
Abstract
Methods to discover biologically active small molecules include target-based and phenotypic screening approaches. One of the main difficulties in drug discovery is elucidating and exploiting the relationship between drug activity at the protein target and disease modification, a phenotypic endpoint. Fragment-based drug discovery is a target-based approach that typically involves the screening of a relatively small number of fragment-like (molecular weight <300) molecules that efficiently cover chemical space. Here, we report a fragment screening on TbrPDEB1, an essential cyclic nucleotide phosphodiesterase (PDE) from Trypanosoma brucei, and human PDE4D, an off-target, in a workflow in which fragment hits and a series of close analogs are subsequently screened for antiparasitic activity in a phenotypic panel. The phenotypic panel contained T. brucei, Trypanosoma cruzi, Leishmania infantum, and Plasmodium falciparum, the causative agents of human African trypanosomiasis (sleeping sickness), Chagas disease, leishmaniasis, and malaria, respectively, as well as MRC-5 human lung cells. This hybrid screening workflow has resulted in the discovery of various benzhydryl ethers with antiprotozoal activity and low toxicity, representing interesting starting points for further antiparasitic optimization.
Keywords
Introduction
Phenotypic screening plays a pivotal role in the identification of novel biologically active compounds. 1 Analysis of first-in-class small-molecule drugs approved by the U.S. Food and Drug Administration between 1999 and 2008 revealed that 56% were discovered using phenotypic screening methods, whereas 34% originated from target-based approaches. 2 Placing a focus on efficacy screening is a clear driver for clinical success.
One of the challenges in phenotypic drug discovery is the distinct definition of the phenotype. 1 In antimicrobial drug discovery, the desired phenotype is well defined and often comprises inhibition of proliferation or cell death. Furthermore, issues such as cell membrane penetration, efflux mechanisms, and unique cellular responses are difficult to incorporate in target-based approaches that focus on a single protein target. Even though hit finding and optimization can be fairly efficient for target-based approaches, it is often found that the biochemical activity of such compounds does not translate into cell-based phenotypic activity.1,3
When considering these issues of translating activity on a single protein target into phenotypic antimicrobial activity, fragment-based drug discovery (FBDD) presents an even bigger challenge, as the hit fragments are often detected using highly sensitive biochemical or biophysical assays. 4 The affinity and activity levels of these low-molecular-weight (MW) compounds are so low that a phenotypic effect may not be detectable. One approach to this problem is to rank fragment hits by ligand efficiency (LE), or comparable metrics, on the assumption that hits with the highest LE represent the most promising starting point for optimization programs. 5 By growing or linking low-affinity fragments, optimized compounds with a higher MW and druglike properties can be obtained, and these are expected to have the desired biological activity. 6
In this study, we explored a fragment-based approach in which fragments identified as hits in a biochemical assay were promptly propagated for evaluation of antiprotozoal activity using a phenotypic screening panel. In addition, the initial structural exploration of the hit fragments was evaluated using a phenotypic readout. As an antimicrobial target, we focused on 3′,5′-cyclic nucleotide phosphodiesterase (PDE) type B1 from the parasite Trypanosoma brucei (TbrPDEB1). This PDE is an essential protein for the proliferation of the parasite that causes human African trypanosomiasis, also called African sleeping sickness. Inhibition of this enzyme, which regulates cyclic nucleotide levels, leads to an arrest of parasite cell division, lysis of the parasites, and elimination of the infection in an infected mouse model.7–9 For this particular parasite target, human PDE type 4D (hPDE4D) is considered an off-target as its inhibition has been suggested to lead to nausea and emesis,10,11 which is highly undesirable for patients who are already severely ill. Therefore, hPDE4D activity was also monitored during the evaluation of fragments. For fragment screening, a commercially available fragment library 12 was evaluated on both TbrPDEB1 and hPDE4D using a biochemical assay. Four interesting hits and a selection of 32 close analogs were identified and then screened for antiparasitic activity in a panel containing T. brucei, as well as three other protozoan parasites, Trypanosoma cruzi, Leishmania infantum, and Plasmodium falciparum, the causative agents of Chagas disease, leishmaniasis, and malaria, respectively. Cytotoxicity to mammalian cells was monitored using MRC-5 human lung fibroblasts.
