
Abstract
Secreted proteins and their cognate plasma membrane receptors regulate human physiology by transducing signals from the extracellular environment into cells resulting in different cellular phenotypes. Systematic use of secretome proteins in assays enables discovery of novel biology and signaling pathways. Several secretome-based phenotypic screening platforms have been described in the literature and shown to facilitate target identification in drug discovery. In this review, we summarize the current status of secretome-based screening. This includes annotation, production, quality control, and sample management of secretome libraries, as well as how secretome libraries have been applied to discover novel target biology using different disease-relevant cell-based assays. A workflow for secretome-based screening is shared based on the AstraZeneca experience. The secretome library offers several advantages compared with other libraries used for target discovery: (1) screening using a secretome library directly identifies the active protein and, in many cases, its cognate receptor, enabling a rapid understanding of the disease pathway and subsequent formation of target hypotheses for drug discovery; (2) the secretome library covers significant areas of biological signaling space, although the size of this library is small; (3) secretome proteins can be added directly to cells without additional manipulation. These factors make the secretome library ideal for testing in physiologically relevant cell types, and therefore it represents an attractive approach to phenotypic target discovery.
Introduction
The human proteome consists of protein products from approximately 20,000 protein-coding genes. 1 Based on predictions of signal peptides and transmembrane (TM) regions around two-thirds of the protein-coding genes code for proteins that primarily have an intracellular location, whereas one-third code for proteins that are destined for the secretory pathway. The latter group includes proteins destined to different membranes (e.g., endoplasmic reticulum, Golgi, lysosome, or plasma membrane), or intracellular compartments, as well as proteins secreted into the extracellular environment. Proteins can also be secreted to the extracellular environment via alternative, nonsecretory pathway mechanisms. 2
Secreted proteins, and their cognate receptors, are responsible for communication between cells, tissues, and organs within the body. Regulation of these communication pathways is critical for normal human homeostasis, with dysregulation playing a major role in pathophysiology and disease. Secreted signaling proteins can be destined to the blood (endocrine) or to the local environment (paracrine or autocrine). The expressed secretome varies with cell type, differentiation state, and environmental cues. Experimentally verified and/or described secretomes include, for example, cardiac, 3 adipocyte, 4 immunocell, 5 cellular senescence, 6 stem cell, 7 cancer cell, 8 and the blood secretome. 9 There are also descriptions of therapy-induced secretomes where inhibition of receptor tyrosine kinases leads to drug-stressed cells with large changes in the expressed secretome and increased drug resistance. 10
Phenotypic drug discovery (PDD) is based on the principle of inducing or preventing a functional readout in a biologically relevant cell system using a sample library of choice.11,12 Use of large compound libraries for PDD has been described, 13 but it is more common to use a smaller set of well-annotated, structurally diverse compounds that are known to act on certain biological targets (i.e., have an annotated mechanism of action [MoA]). 14 A number of small-molecule phenotypic screens for the identification of novel targets regulating various biological phenotypes, for example, epigenetic processes, 15 stem cell proliferation, 16 cardiac cell proliferation,17,18 tumor cell suppression, 19 and regulatory T-cell stability, 20 have been reported. However, there are technical challenges associated with the identification/verification of the molecular target of an interesting small-molecule hit, termed “target deconvolution.”12,21 Although recent advances in chemoproteomics, machine learning, cell microarray, and other methods have been adopted to facilitate target deconvolution,22–27 there remain few examples of novel targets identified through this approach. 28 Thus, there is a need to explore the benefits of additional screening modalities for target discovery. Phenotypic screening using functionalized fragment sets has been described to enable straightforward target deconvolution since fragments will be irreversibly bonded to target proteins. 29 Small interfering (si) RNA and lately CRISPR libraries have allowed functional genomics screens for the identification of novel drug targets.30–33
