiss Tropical and Public Health Institute (Swiss TPH), Basel, Switzerland, against T. b. rhodesiense in duplicate and at varying concentrations to obtain a dose-response curve. The STIB 900 strain was isolated in 1982 from a human patient in Tanzania and after several mouse passages cloned and adapted to axenic culture conditions. 23 MEM (50 µL) supplemented with 25 mM HEPES, 1 g/L glucose, 1% MEM nonessential amino acids (100×), 0.2 mM 2-mercaptoethanol, 1 mM Na-pyruvate, and 15% heat-inactivated horse serum was added to each well of a 96-well plate. Serial drug dilutions of 11 threefold dilution steps covering a range from 100 to 0.002 µM were prepared. Then, 4 × 103 bloodstream forms of T. b. rhodesiense STIB 900 in 50 µL were added to each well, and the plate was incubated at 37 °C under a 5% CO2 atmosphere for 70 h. Ten microliters of Alamar Blue (resazurin, 12.5 mg in 100 mL double-distilled water) was then added to each well and incubation continued for a further 2 to 4 h. 19 The plates were read with a Spectramax Gemini XS microplate fluorometer (Molecular Devices Corporation, Sunnyvale, CA) using an excitation wave length of 536 nm and an emission wave length of 588 nm. The IC50 values were calculated by linear regression 24 from the sigmoidal dose inhibition curves using SoftmaxPro software (Molecular Devices Corporation). For the retests, newly delivered solid compound stocks were used. Melarsoprol (Arsobal Sanofi-Aventis, received from the World Health Organization) was used as control (IC50: 0.008 µM).
To investigate the effect of serum on compound activity, the serum concentration in vitro was varied at 5%, 15%, and 30%. IC50 and IC90 values were determined as described above.
Examination of Drug Action
Microcalorimetry Studies Using T. b. rhodesiense
In vitro time of drug action was monitored using isothermal microcalorimetry. Previous experiments have shown that heat flow data can be used as a proxy for the number of viable cells, 25 defined as trypanosomes still moving under the microscope. With this method, the time to the onset of drug action and the time to kill can be determined on a real-time basis. 26 For experiments with permanent drug exposure, bloodstream trypanosomes (2 mL at 1 × 105 organisms/mL per ampoule) were placed in 4 mL ampoules and spiked with the appropriate test compound at concentrations of 1×, 3×, 10×, and 30× IC50 with a final DMSO concentration of 0.1% (vol/vol). The same culture medium with 15% horse serum was used as described above. Trypanosome-free negative controls contained only culture medium. After the ampoules had been inserted into the isothermal microcalorimetry instrument (Thermal Activity Monitor, model 249 TAM III; TA Instruments, New Castle, DE), heat flow was continuously measured (1 reading/s) at 37 °C. Each experiment with permanent compound exposure was set up in duplicate and repeated two times. The IC50 values against T. b. rhodesiense were first determined at Swiss TPH as described above.
Reversibility of Trypanocidal Effects
Trypanosomes were seeded in clear 96-well V-bottom plates at a density of 1 × 105 parasites per well and incubated with the serially diluted test compound. One plate was prepared for each time point. At the designated time, 1, 6, and 48 h, a plate was removed from the incubator and spun at 4.4 × 103 rpm for 5 min to collect the parasites. The supernatant was aspirated, and 100 µL of warmed medium (37 °C) was added to each well. The wash was repeated twice more. The parasites were resuspended in 100 µL of warmed medium, and 20 µL of this suspension was added to 80 µL of medium in triplicate plates. Following 72 h of incubation, resazurin was added and trypanocidal activity determined.