Materials and Methods
Fragment Library
The commercially available IOTA Pharmaceuticals fragment library containing 1040 diverse fragments with an average MW of 213.9 and an average predicted lipophilicity (SlogP) of 1.96 was used in our search for TbrPDEB1 inhibitors. The complexity of this library is slightly higher than that of rule-of-three–compliant fragment libraries, which is hypothesized to be ideal for biochemical fragment screening approaches and for keeping nonspecific binding to a minimum. 12 Starting from stock solutions of 20 mM in DMSO, fragments were diluted to concentrations used in the biochemical assays. Molecular properties and descriptors were calculated with MOE 2012.10 (Chemical Computing Group Inc., Montreal, Quebec, Canada). The predicted 1-octanol/water partition coefficient SlogP was calculated from an atomic contribution model, as implemented in MOE 2012.10. Structural diversity analyses were performed using a scaffold-based classification approach (SCA) algorithm, 13 as implemented in a SVL script (sca.svl, version 2005.11) obtained through SVL Exchange (http://svl.chemcomp.com/). Briefly, the procedure included the calculation of a cyclicity score that represents the ratio between atoms present in ring structures versus noncyclic atoms. The complexity score was based on descriptors of scaffold size and shape, taking into account the smallest set of smallest rings, the number of heavy atoms, the number of bonds between the heavy atoms, and the sum of the atomic number of the heavy atoms. LE was calculated as
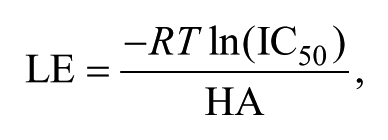
wherein the universal gas constant (R) is in kcal K−1 mol−1, the absolute temperature (T) in Kelvin, the half-maximal inhibitory concentration (IC50) in M, and HA represents the number of heavy atoms, defined as nonhydrogen atoms. Analogs of the fragment hits were selected from in-house compound libraries with ROCS v2.3.1 (OpenEye Scientific Software Inc., Santa Fe, NM), which was used as a three-dimensional shape- and chemistry-matching method to perform similarity searches.
PDE Cloning, Expression, and Purification
The catalytic domain of hPDE4D (amino acids 316–692) was amplified by PCR and subcloned into expression vector pET15b (Invitrogen, Carlsbad, CA), which encodes an N-terminal His-tag. Using previously reported methods, 14 the plasmid pET-PDEB1 was used for expression of the catalytic domain of His-tagged TbrPDEB1 (amino acids 576–918). The vectors were transferred into Escherichia coli strain BL21 (Codonplus) and grown in LB medium at 37 °C to an absorption of A600 = 0.7, and then 0.2 mM isopropyl β-D-thiogalactopyranoside was added to induce overexpression at 15 °C for 24 h. The expressed proteins were purified with an Ni-NTA column (Qiagen, Venlo, the Netherlands) using standard methods as described in the instruction manual “The Qiaexpressionist” (fifth edition, Qiagen, 2003).
PDE Luminescence Assay
Fragment screening at the two PDE enzymes and determination of half-maximal inhibitory concentrations (IC50) of fragment hits were performed with the commercial luminescence-based assay PDELight HTS cAMP PDE kit (Lonza Group Ltd., Basel, Switzerland), using procedures from the instruction manual (Inst-LT07-600 07/11, Lonza Walkersville Inc., 2011). Briefly, hydrolysis of cAMP was measured in assay buffer containing 10 mM HEPES (pH = 7.4), 100 mM NaCl, 5 mM MgCl2, 5% glycerol (v/v), and a final concentration of 1% DMSO (v/v) in deionized water. As substrate 0.5 µM and 1.0 µM cAMP was used for TbrPDEB1 and hPDE4D activity measurements, respectively. Fragments were screened at a single concentration of 200 µM for TbrPDEB1 and at 100 µM for hPDE4D. The reaction volume was 50 µL, and the assay was run in 96-well plate format. Assay output was monitored every 1 min at 25 °C for a total time of 20 min on a VICTOR 3 1420-012 multilabel counter (PerkinElmer Inc., Waltham, MA) with the emission filter centered at 535 nm and a measurement time of 0.1 s. Activity of PDEs results in the hydrolysis of cAMP to AMP, and the AMP production rate was linear with light output. Reaction rates were calculated from the rate of light intensity change. Concentration-response curves were generated by plotting the percentage inhibition of PDE activity against inhibitor concentrations, and IC50 values were obtained by nonlinear regression analysis using GraphPad Prism 5.01 (GraphPad Software Inc., La Jolla, CA).
Cellular Assays
For the cellular assays, the following reference drugs were used as positive controls: suramin for T. brucei, benznidazole for T. cruzi, chloroquine for P. falciparum, miltefosine for L. infantum, and tamoxifen for MRC-5 cells. The reference drugs suramin and tamoxifen were obtained from Sigma-Aldrich (Taufkirchen, Germany), and benznidazole, chloroquine, and miltefosine were obtained from the World Health Organization (WHO) Special Programme for Research and Training in Tropical Diseases (WHO-TDR, Geneva, Switzerland). The integrated panel of microbial screens and standard screening methodologies were adopted as previously described. 15 All compounds were tested at five concentrations (64, 16, 4, 1, and 0.25 µM) to establish a full dose-titration and to determine the IC50. The final concentration of DMSO did not exceed 0.5% in the assays.
Antitrypanosomal Assays
T. brucei Squib-427 strain (suramin-sensitive) was cultured at 37 °C and 5% CO2 in Hirumi-9 medium, supplemented with 10% fetal calf serum (FCS). About 1.5 × 104 trypomastigotes were added to each well, and parasite growth was assessed after 72 h at 37 °C by adding resazurin. 16 For Chagas disease, T. cruzi Tulahuen CL2 (benznidazole-sensitive) was maintained on MRC-5 cells in minimal essential medium (MEM) supplemented with 20 mM L-glutamine, 16.5 mM sodium hydrogen carbonate, and 5% FCS. In the assay, 4 × 103 MRC-5 cells and 4 × 104 parasites were added to each well. After incubation at 37 °C for 7 d, parasite growth was assessed by adding the α-galactosidase substrate chlorophenol red-α-D-galactopyranoside. 17 The color reaction was read at 540 nm after 4 h, and absorbance values were expressed as a percentage of the blank controls.