Secretome-based screening builds on the concept that the secreted proteins are biologically active signaling molecules. When a secretome resource/library is used in combination with PDD, novel targets and signaling pathways can be identified. In this review article, we describe the concept of secretome-based screening using human secreted proteins in combination with different cellular readouts. We give a summary of the field, including industrial efforts, for example, EMDSereno, 34 FivePrime Therapeutics,35,36 Genomics Institute of the Novartis Research Foundation (GNF),37–40 Novartis, 41 and academic efforts.42,43 We describe the screening platform that has been set up at AstraZeneca, in collaboration with the Royal Institute of Technology (KTH) in Stockholm, in more detail. Learnings, insights, and challenges are shared. In addition, our view on future applications of secretome-based screening in drug discovery, as well as accessibility to a secretome library, is discussed.
Antibody-based phenotypic screening or methods to study interactions between secreted proteins and receptors will not be covered and readers are referred to other reviews.44–46
The Concept of Secretome-Based Screening
Secretome-based screening was first described in the postgenomics era when secreted proteins could be identified by a bioinformatics approach, produced by recombinant expression in high-throughput fashion, and tested in different cell-based assays to identify an effect34,35,37,41 (
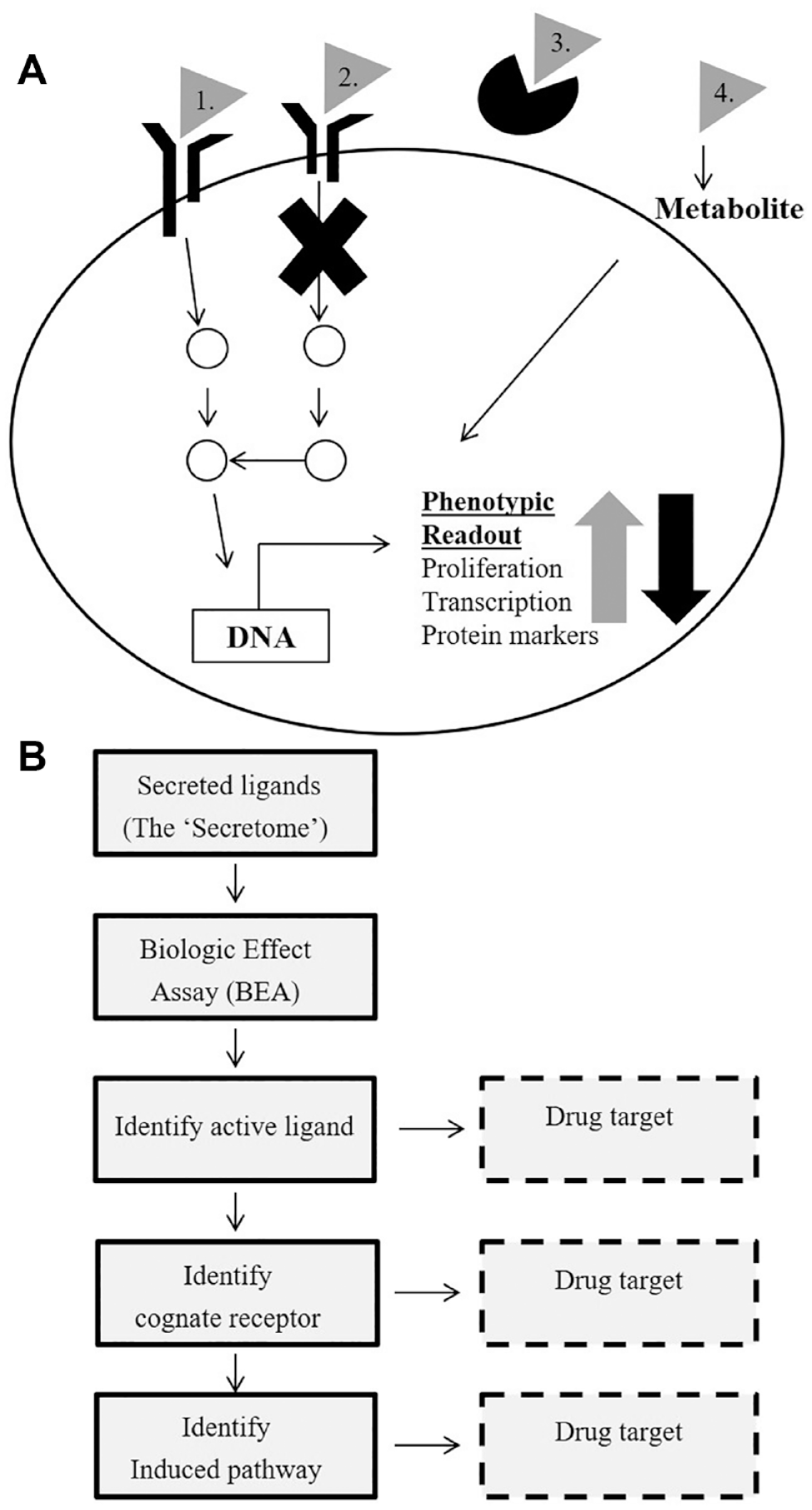
Concept of secretome-based screening—the combination of a secretome library and a cell-based assay with a disease-relevant readout results in the identification of novel targets and elucidation of signal transduction pathways. (
Affinities and/or Kd values between ligand–cognate receptor pairs reported in the literature vary between low picomolar and low micromolar.35,54–56 It is critical that the concentration of individual samples in the secreted protein library is determined before doing an unbiased secretome screen. This avoids false negatives due to low concentrations of proteins in the assay. This is why most efforts have established methods to quantify the amount of protein produced. However, a low-affinity interaction is sometimes accompanied by a high-affinity interaction, within a ligand–heterodimeric receptor complex, enabling fine-tuning and specificity of a particular signaling response. 56
A secretome library offers various benefits over other libraries used for PDD. First, the size of the library is small compared with other libraries, enabling screening with precious and disease-relevant cells. Second, cells used for the secretome-based screens do not need to be manipulated via transfection as needed for siRNA screens or to be manipulated to enable Cas9 expression as needed for CRISPR functional genomics screens. This enables screening with primary cells without manipulations. Third, the constituents of the secretome library are biologically relevant. This should make target identification potentially easier than for small molecules. Finally, the secretome library has better coverage of agonistic MoAs than the small-molecule compound set. However, the secretome library also has certain drawbacks. It primarily captures biology that is orchestrated from the plasma membrane and thus is less ideal when probing biology that is mechanistically initiated from the intracellular environment, for example, DNA-repair mechanisms and metabolic events. It has less coverage of antagonistic MoAs than the small-molecule set and CRISPR library and the inherent selectivity may lead to a low hit rate. Therefore, ideally, a secretome library should be used as a complementary approach together with small-molecule, siRNA, and CRISPR libraries. Various secretome libraries and outcomes from secretome-based screens are summarized in
Summary of Different Secretome-Based Efforts That Have Been Described in the Literature.
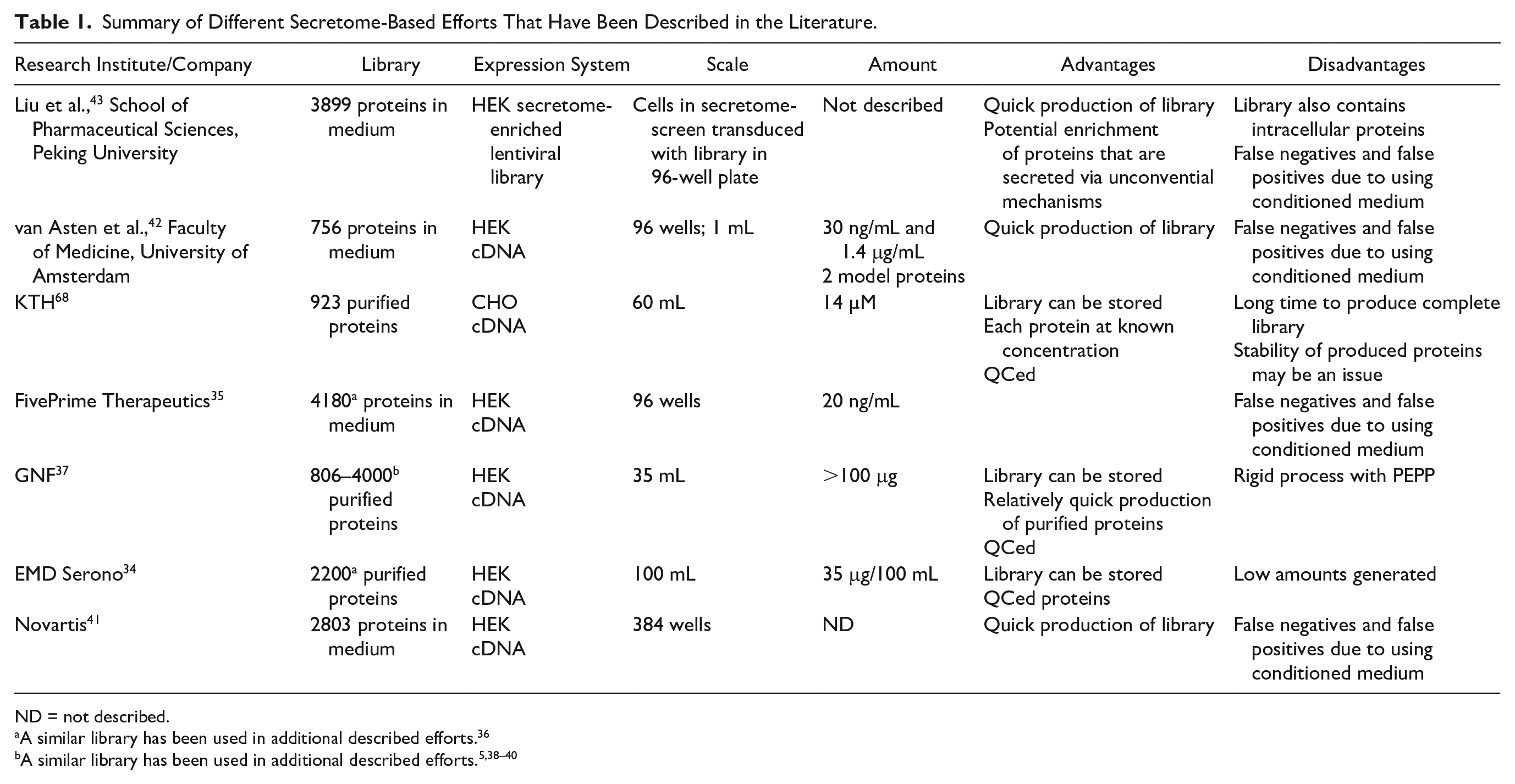
ND = not described.