In Vivo T. b. rhodesiense (STIB 900) Acute Mouse Model
The STIB 900 acute mouse model mimics the first stage of the disease. Four female NMRI mice were used per experimental group. Each mouse was inoculated intraperitoneally with 104 bloodstream forms. Heparinized blood from a donor mouse with approximately 5 × 106/mL parasitaemia was suspended in PSG buffer to obtain a trypanosome suspension of 1 × 105/mL. Each mouse was injected with 0.25 mL. Compounds were formulated in 100% DMSO, diluted 10-fold in 1% HPMC. Compound treatment was on 4 consecutive days, day 3 to 6 postinfection. The compounds were administered intraperitoneally or orally by gavage in a volume of 0.1 mL/10 g. Four mice served as infected-untreated controls. Parasitaemia was monitored using smears of tail-snip blood twice a week after treatment. Mice are considered cured when no parasitaemia relapse was detected in the tail blood over the 60-day observation period. Mean relapse days are determined as day of relapse postinfection of mice. Mice were euthanized when high levels of parasitemia were evident (107 trypanosomes/mL blood) on peripheral blood slides. In vivo efficacy studies in mice were conducted at the Swiss Tropical and Public Health Institute (Basel, Switzerland) according to the rules and regulations for the protection of animal rights (“Tierschutzverordnung”) of the Swiss “Bundesamt für Veterinärwesen.” They were approved by the veterinary office of Canton Basel-Stadt, Switzerland.
Organization and Responsibilities of the Collaborative Framework
The collaborative frame included the following contributing partners: MMV provided the physical supply of 400 compounds of the Open Access Malaria Box, including chemical structures, physiochemical properties, and their associated in vivo pharmacokinetic (PK) profiles. The Laboratory of Microbiology, Parasitology and Hygiene from University of Antwerp undertook the primary screening of the Open Access Malaria Box against the kinetoplastid assay panel as well as the cytotoxicity