Antileishmanial Assay
L. infantum MHOM/MA(BE)/67 amastigotes were collected from the spleen of an infected donor hamster and used to infect primary peritoneal mouse macrophages. To determine in vitro antileishmanial activity, 3 × 104 macrophages were seeded in each well of a 96-well plate. After 2 d of outgrowth, 5 × 105 amastigotes/well were added and incubated for 2 h at 37 °C. Solutions with or without test compound were subsequently added, and the plates were further incubated for 5 d at 37 °C and 5% CO2. Parasite burdens (mean number of amastigotes/macrophage) were microscopically assessed on 500 cells after Giemsa staining of the test plates and expressed as a percentage of the blank controls without test compound. In the case of observed toxicity to the macrophages, the lowest concentration was recorded at which the toxicity was observed, and this was used as a qualitative phenotypic assessment.
Antiplasmodial Assay
Chloroquine-resistant P. falciparum K1-strain was cultured in human erythrocytes O+ at 37 °C under a low oxygen atmosphere (3% O2, 4% CO2, and 93% N2) in RPMI-1640, supplemented with 10% human serum. Infected human red blood cells (200 µL, 1% parasitaemia, 2% haematocrit) were added to each well and incubated for 72 h. After incubation, test plates were frozen at −20 °C. Parasite multiplication was measured using the Malstat assay, a colorimetric method based on the reduction of 3-acetyl pyridine adenine dinucleotide by parasite-specific lactate-dehydrogenase. 18
MRC-5 Cytotoxicity Assay
MRC-5 SV2 cells, originally from a human diploid lung cell line, were cultivated in MEM, supplemented with L-glutamine (20 mM), 16.5 mM sodium hydrogen carbonate, and 5% FCS. For the assay, 104 MRC-5 cells/well were seeded onto the test plates containing the prediluted sample and incubated at 37 °C and 5% CO2 for 72 h. Cell viability was assessed fluorimetrically 4 h after the of addition of resazurin. Fluorescence was measured (excitation 550 nm, emission 590 nm), and the results were expressed as percentage reduction in cell viability compared with control.