A similar library has been used in additional described efforts. 36
Summary of Targets That Have Been Generated from Secretome-Based Screens and Additional Validation Assays.
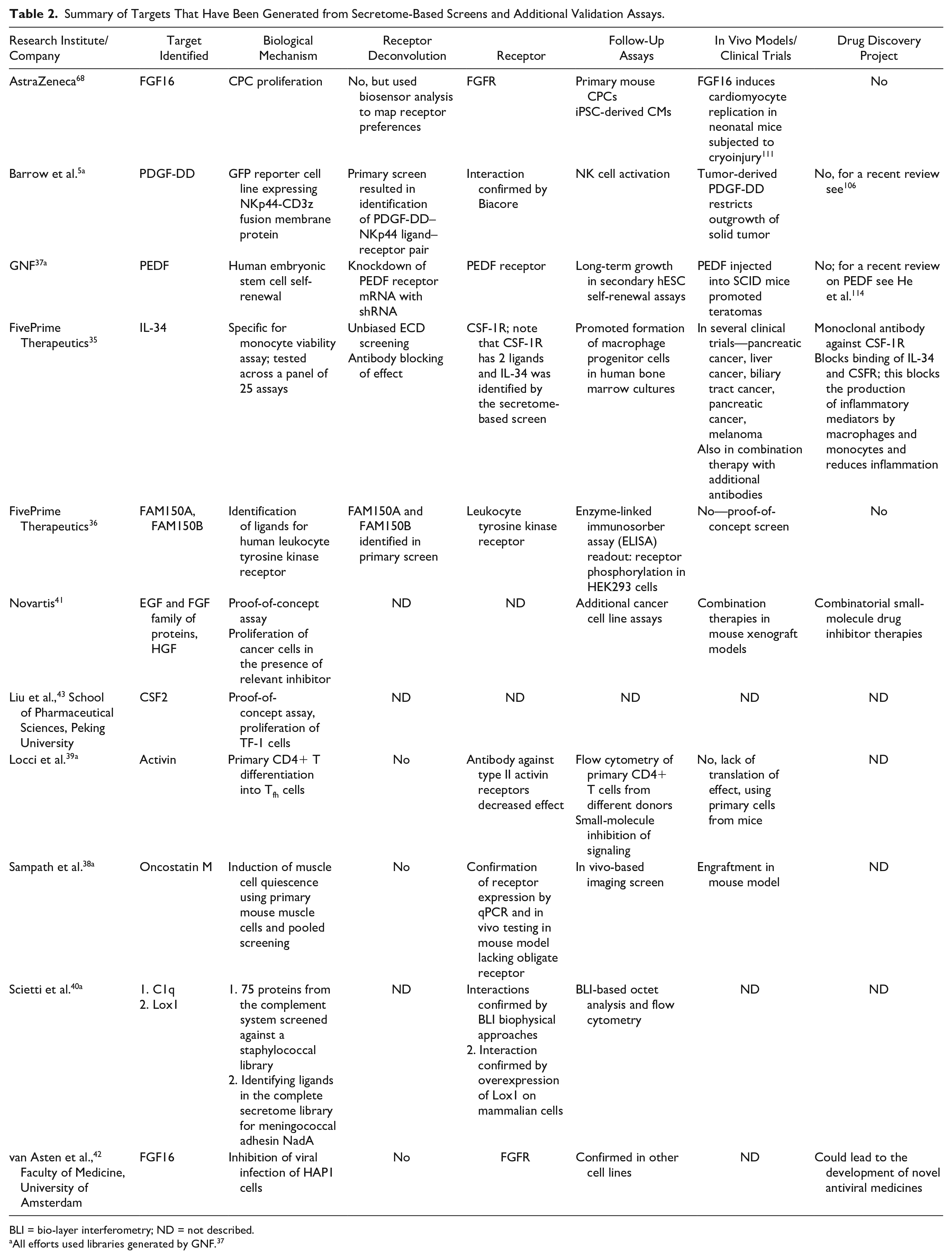
BLI = bio-layer interferometry; ND = not described.
All efforts used libraries generated by GNF. 37
Secretome Libraries
Annotation of the Library
There are several different databases available for annotation of the library, including Uniprot (https://www.uniprot.org/), 57 Ensembl (http://www.ensembl.org/), 58 pfam (http://pfam.xfam.org/), 59 and the NCBI Reference Sequence Database (https://www.ncbi.nlm.nih.gov/refseq/). 60
When a gene list for production of a secretome library is assembled, secreted proteins in the genome need to be stratified from the remaining human genome consisting of intracellular proteins and membrane proteins. Underlying the stratification is prediction of cellular location by, for example, identification of sorting sequence 61 or membrane region.62,63 There are several databases focusing on identification of secreted proteins including the Secreted Protein Discovery Initiative, 64 the Secreted Protein Database, 65 and the more recent MetazSecKB and VerSeDa66,67 databases. In Uniprot, secreted proteins are also annotated with a function according to keywords for molecular function and/or assigned to a biological process. These include growth factors, cytokines, hormones, regenerative factors, coagulation factors, and different classes of enzymes. The largest annotated group is enzymes, followed by proteins related to immunity, growth factors, and cytokines.
Different secretome-based screening initiatives have stratified their libraries slightly differently34,35,37,68 but basically using the same principle of predictions of a signal peptide and TM regions, based on algorithms and public information available.
In Jennbacken et al., 68 we describe how we stratified our library. Secreted proteins were defined as all Uniprot entries having the subcellular location “Secreted” in addition to all genes with at least one transcript predicted to be secreted according to the Human Protein Atlas (HPA). For prediction of secreted proteins in HPA, three different signal peptide prediction algorithms,69–71 in combination with seven different TM region prediction algorithms, 72 were used. To be categorized as secreted, a transcript must have a signal peptide predicted by at least two of three methods, and no TM region predicted by four or more methods. Selected ECDs were also included in the secretome library. One-pass TM proteins for the production of ECDs were selected from Uniprot entries with subcellular location “one-pass TM proteins,” as well as from HPA TM region predictions. 72
Additional stratification of a secretome library is useful to be able to prioritize in which order the library should be produced. Gonzalez et al. describes 37 how they use genome-wide association scans of different traits that are relevant to disease in human samples73,74 and traits of inbred mice 75 to stratify their library. At AstraZeneca we used additional databases such as GeneOntology: “Extracellular space” annotation, Ingenuity Pathway Analysis (IPA) analysis, and an in silico survey of relevant literature, including the “Human Secretome Atlas” 76 and specific stratification for cardiac cells,77–81 for the library described above.