counterscreening assay. The Parasite Chemotherapy Unit of the Swiss Tropical and Public Health Institute was responsible for hit reconfirmation against T. b. rhodesiense, secondary assays against T. b. rhodesiense (as listed above), as well as the execution of the in vivo assays in the acutely infected T. b. rhodesiense mouse model. The DNDi was responsible for the coordination of the research collaboration, guidance with respect to hit and lead selection criteria, and management of data from the collaboration.
Results
The 400 compounds of the Open Access Malaria Box were evaluated in duplicate in an in vitro dose-response screening scheme against the bloodstream forms of T. b. rhodesiense and T. b. brucei as well as against the intracellular amastigote forms of T. cruzi and L. infantum. A counterscreen using MRC-5 cells was used to determine a selectivity index (ratio of IC50s). All data generated in the primary screening campaign against the T. b. brucei (CHEMBL2028074), T. b. rhodesiense (CHEMBL2028075), L. infantum (CHEMBL2028076), T. cruzi (CHEMBL2028073), and the MRC-5 cell line (CHEMBL2095143) are available from the CHEMBL Web site (https://www.ebi.ac.uk/chembl) together with the 400 chemical structures of the Open Access Malaria Box. The very good correlation (r2 of 0.80,
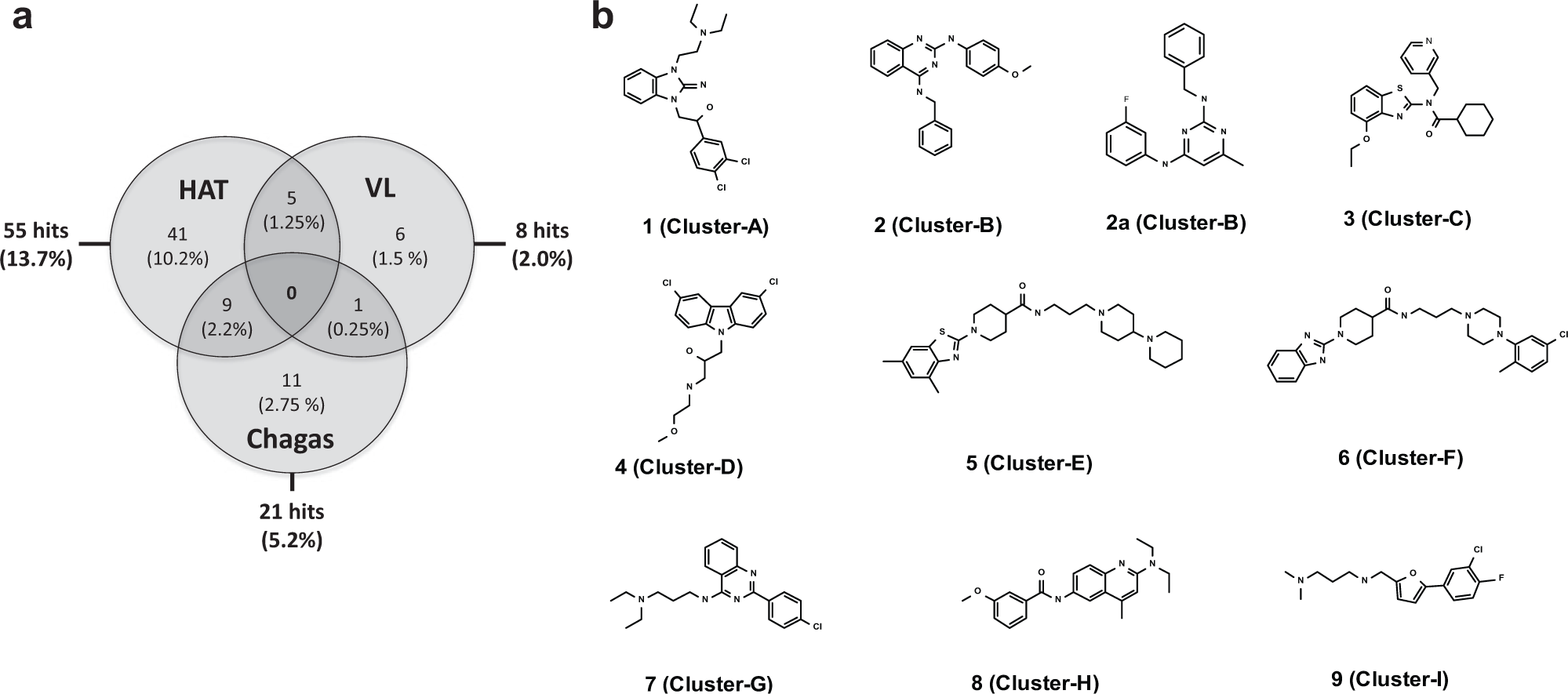
(
From an initial list of active clusters (4 for L. infantum, 5 for T. cruzi, and 15 for T. b. rhodesiense) based on the distribution of the active compound scaffolds identified from cluster analysis, the application of the aforementioned set of criteria led to the selection of two priority hits (
Compound
In Vitro Activity against T. b. rhodesiense (STIB 900) and Pharmacokinetic Profile of Four Selected Compounds.
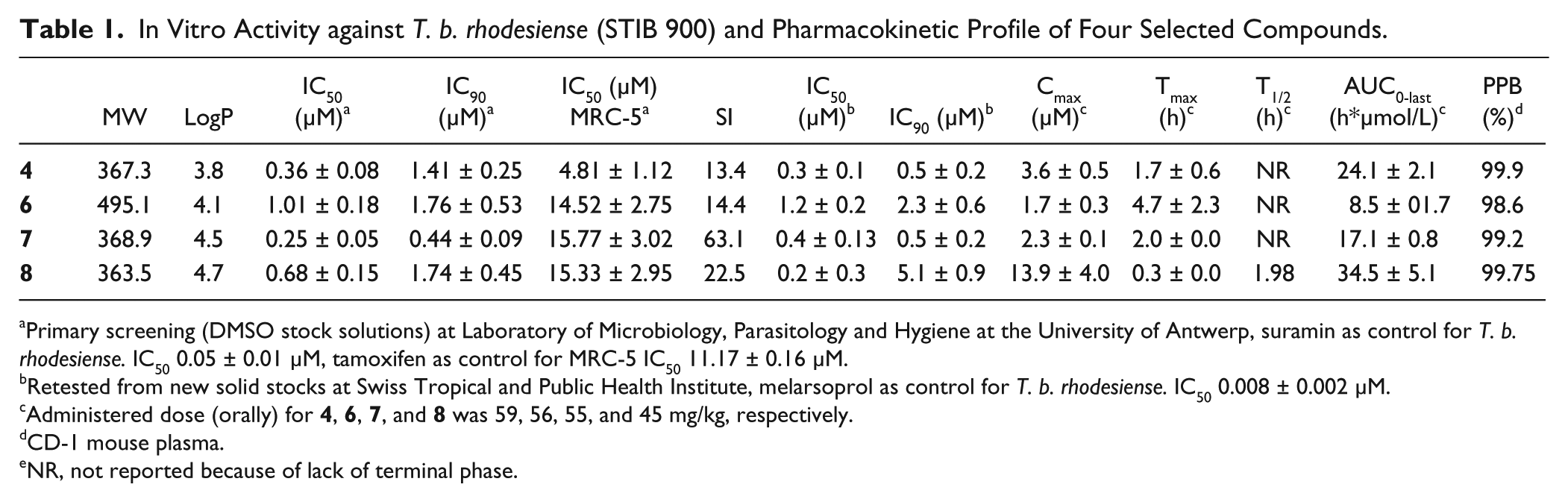
Primary screening (DMSO stock solutions) at Laboratory of Microbiology, Parasitology and Hygiene at the University of Antwerp, suramin as control for T. b. rhodesiense. IC50 0.05 ± 0.01 µM, tamoxifen as control for MRC-5 IC50 11.17 ± 0.16 µM.