Results and Discussion
The IOTA fragment library comprising 1040 compounds was screened in a bioluminescence assay on TbrPDEB1. A total of 12 fragments inhibited TbrPDEB1 activity by more than 90%, which constitutes a hit rate of 1.2% ( Fig. 1A ). A total of 26 fragments were found to inhibit TbrPDEB1 activity by more than 70%. To identify fragments with selectivity for TbrPDEB1 over hPDE4D, the fragment library was also screened for inhibition of hPDE4D. A total of 41 fragments were identified that inhibited hPDE4D activity by more than 90%, constituting a hit rate of 3.9% ( Fig. 1B ). A preliminary comparison of PDE inhibition potency in the fragment screening assay suggested that at least seven fragments displayed selectivity for TbrPDEB1 inhibition over hPDE4D. The hits were of diverse structural complexity and cyclicity as indicated by the SCA plots ( Fig. 1A, B ). In addition, the MW and predicted lipophilicity of the hits on both targets were well distributed throughout the fragment library ( Fig. 1C ), indicating that the hit finding results were not biased toward more bulky or lipophilic compounds. The average MW and lipophilicity of hits that inhibited more than 90% of the activity of TbrPDEB1 (MW = 229.2; SlogP = 2.63) were on average very similar to those that inhibited hPDE4D (MW = 233.4; SlogP = 2.59). The low MW of these hits is noteworthy, and the hits represent promising starting points for fragment optimization toward more potent inhibitors.
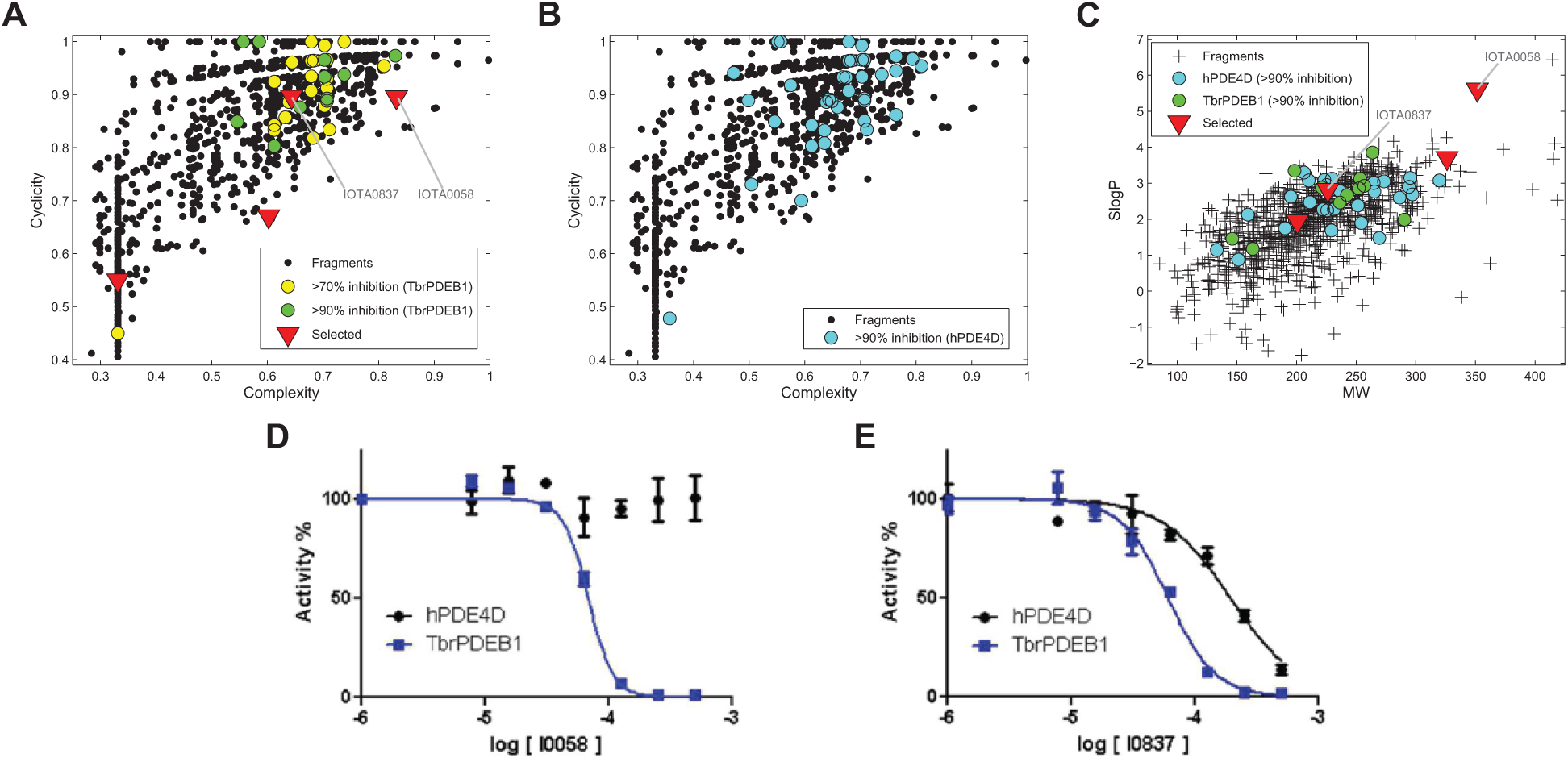
Results of the fragment screening performed on TbrPDEB1 and hPDE4D. (
In this study, we have focused on the scaffolds of four compounds selected from the primary screening based on their similarity to known active drug scaffolds ( Fig. 2 ). The compounds selected for follow-up were all inhibitors of TbrPDEB1 in the low micromolar range ( Table 1 ). The most selective hit was IOTA0058 (MW = 351.5; SlogP = 5.61), which displayed more than 15-fold selectivity for TbrPDEB1 over hPDE4D ( Fig. 1D ). Some selectivity for TbrPDEB1 ( Fig. 1E ) was also shown by IOTA0837 (MW = 226.3; SlogP = 2.82). The most potent inhibitor of TbrPDEB1 was IOTA0372 (MW = 326.2; SlogP = 3.70), but this compound inhibited hPDE4D to the same extent. The highest LE value (0.45) was observed with IOTA0771 (MW = 200.7; SlogP = 1.94), providing an interesting fragment for further optimization as a PDE inhibitor in a FBDD effort.
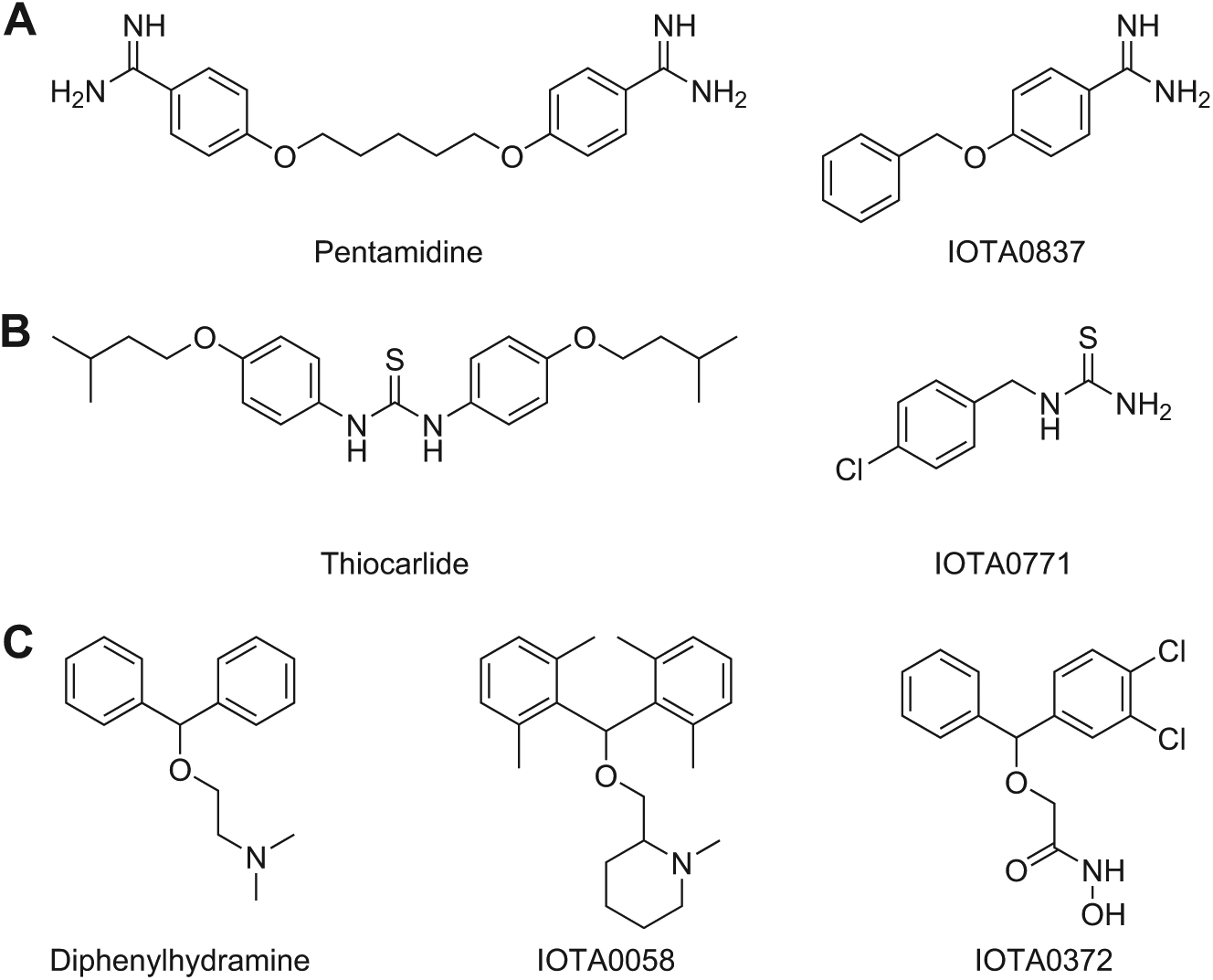
Structures of known drugs and the selected hits from the fragment library. (
Examination of TbrPDEB1 and hPDE4D inhibition.

Structures given in Figure 2 .
Inhibition (pIC50) of TbrPDEB1 by selected hits.
Ligand efficiency (LE) of compounds at TbrPDEB1.
Inhibition (pIC50) of hPDE4D by selected hits.
Selectivity index (SI) calculated as hPDE4D (IC50)/TbrPDEB1 (IC50).
Preliminary structure-activity relationships (SAR) were derived for the antiparasitic activity of fragments and compounds with similarity to known drug chemotypes, which were selected from in-house libraries containing >15,000 druglike compounds.
Upon inspection, overt structural similarities were noted between IOTA0837 and pentamidine ( Fig. 2A ), which is a classical aromatic diamidine with antiprotozoal activity. 19 The biochemical screening assays revealed that IOTA0837 was slightly more potent on TbrPDEB1 than on hPDE4D ( Fig. 1E ; Table 1 ). The antiparasitic activity of IOTA0837 revealed weak antitrypanosomal and antiplasmodial effects ( Table 2 ). Other more potent fragments were prioritized, but the presence of substructures of existing drugs in the identified hit set was noted.
Antiparasitic activity and cytotoxicity of reference compounds, IOTA0837, IOTA0771, and analogs.
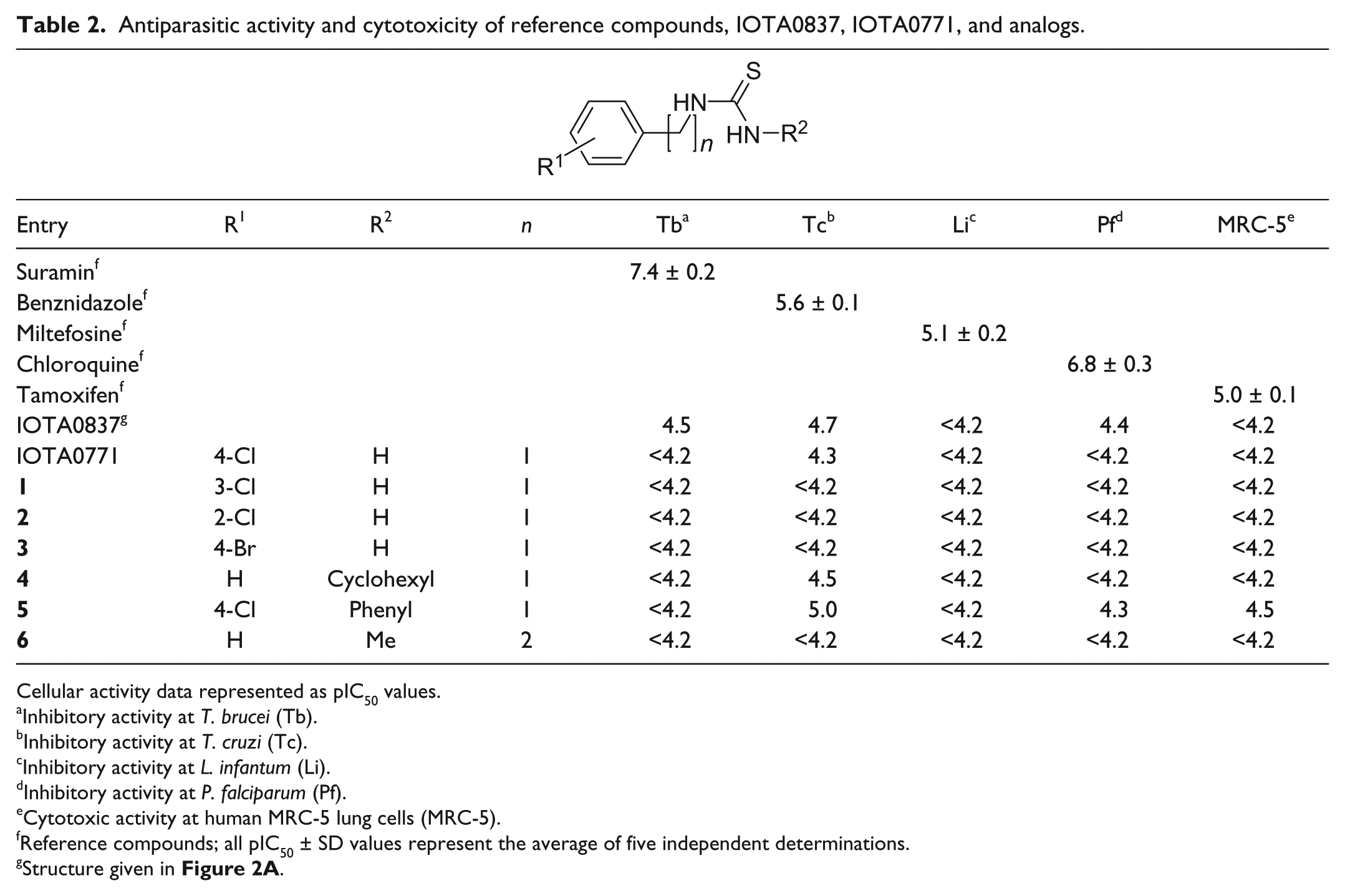
Cellular activity data represented as pIC50 values.
Inhibitory activity at T. brucei (Tb).
Inhibitory activity at T. cruzi (Tc).
Inhibitory activity at L. infantum (Li).
Inhibitory activity at P. falciparum (Pf).
Cytotoxic activity at human MRC-5 lung cells (MRC-5).