A functional analysis of the library, using the Uniprot keywords (
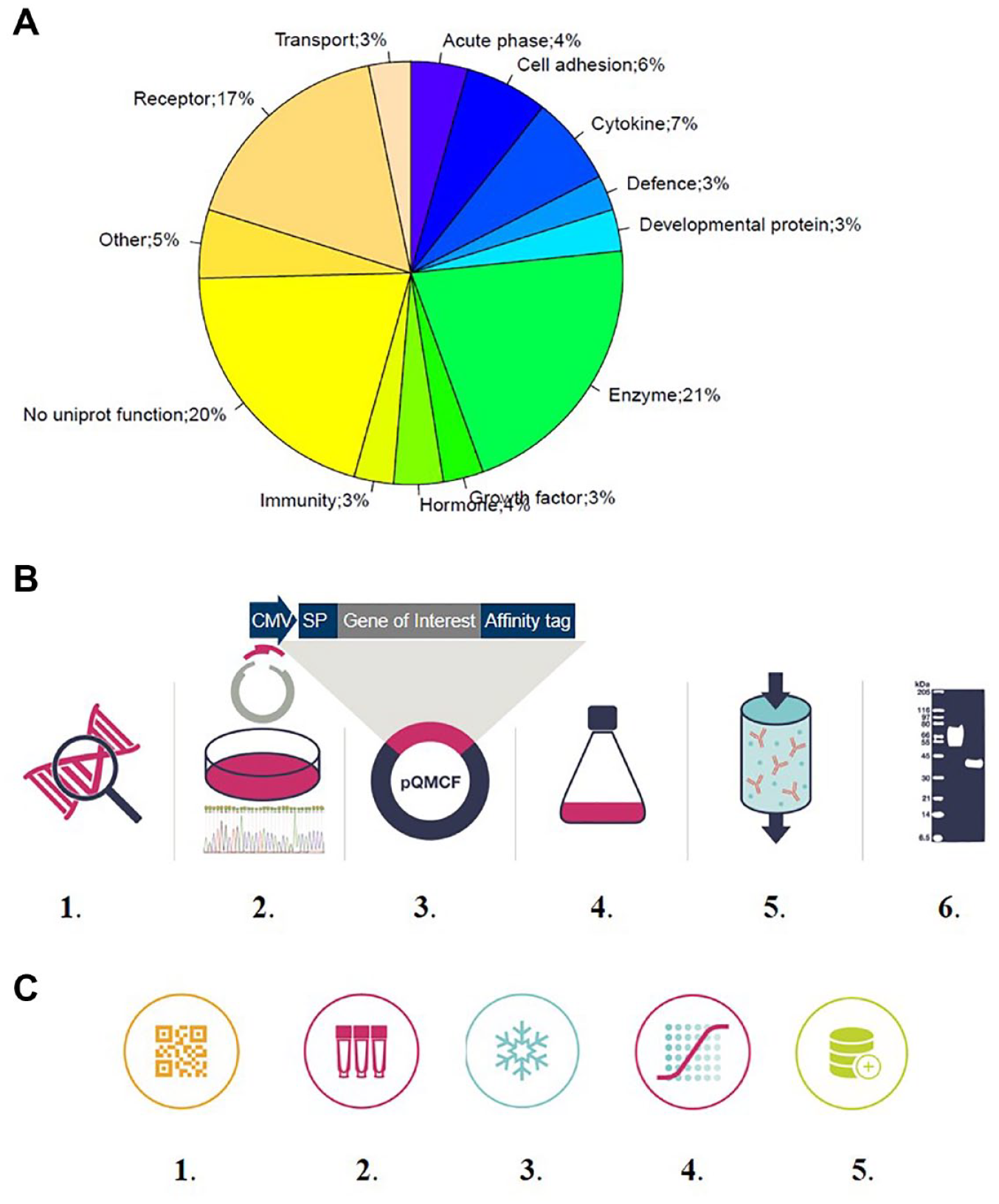
The secretome library—the constituents, how to produce it, information flow, and sample management. (
Recently, Uhlén et al. 9 published an extensive additional annotation of the human secretome where the actively secreted proteins in humans were identified. Starting with a bioinformatics-based definition of the secretome, a set consisting of 2641 genes with at least one predicted secreted isoform was manually annotated and classified into three major categories: (1) the blood proteins, (2) the locally secreted proteins, and (3) the intracellular or membrane-associated proteins. Groups 1 and 2 were defined as the secreted genes and consisted of 1709 genes with at least one secreted protein isoform (available at the http://www.proteinatlas.org/blood). The remaining 932 genes in group 3 were annotated as having an intracellular or membrane-associated location.
Liu et al. do the stratification in an entirely different way. 43 The practical work is started with the complete set of open reading frames in the human genome (ORFeome V8.1 library; http://horfdb.dfci.harvard.edu/) 82 in frame with a truncated form of human CD4 containing the membrane-spanning part. The resulting lentiviral library is transduced into HEK293 cells and sorted using a CD4 antibody. Genes that correspond to proteins that are secreted will be displayed on the surface and constitute the basis of the secretome library. Almost 4000 DNA sequences were found in the secretome-enriched library. Nine hundred sequences correspond to secreted proteins according to database annotation, and these account for ~80% of the produced proteins in the library based on copy number. Around 1000 genes are annotated as intracellular proteins and 800 genes have an unknown function. This method to produce the library is interesting since it could possibly be more effective in enriching for proteins that are secreted via unconventional mechanisms. 2 However, this will require more data to be generated using the secretome-enriched library to be conclusive.
The library generated by FivePrime Therapeutics is based on generation of cDNAs from human tissue material from diverse sources such as fetal, normal adult, cancer adult, and inflamed adult human tissue. 35 The library is highlighted in an investment report from 2014 (http://investor.fiveprime.com/static-files/c4ea6f83-e6ed-4334-accb-7ffb002a0122; p 81) and described to comprise a more comprehensive collection of “full-length” cDNA clones based on the use of proprietary technology to capture additional mRNAs with intact 5′ ends.