Retested from new solid stocks at Swiss Tropical and Public Health Institute, melarsoprol as control for T. b. rhodesiense. IC50 0.008 ± 0.002 µM.
Administered dose (orally) for
CD-1 mouse plasma.
NR, not reported because of lack of terminal phase.
Before proceeding with in vivo efficacy assays, the in vitro activity of
Possible causes of failure, including influence of serum protein binding on the in vitro activity, the time course of drug action, and reversibility of the drug effect, were examined to explain these disappointing in vivo results. As all 4 compounds (
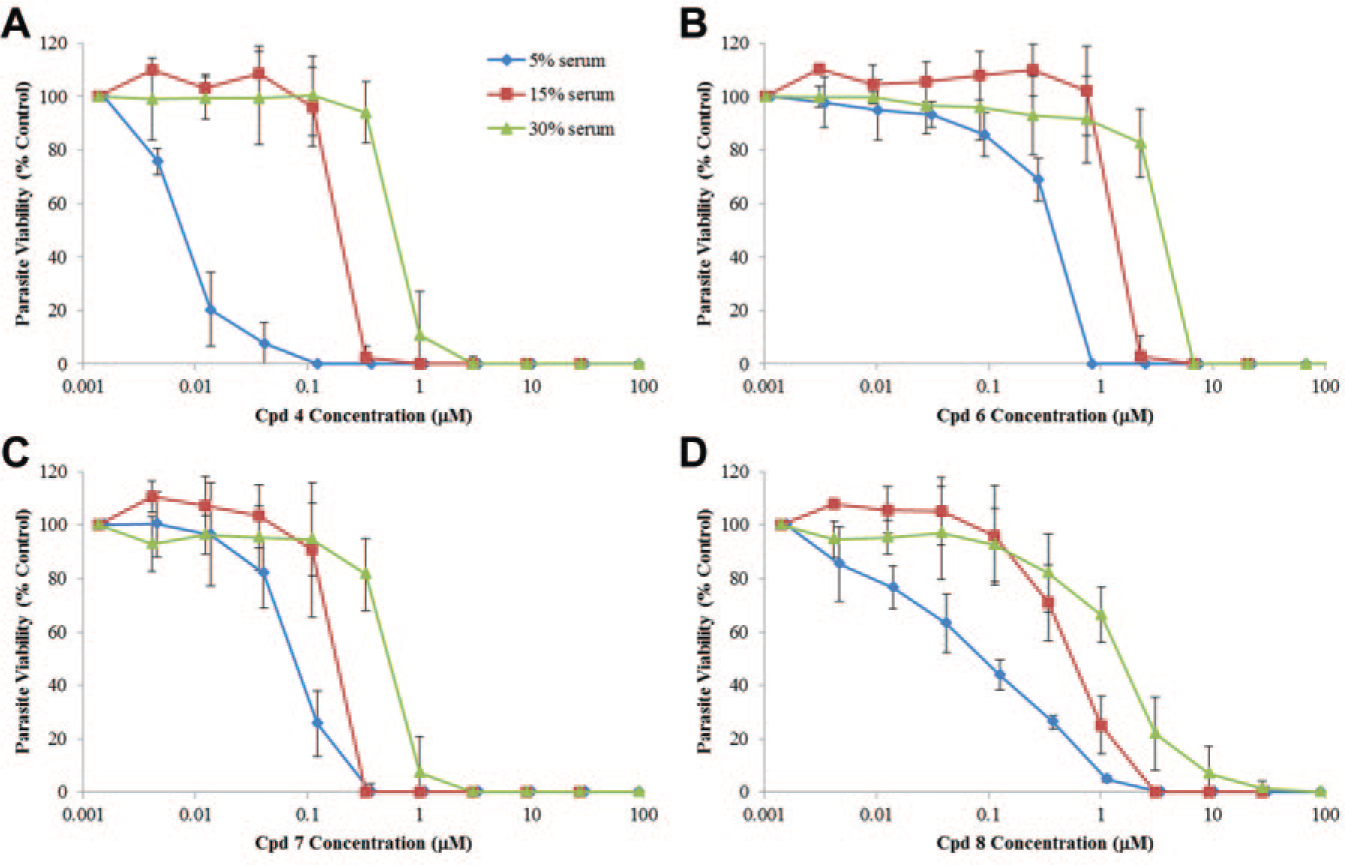
In vitro dose-response curve of compounds
To obtain a better understanding of the PK/pharmacodynamic parameters of the in vitro activity of compounds
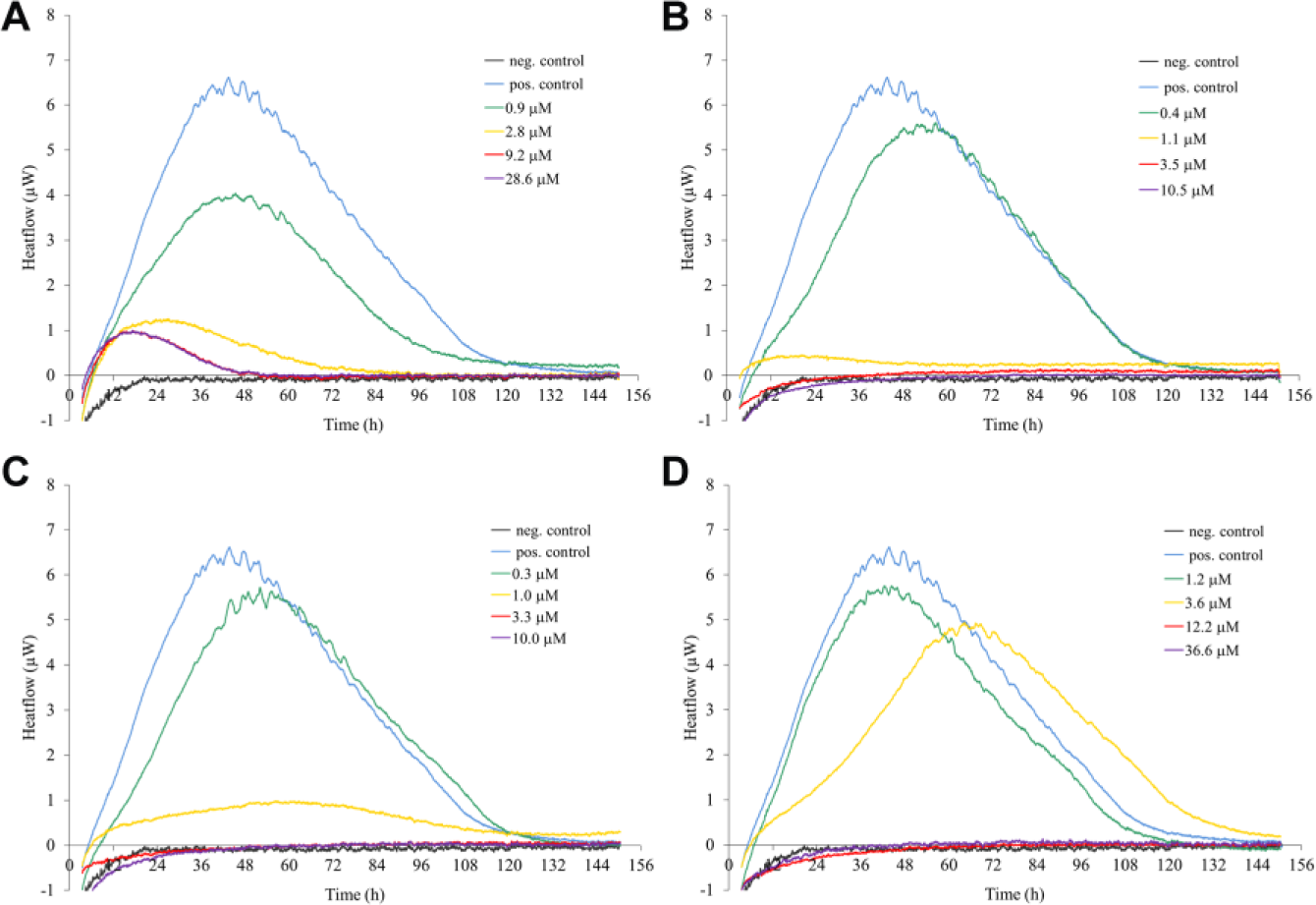