Reference compounds; all pIC50 ± SD values represent the average of five independent determinations.
Structure given in Figure 2A .
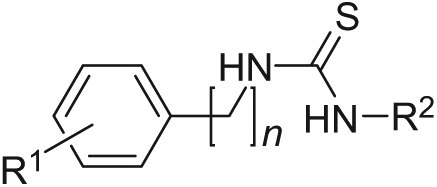
Fragment hit IOTA0771 contains a thiourea functional group, which is also found in the antimycobacterial drug thiocarlide (isoxyl;
Fig. 2B
). Thiocarlide is a second-line treatment against Mycobacterium tuberculosis, the causative agent of multidrug-resistant tuberculosis (MDR-TB), which acts as a prodrug requiring activation by mycobacterial monooxygenases and has been suggested to modulate lipid metabolism.
20
Given its high LE value (
Table 1
), IOTA0771 was considered to be an interesting fragment for validation and optimization in an FBDD effort. Several analogs (
From analyses of drug discovery programs, it becomes clear that lipophilicity tends to increase as initial hits are optimized to lead compounds. 21 Two hits, IOTA0058 and IOTA0372, had a relatively high MW as well as a relatively high predicted lipophilicity ( Fig. 1C ). These hits showed structural similarity to diphenhydramine (Benadryl) and related benzhydryl ethers ( Fig. 2C ), which are first-generation antihistamines. 22 This class of drugs was discovered in the 1940s 23 and has been on the market for the treatment of allergic syndromes, but it also displays anticholinergic, antiserotonergic, and antimuscarinic side effects. 24 Interestingly, chlorpheniramine, which is a structurally similar first-generation antihistamine, has recently been reported to resensitize chloroquine-resistant malaria parasites to chloroquine. 25
Hit compound IOTA0058 was found to be a selective inhibitor of TbrPDEB1, with hPDE4D inhibitory activity outside the measured concentration range (
Fig. 1D
), although its LE was fairly low (
Table 1
). The activity of IOTA0058 against Leishmania donovani intracellular amastigotes has recently been reported using a new colorimetric trypanothione reductase-based assay as well as classical microscopic assessment.
26
Seven close analogs (
Antiparasitic activity and cytotoxicity of IOTA0058 and analogs.