Production and Quality Control of the Library
Secretome libraries can be produced in different ways, including de novo produced recombinant proteins in conditioned medium,35,37,41,42 purified proteins,34,37,68 or a lentiviral secretome-enriched open reading frame library. 43
The quality and the concentration of the secreted protein library to be used in the functional assay is critical. Therefore, it is very important to choose a eukaryotic expression host, preferably a mammalian host, for production of the secretome library.83,84 This should enable correct folding and posttranslational modifications of produced human secretome proteins. Posttranslational modifications include glycosylations 85 and removal of pro-peptides that are present in some secreted protein families such as the TGF-β superfamily 47 and the PDGF family. 86 Proteins that contain pro-peptides will most likely be inactive in a secretome-based screen if they are not processed properly. The most frequently described expression host for production of a secretome library is human embryonic kidney (HEK) cells, followed by Chinese hamster ovary (CHO) cells, also used for the production of most biological drugs. 87
It is useful to establish a general process for production when making a secretome library (illustrated in
Table 1 lists different human secretome libraries that have been published. The libraries can be divided into three categories: (1) conditioned medium libraries containing expressed secretome proteins, but also metabolites, growth factors, and extracellular matrix proteins secreted by the cells; (2) purified protein libraries; and (3) a secretome-enriched lentiviral library that starts with the full human ORFeome. 43
Conditioned Medium Libraries
Generation of a conditioned medium library is described in the seminal article by Lin et al. 35 After the initial stratification for production, human cDNAs were generated from different human tissues, resulting in a cDNA collection consisting of a total of 4180 constructs for protein production. All proteins were expressed in HEK293T suspension cells in 96-well plates. A generic signal peptide was used for expression of the ECDs. Proteins were expressed with and without a C-terminal V5-His affinity tag to allow for the detection and quantification of secreted protein. According to Lin et al., 90% of clones secreted detectable protein into the medium, with the median concentration of protein produced being 20 ng/mL.
Harbinski et al. 41 and van Asten et al. 42 used a similar setup except for that genes were sourced commercially; there was no affinity tag included for detection and, in the case of van Asten et al., cultures were at the larger 24-well scale. 42
A serum-free defined medium is preferred when proteins are used directly for screening, without purification, since it cannot be excluded that, for example, growth factors and other agents could contribute to a polypharmacological effect on the functional readout. Lin et al. used a serum-containing medium, 35 and all conditioned medium samples were used in screens within 12 h. This also avoids degradation or modification of produced proteins by components that are present in the medium.
It usually takes a few weeks to produce a complete secretome-conditioned medium library containing a few thousand proteins if proteins are produced in parallel in 96-well plates.
Purified Protein Libraries
The first description of a purified human secretome library was published in 2006. 34 In this example, all proteins were expressed in HEK293-EBNA cells at 100–500 mL culture scale. In this large project that lasted for 4 years, 2200 protein batches were purified in total. The success rate of production was quite low (30%). 34 This may be explained by choice of cells for expression and use of native signal peptide instead of generic. 88 Also, recent advances in mammalian cell culture83,84 have probably contributed to higher success rates in later efforts.
Gonzalez et al. 37 described a fully automated Protein Expression and Purification Platform (PEPP) robot for the expression and purification of 24 proteins in parallel at <100 mL scale. Success rates were as high as 70%, with average yields reported to be 3 µg/mL. 37 In this example, proteins were secreted with and without an FC tag enabling immobilization of the library for different downstream applications, such as the identification of ligand–receptor pairs. 5 Throughput for the production of a purified protein library varies between 25 proteins/week 68 and 200–400 proteins/week using the PEPP platform. 37
For purified proteins, a more rigorous quality package should be established.34,37,68 It is vital to implement a sample management process that reduces the number of freeze–thaw cycles and to control how protein stability is affected by storage. As a consequence, a separate vial of protein should be used for the quality check (QC) analysis.
Generation of the KTH Library
We use the KTH library as an example to illustrate in detail how a purified secretome library is generated. A standardized pipeline has been established, including construct design, gene synthesis, protein production, protein purification, and quality control, with the aim of producing pure protein samples (illustrated in
Sample Management and Protein Storage
There are different requirements for the handling of secretome samples depending on if conditioned medium or purified proteins are used for screening. In the former case, when screening takes place immediately after production of the library, a liquid handling robotic system is needed. If a purified protein secretome library is produced within a few weeks, 37 long-term storage of proteins may not need to be considered. However, when library production takes several months to years, a process needs to be established.34,68 Our process for sample management is outlined in Figure 2C and is similar to the one described by Battle et al. 34 After production, each protein batch was thawed once and divided into smaller aliquots before snap-freezing and long-term storage. Before each new screen, an aliquot was thawed and dispensed at the desired concentration in a deep well plate before adding to the cell-based assay. All information about the individual proteins in the library (i.e., gene name, sequence, concentration, and QC report) is maintained in a laboratory information system at KTH and exported to AstraZeneca’s Labguru application (BioData; http://www.labguru.com//), which is used to share information about preclinical bioreagents. The library of proteins is also registered in AstraZeneca’s compound management databases (internal AstraZeneca software and Mosaic; https://www.titian.co.uk), originally used for small molecules but now expanded to handle proteins. This allow for seamless integration between compound handling, assay screening, and data analysis. Cross-referencing of the databases allows full traceability of results and information.
A Secretome-Based Screening Workflow
In Figure 3A a secretome-based workflow is illustrated for a flow cytometry secretome-based screen in 384-well format using two different marker readouts. Similar cell-based screening setups have been published.20,39 Production of the library should occur in the same location as the screening if conditioned medium is used. Ideally, each individual protein in the secretome library should be tested in duplicate at several different concentrations, 68 provided that a sufficient number of cells are available. One rationale for testing the library at multiple concentrations is that response curves can be bell-shaped due to receptor desensitization, counterregulatory mechanisms, or self-inhibition of a receptor at higher concentration.92–94 The library should be screenable in a small number of plates (<20 plates) due to its size and the compatibility of cell-based assays with the 384-well format. Cells are usually incubated with secretome proteins for 1–5 days to capture cell proliferation, differentiation, and de novo expression of specific marker proteins. Cells are usually fixed before analysis. Ideally, positive and negative controls should be included on all plates. Z′ calculated from positive and negative controls is normally used for characterizing assay performance. Assay quality criteria similar to those of other PDD approaches apply to secretome-based screens. Primary actives are identified by applying an activity threshold cutoff.