Microcalorimetric growth profiles of T. b. rhodesiense strain STIB900 in the presence of various concentrations of
Compound
Compound
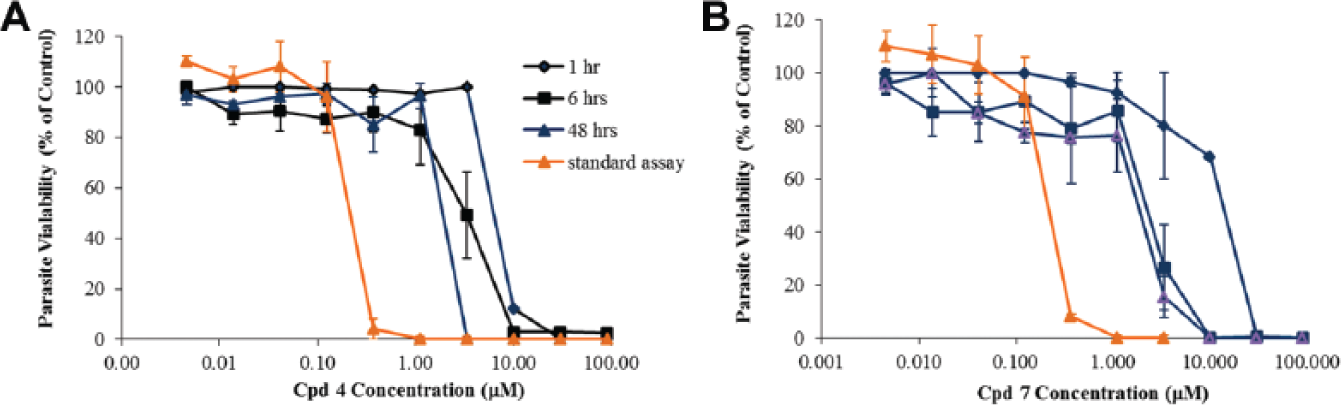
Irreversibility of trypanocidal effect A (
Compound
A concentration of 1.2 µM (1× IC50) of
Discussion
Figure 5
provides an overview of the sequence of in vitro and in vivo experiments run in the frame of this collaborative work in association with the criteria used to assist the decision-making process. The respective cutoff values used for hit qualification (IC50 <5 µM and SI >10 for T. b. rhodesiense and T. cruzi and IC50 <10 µM and SI >10 for L. infantum) have been arbitrarily set on the basis of the DNDi empirical experience to progress active starting points identified from in vitro phenotypic screening into drug development programs. The resulting hit rates of 13.7% for T. b. rhodesiense, 5.2% for intracellular amastigote form of T. cruzi, and 2.0% for the intracellular amastigote form of L. infantum were significantly higher than the ones associated with the screening of unbiased selected compound collections, also frequently referred to as “diversity collections.” Indeed, recent figures based on previous screening campaigns of diversity collections against the bloodstream form of T. brucei subspecies (average 0.45%, ranging from 0.1%–1.2%, approximately 1.3 million compounds assayed), T. cruzi (average 0.55%, ranging from 0.03%–0.85%, approximately 500,000 compounds assayed) as well as L. donovani 0.06% (ranging from 0.02% to 0.09%, 500,000 compounds assayed) yielded much lower hit rates.