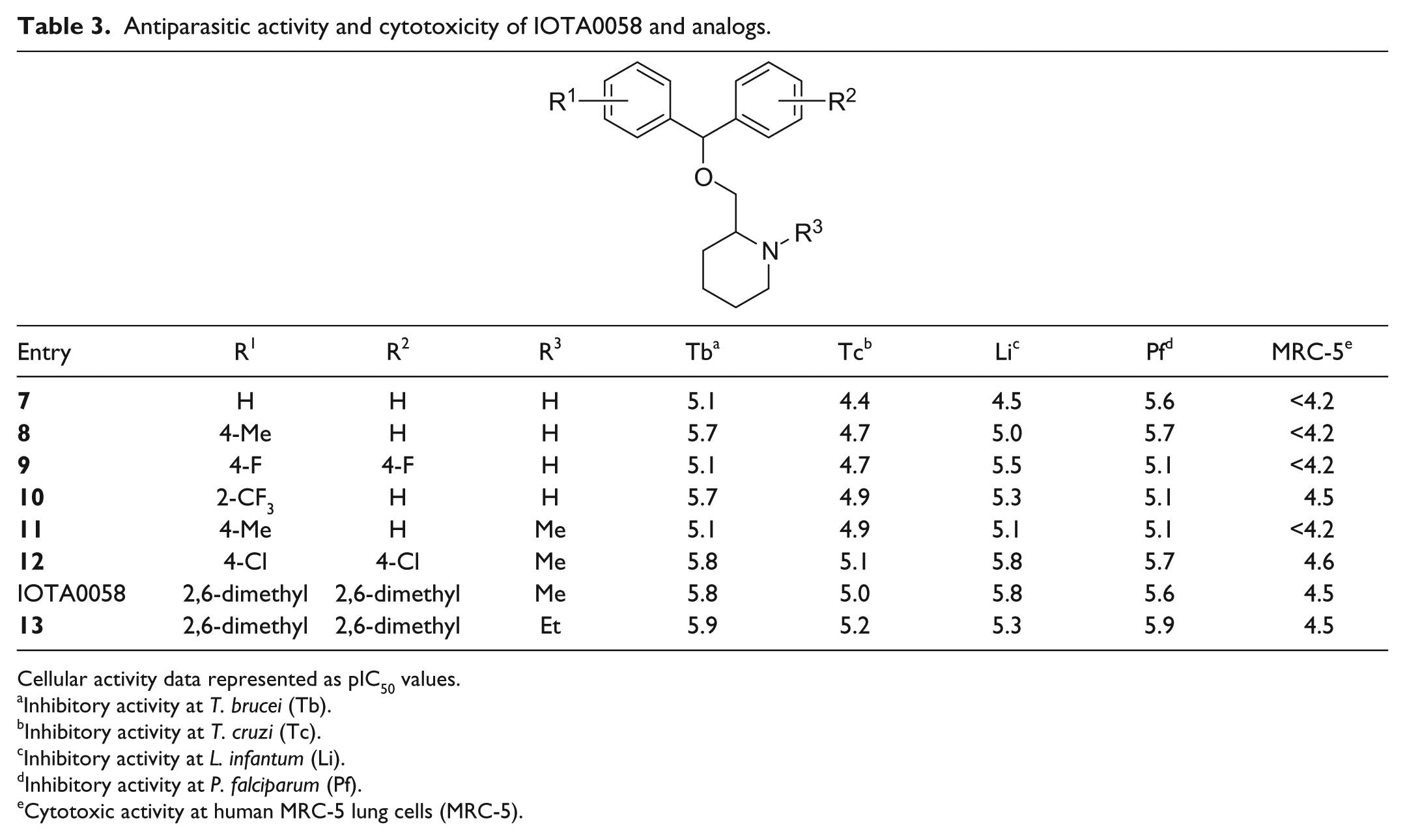
Cellular activity data represented as pIC50 values.
Inhibitory activity at T. brucei (Tb).
Inhibitory activity at T. cruzi (Tc).
Inhibitory activity at L. infantum (Li).
Inhibitory activity at P. falciparum (Pf).
Cytotoxic activity at human MRC-5 lung cells (MRC-5).
Benzhydryl ethers
Antiparasitic activity and cytotoxicity of IOTA0372 and analogs.
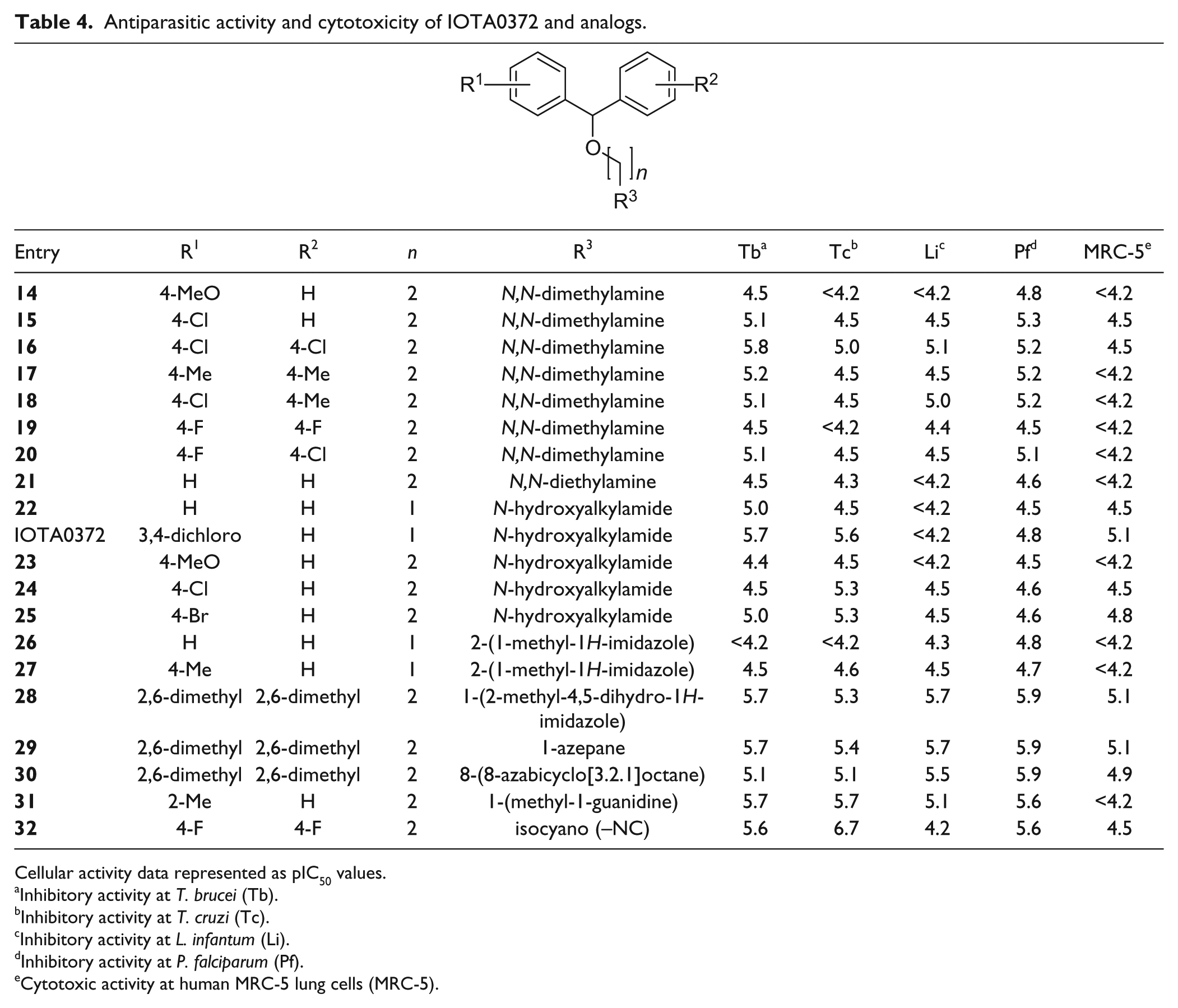
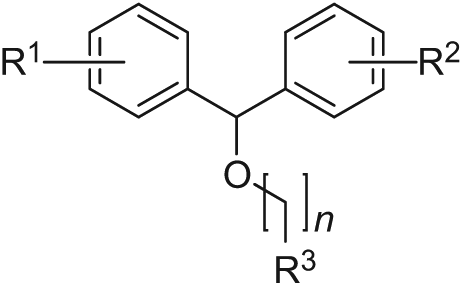
Cellular activity data represented as pIC50 values.
Inhibitory activity at T. brucei (Tb).
Inhibitory activity at T. cruzi (Tc).
Inhibitory activity at L. infantum (Li).
Inhibitory activity at P. falciparum (Pf).
Cytotoxic activity at human MRC-5 lung cells (MRC-5).
Belonging to the class of hydroxamic acids, IOTA0372 was found to be the most potent TbrPDEB1 inhibitor, but it did not display any selectivity over hPDE4D (
Table 1
). As a functional group, hydroxamic acids have metal chelating properties and are found in drugs such as vorinostat (Zolinza), an approved anticancer histone deacetylase inhibitor.
27
In addition to IOTA0372, hydroxamic acids
The molecular properties for antiparasitic activity were further probed by varying the substituents of compounds
The expected polypharmacological profile of the benzhydryl ethers can be considered an advantage as multiple mechanisms of action might be operating in concert, resulting in an increased efficacy or a decreased chance of parasite resistance. 