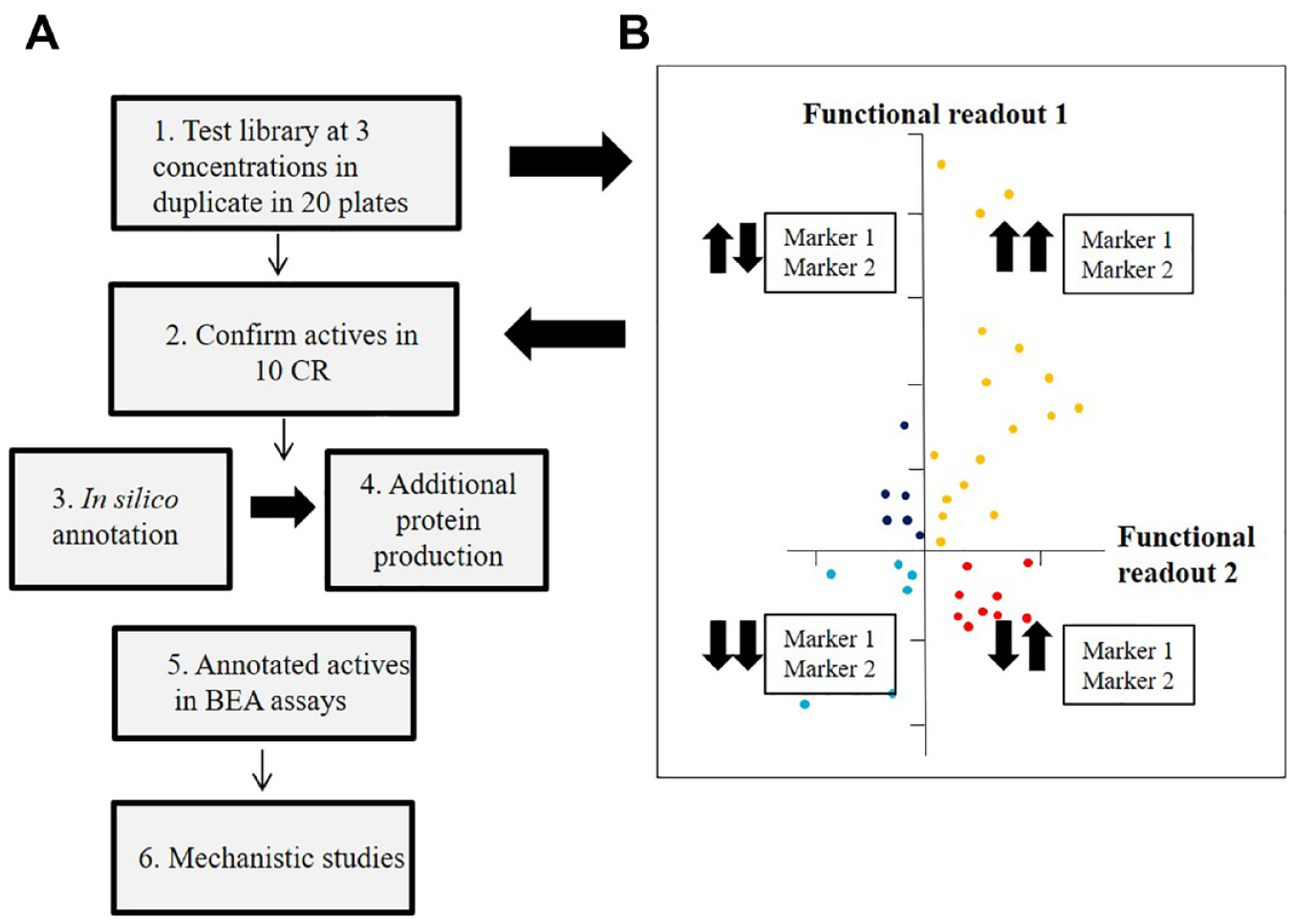
The secretome-based workflow from initial screen to confirmed active. (
The output from a primary screen, with independent measurement of more than one marker, can be quite complex; for example, four types of actives are identified if two markers are measured (
Once initial actives have been confirmed in CR, the list of confirmed active proteins should be annotated in silico, including information concerning cognate receptor and signaling. In addition, expression data, disease relevance, and human target validation should be considered. Prioritization of the annotated proteins will generate a list to guide additional protein production, to avoid depletion of the secretome library. Commercially available proteins5,39,41 or internally purified proteins 35 are usually used at this stage if conditioned medium is used for primary screening. This enables the testing of a few selected candidate proteins in additional biologic effect assays, biophysical experiments, and combinatorial screening with compounds to elucidate receptor preferences.
In the 2008 article from Lin et al., 35 note that some proteins with “low selectivity” emerge as actives across the 25 different assays. Proteins denoted as low-selectivity proteins include the interferon-α family, FGF2, FGF3, and some of the interleukins. In contrast, other proteins, such as interleukin 34 (IL-34), are active for a specific cell type and thus display a “high functional selectivity.” We have made similar observations when applying the KTH library to different assays (unpublished). 68 For example, FGF9 is active on different cell types, including cardiac progenitor cells (CPCs) and cardiac fibroblasts (CFs), whereas FGF16 is specific for the CPCs. 68 It should also be noted that FGF16 and other members of the FGF family were identified as a potent inhibitor of viral replication in a secretome-based screen using the near-haploid cancer cell line HAP-1. 42
After an active has been identified in a phenotypic screen, the next step is to determine whether the effect is mediated via a ligand–receptor interaction or via some other mechanism (different scenarios are illustrated in
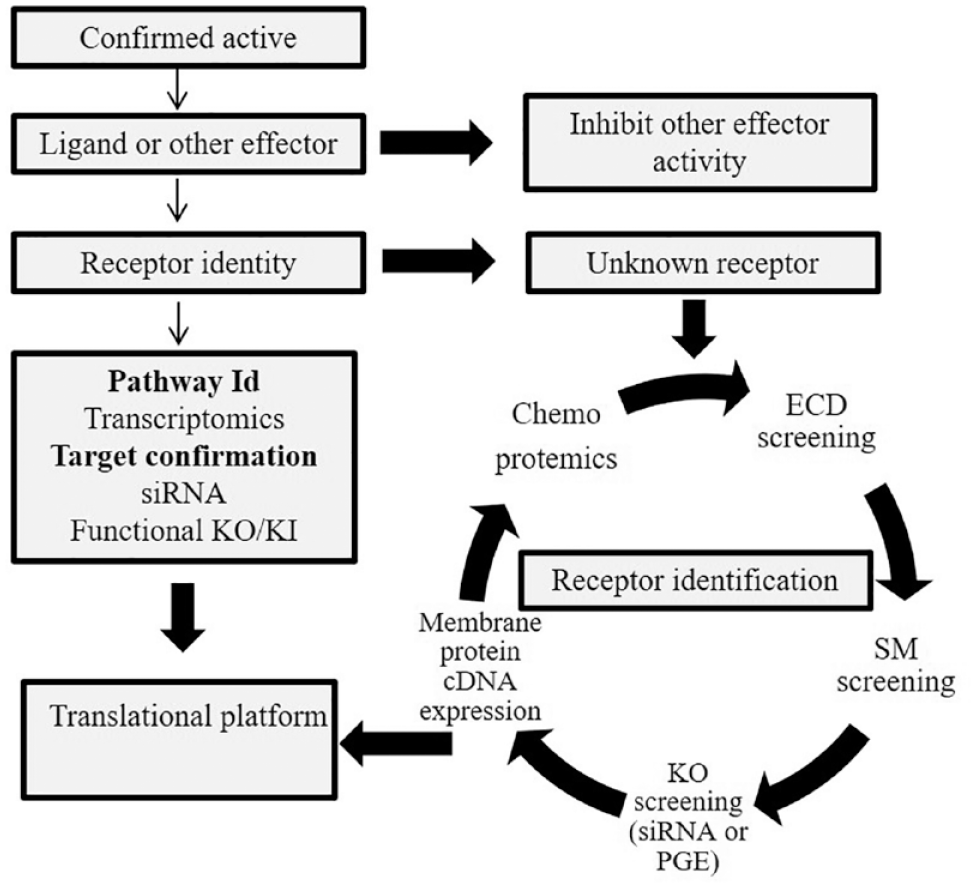
A summary of different steps needed to identify a receptor and signaling pathway induced by a secreted ligand. When an active has been identified from a secretome-based screen, the next step is to identify the cognate receptor and/or enzymatic activity that is needed to transduce the signal into the cells. There are several methods available to establish the identity of the receptor as described in the main text. Also, gene expression analysis can be utilized to profile the transcriptional events that are induced by the active secretome proteins. Finally, this can be confirmed by siRNA or precise genome editing (PGE). See text for more details.