10
Although based on a rather limited number of compounds, this observed enrichment of the hit rate (about 30 times for T. brucei and L. donovani, 10 times in the case of T. cruzi) can be explained by the biological selection bias as well as the druggability properties of the compounds included in the Malaria Box collection. All compounds have indeed previously been demonstrated as being active against the blood-stage form of P. falciparum in vitro,
14
suggesting that the hits have favorable properties to reach their biological target in a whole-cell assay. A potential commonality of targets shared between P. falciparum and kinetoplastids might additionally contribute to the improved hit rates, as seen for the T. cruzi and L. infantum hits related to 2,4-diaminopyrimidine and 2, 4-diaminoquinazoline scaffolds (cluster 2;
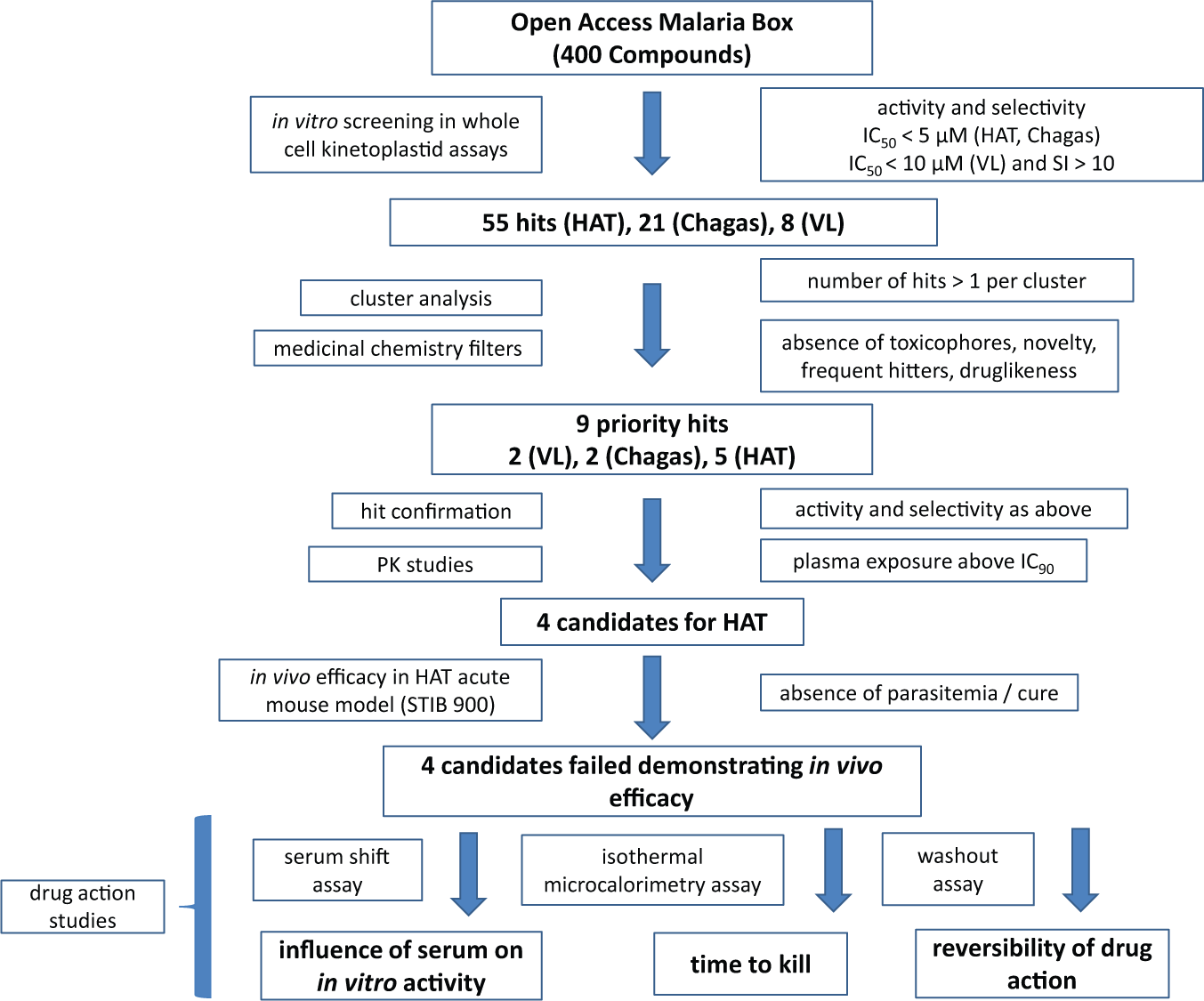
Workflow used to identify and progress hits of the Open Access Malaria Box including key criteria considered in the decision-making process. Human African trypanosomiasis (HAT), T. b. rhodesiense (STIB 900); visceral Leishmaniasis (VL), L. infantum MHOM/MA(BE)/67); and Chagas disease (T. cruzi, Tulahuen CL2).