28 Also, considerable knowledge is already available on the therapeutic potential and pharmacology of the parent 1,1-diaryl scaffold as well as the features contributing to its cardiotoxicity,22,29 allowing hypothesis-driven optimization. Although target deconvolution and optimization of phenotypic activity are generally considered a challenge, 30 our data suggest that it might be possible to optimize the antiprotozoal activity of these compounds while minimizing toxicity to human cells.
In summary, a fragment-based screening assay on an enzyme drug target was coupled with a phenotypic screening assay in a workflow that rapidly provided information on the antiparasitic activity of fragment hits. This workflow provided cellular activity data on four protozoan parasites and a human cell line, of assistance in selecting and prioritizing chemotypes for further optimization. This screening cascade has identified small molecules with antiparasitic activity that might be interesting starting points for further antiparasitic optimization. Although it is not established that the phenotypic activities are downstream of inhibition of parasitic PDEs, the results do suggest that it is useful to start phenotypic screening early in the fragment hit validation and exploration stages. Whatever the mechanism of action of these compounds might be, they represent small and high LE starting points with a lot of potential to grow into antiparasitic compounds with favorable properties such as selectivity and hydrophobicity.
Footnotes
Acknowledgements
We gratefully acknowledge the technical assistance of An Matheeussen, Margot Desmet, and Khaled A. Attia.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by TI Pharma grant T4-302 and the European Commission 7th Framework Programme FP7-HEALTH-2013-INNOVATION-1, PDE4NPD (No. 602666).