Different ways of receptor deconvolution are illustrated in Figure 4 . Ligand–receptor pairs can be identified via ECD screening, as discussed previously. As a result, the functional effect will be antagonized by the ECD binding to the ligand. Ligand–receptor pairs can also be identified in the primary screen by using a target-based approach as described by Zhang et al. 36 and Barrow et al. 5 Also, a small-molecule library annotated for plasma membrane receptor can be used. Arrayed CRISPR and siRNA libraries comprising the plasma membrane proteome can also be applied. 54
The receptor of the GDF15 ligand, which regulates appetite, was identified by several different groups by overexpression of a cDNA library comprising the plasma membrane proteome,55,96–98 or by bespoke pull-down experiments. 99 More involved methods include chemoproteomic approaches where the purified ligand is labeled with a reactive group that can be crosslinked to, for example, live cells. 100
Pathway biology can be interrogated via transcriptomics experiments independent of whether receptor identification experiments were successful. Once putative genes have been identified, this can be followed by targeted siRNA or CRISPR knockout experiments. This should be finalized by translational experiments, for example, in vivo model experiments looking at a therapeutical benefit in a relevant disease model.
In summary, access to a high-quality secretome library as well as disease-relevant cells and assays enables the identification of secreted proteins that regulate a phenotype of interest. Target identification for secretome actives with known receptors can be easier than for small-molecule actives. However, for secretome actives with unknown receptors, the deconvolution work will require bespoke approaches that may not result in the identification of the receptor/target.
Application of Secretome-Based Screening for Target Identification
A secretome-based screening approach was first described by Merck Serono/EMD Serono. 34 However, no details were given on therapeutic alignment or cellular readouts applied with the generated secretome library. FivePrime Therapeutics 35 reported the development of a proprietary secretome-based platform that facilitated the discovery of IL-34 as a target for cancer treatment (described in more detail below). The GNF library 37 enabled the identification of PEDF as a regulator of stem cell renewal. 37 Collaborations between GNF and other groups have resulted in the identification of novel target biology (summarized in Table 2 ). For example, Locci et al. 39 describes the identification of activin as a regulator of differentiation of follicular helper T (Tfh) cells for the treatment of autoimmune diseases. Sampath et al. describe the identification of oncostatin M as an inducer of muscle stem cell quiescence, which could be of interest to stem cell engraftment. 38 Barrow et al. reported the identification of ligand–receptor pairs by screening a secretome library 5 (described in more detail below). Finally, Scietti et al. uses the library to interrogate host–pathogen interactions relevant to infection diseases. 40 Novartis 41 has also used commercial cDNA libraries to identify signaling pathways that are involved in drug-induced resistance in different cancers. Independent academic efforts include the identification of secreted factors that inhibit virus infection, in addition to well-known secreted proteins such as the interferons. 42
At AstraZeneca there is a growing interest in regenerative aspects of biology in several therapeutic areas, for example, in heart failure, in which there is an ambition to regenerate cells in the heart to treat heart failure. In the respiratory therapeutic area, there is an ambition to repair lung damage by regrowth of lung tissue, potentially restoring lost lung function in obstructive lung disease. The role of the human secretome, including growth factors, cytokines, and additional factors, in regulating regenerative processes is attractive for target discovery in these therapy areas. FGF16 was identified as an interesting candidate for cardiac regeneration and repair 68 (described in more detail below).
Identification of the Novel Cytokine IL-34 That Regulates Monocyte Viability
Tumor-associated macrophages (TAMs) inhibit antitumor T-cell activity in the tumor microenvironment. In pancreatic and other cancers, high levels of TAMs are associated with poor prognosis. Signaling through the CSF-1R promotes the maintenance and function of TAMs (https://www.fiveprime.com/file.cfm/16/docs/CB_2017_11_SITC_Oral_CSF-1R.pdf).101,102 IL-34 was first discovered in the secretome-based screen performed by Lin et al.