Taken overall, the hit rates recorded in the screening of the Malaria Box advocate for the larger-scale screening of antiplasmodial activity-biased compound collections against kinetoplastids. Finally, the fact that compounds qualify as P. falciparum hits with activity ranging from the low-nanomolar to micromolar range in respect to IC50 in whole-cell screening assays but have not yet been engineered out to act against any specific Plasmodium falciparum target may also account for the increased hit rate.
Free access to chemical structures made public domain before undertaking any screening activities enabled a fast initial triage of the identified hits from primary screening based on activity, selectivity, structural similarity, and drug-likeness. Furthermore, upon request, access to key information generated as part of the Malaria Box Open Access collaboration, such as the in vivo levels of drug exposure in murine noninfected models, greatly supported a quick and scientifically based rationale to move forward a short list of 7 hits (
Overall, the Open Access Malaria Box has offered a fast, transparent, and cost-effective collaborative framework to advance a set of compounds directly from screening to proof of concept of in vivo efficacy with a clear scientific rationale. The upfront availability of chemical structures devoid of any intellectual property rights, immediate access, and resupply of physical quantities of material for screening and follow-up experiments, as well as the availability of cytotoxicity data related to the compounds and in vivo PK profile, have enabled a quick and rational progression of this interlaboratory collaborative work and greatly facilitated the associated decision-making process. This collaborative framework has also permitted avoiding any work duplication and the concomitant waste of resources by prioritizing the most promising molecules while working on a slower pace or discontinuing any discovery activities on others. Currently, lower priority should be given to those compounds associated with undesirable mechanisms of action, such as static or slow-killing profiles. For other compounds, novel data on structurally related chemical analogs—such as those associated with significantly higher in vitro activity, short onset of drug action, lower protein binding profile, or superior drug exposure in vivo—may lead to a revival of one of more of those active scaffolds in the future.
In summary, the failure to identify to date an in vivo active candidate for kinetoplastid diseases at the end of this collaborative process may simply reflect the relatively high bar to be cleared without investing more intensively into a medicinal chemistry program, rather than calling into question the adequacy of this specific compound collection and collaborative model to identify new active starting points.
Although the prioritized compounds showed no in vivo efficacy, further investigation of the chemistry of these scaffolds may improve the in vitro activity and/or the PK profiles. Research groups are encouraged to contact the authors should they be interested in these hits. The current literature available regarding hits
In conclusion, the Open Access Malaria Box initiative was able to create an original open collaborative working environment suitable for the discovery and early progression of active candidates for neglected diseases in a timely manner and within an IP-free framework while working with limited resources. The forthcoming Open Access Pathogen Box initiative (www.pathogenbox.org), which will make 400 diverse, druglike molecules with demonstrable activity against a broad panel of neglected diseases available by the end of 2015, is eagerly awaited.
Footnotes
Acknowledgements
The authors thank An Matheeussen (LMPH), Monica Cal, Maja Jud, Christiane Braghiroli, and Guy Riccio (all Swiss TPH) for technical assistance. We thank Pascal Mäser (Swiss TPH) for discussions of and input into the manuscript. The authors also wish to thank Dr. Susan Wells for the critical reading as well as editing of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was primarily funded by the Drugs for Neglected Diseases initiative. For the work described in this article, the Drugs for Neglected Diseases initiative received financial support from the following donors: Department for International Development (UK), Reconstruction Credit Institution–Federal Ministry of Education and Research (KfW-BMBF; Germany), Bill & Melinda Gates Foundation (United States), and Médecins Sans Frontières. The donors had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the article apart from those disclosed.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.