35
(summarized in
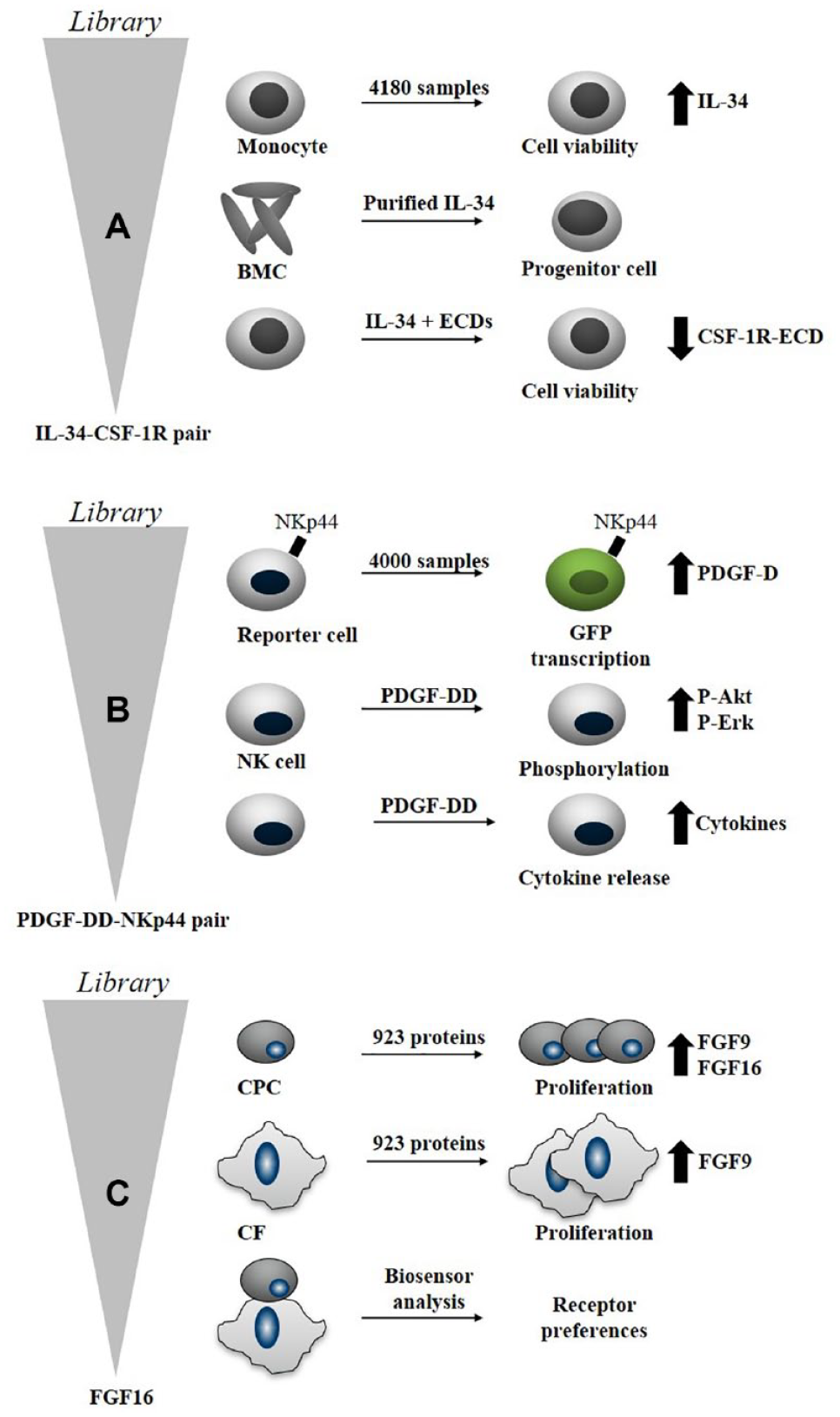
Examples of targets discovered by secretome-based screening.5,35,68 (
Based on this initial discovery, the cabiralizumab antibody, which inhibits the signaling via CSF-1R, was developed. It is now in several different phase 2 clinical trials, in combination therapy with a PD-1 antibody, for treatment of different cancer indications. The humanized monoclonal antibody is directed against the CSF-1R expressed on monocytes, macrophages, and osteoclasts, and it inhibits the binding of macrophage CSF-1 and IL-34 to CSF-1R (https://www.cancer.gov/publications/dictionaries/cancer-drug/def/cabiralizumab; https://www.fiveprime.com/programs/cabiralizumab/). 105
Identification of PDGF-DD as the Ligand of the NKp44 Receptor
Natural cytotoxicity receptors are potential targets for autoimmune diseases and, together with their ligands, can be successfully targeted for cancer immunotherapy.
106
Natural cytotoxicity triggering receptor 2 (NKp44) is a receptor found on natural killer (NK) cells. It was originally identified in 1999 as a novel receptor.
107
However, the ligands of the receptor have remained elusive.
5
To identify ligands, a secretome-based screening approach was performed using a gene reporter assay and a library similar to the one described in Gonzalez et al. (consisting of 806 proteins) and described to contain more than 4000 mouse and human proteins
5
(
Identification of FGF16 That Induces Proliferation of CPCs
Heart failure after myocardial infarction is a clinical condition that causes high morbidity and mortality.
108
There is a large unmet medical need for treating heart failure driven by the loss of functional cardiomyocytes that occurs during myocardial infarction. It has been shown that CPCs present in the heart contribute to repair of the myocardium and are promising candidates for cardiac repair/regenerative therapies.
109
A subset of the KTH secretome library (923 proteins) was screened with the aim to identify proteins that stimulate proliferation (
Summary
Secreted proteins regulate numerous physiological functions in humans and have become attractive tools for drug discovery. Screening using secretome libraries in disease-relevant assays has been successfully applied in phenotypic target discovery. There are many examples of successful identification of novel targets regulating various biological phenotypes, for example, cell proliferation, tumor cell suppression, and viral infection.
Although secretome-based screening has been successfully applied for target discovery, there is a benefit to making further improvements, both regarding the constituents of the library and from a screening perspective. For example, including secreted proteins that are part of the “hidden human proteome” encoded, for example, by “noncoding genes”112,113 will facilitate the study of the unknowns in the secretome. Smaller peptides are highly relevant from a disease perspective but are often difficult to generate using a recombinant approach. Instead, smaller peptides can be generated by peptide synthesis and included in the library. Additional improvements to mammalian expression systems and automation of production should speed up the process to produce a purified and quality-checked library. Co-expression with factors that are required to make the processed and activated secreted protein should also be pursued.
We also expect technology developments when it comes to screening. A combination of multiplexed readouts with miniaturization of assays should generate more value from co-cultures of different cell types. For example, one cell type could be expressing and secreting the sample library and thereby affecting another cell type that expresses the relevant receptors. In addition, “combination” screening, where two or more secreted proteins are combined and used for screening, would facilitate the identification of synergistic and antagonistic interactions.
Development of the secretome screen platform at AstraZeneca in collaboration with KTH has provided new opportunities in our drug discovery process. There is an excellent opportunity for collaborations in this area to maximize the use/value of the secretome library to explore a wide range of biologies as exemplified by the collaborations based on the availability of the library produced by the GNF group (e.g., Barrow et al., 5 Sampath et al., 38 Locci et al., 39 and Scietti et al. 40 ). Similarly, AstraZeneca has recently launched the concept of secretome-based screening via the Open innovation platform (https://openinnovation.astrazeneca.com/).
Finally, even though secretome-based screening is based on a protein sample library, it is agnostic of drug modality. Different approaches can follow after a secretome-based active has been validated. An antibody, or an antisense approach, can be used if an antagonistic effect is desired resulting in inhibition of signaling via the cognate receptor. A protein or RNA therapeutics can be developed to mimic a desired agonist response caused by the endogenous secreted ligand. A small-molecule inhibitor can be applied to inhibit signaling downstream of the ligand–receptor interaction. Thus, a secretome library is a valuable complement to any drug discovery toolbox.
Footnotes
Acknowledgements
The authors would like to thank anonymous reviewers for providing insightful feedback during the revision of this review. The authors would like to thank Jeremie Boucher for critical reading of the manuscript. The authors would also like to thank the “Protein Factory” at KTH for producing the secretome library, which has been funded by the Knut and Alice Wallenberg Foundation, Novo Nordisk Foundation, and AstraZeneca.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Mei Ding, Arjan Snijder, Mats Ormö, Per-Erik Strömstedt, Rick Davies, and Lovisa Holmberg Schiavone are employees of AstraZeneca
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.