
Abstract
Antibody drugs have become an increasingly significant component of the therapeutic landscape. Their success has been driven by some of their unique properties, in particular their very high specificity and selectivity, in contrast to the off-target liabilities of small molecules (SMs). Antibodies can bring additional functionality to the table with their ability to interact with the immune system, and this can be further manipulated with advances in antibody engineering. This review summarizes what antibody therapeutics have achieved to date and what opportunities and challenges lie ahead. The target landscape for large molecules (LMs) versus SMs and some of the challenges for antibody drug development are discussed. Effective penetration of membrane barriers and intracellular targeting is one challenge, particularly across the highly resistant blood-brain barrier. The expanding pipeline of antibody-drug conjugates offers the potential to combine SM and LM modalities in a variety of creative ways, and antibodies also offer exciting potential to build bi- and multispecific molecules. The ability to pursue more challenging targets can also be further exploited but highlights the need for earlier screening in functional cell-based assays. I discuss how this might be addressed given the practical constraints imposed by high-throughput screening sample type and process differences in antibody primary screening.
Keywords
Introduction
Historically, pharmaceutical drug discovery has focused on exploring the molecular pharmacology of small-molecule (SM) entities or exploiting natural products. This has been a successful endeavor, producing a wide range of therapeutic molecules to intervene in a broad spectrum of disease areas. However, advances in the monoclonal antibody field—namely, the ability to produce humanized or fully human antibodies via hybridoma technology1 –3 and the power of display technologies, 4 combined with construction of diverse recombinant antibody libraries5–7—have resulted in antibodies becoming an increasingly significant component of the therapeutic landscape.8,9 Numerous antibodies are approved, undergoing clinical evaluation, or in earlier stages of preclinical development.10–12 At the beginning of 2013, the entire clinical pipeline comprised approximately 350 monoclonal antibodies, although most were in earlier stages of development. As of November 2013, there were 23 antibodies in phase 2 or phase 3 clinical studies for noncancer indications and a further 10 in phase 3 for cancer. This is in addition to the 39 therapeutic antibodies already approved and a significant number at various stages of preclinical research.11,12
Antibodies have utility across a range of therapeutic areas, and their success has been driven by some of their unique properties, in particular their very high specificity and target selectivity, in contrast to some of the off-target liabilities of SMs. Biopharmaceuticals can also offer reduced cycle times from concept to clinic. Initial antibody selection using in vitro display technologies can be achieved against most targets in under 2 weeks 5 and can generate antibodies with properties more difficult to obtain by immunisation. 4 Antibody characteristics can be finely tuned using defined selection strategies, for example, targeting specific antigen conformations or epitopes, including competitors, different antigens, or species homologues. 13 In vitro affinity maturation routinely yields antibodies with picomolar affinities and can even generate femtomolar candidates. 14
This review summarizes what antibody therapeutics have achieved to date and what opportunities and challenges lie ahead. In terms of target landscape, antibodies are well suited to circulating ligands and cell-surface targets, whereas targets commonly addressed by SMs include cell-surface receptors and intracellular targets. Effective penetration of membrane barriers is one of the challenges for large molecules (LMs), particularly across the highly resistant blood-brain barrier. The fact that a large proportion of potential drug targets are in the intracellular environment means they have remained largely inaccessible to conventional antibody approaches to date. More challenging targets can also be further exploited, but this highlights the need for earlier screening in functional cell-based assays. Ways in which this might be facilitated will be discussed, including the challenges of using more physiological and disease-relevant systems and phenotypic versus target-based approaches.
Antibody Structure and Alternative Antibody Fragments
Full antibodies are large molecules around 150 kDa, and their bivalent nature can add an avidity component to binding dynamics ( Fig. 1 ). Besides any target pharmacology mediated by binding via the complementarity determining region (CDR) loops, antibodies can bring additional functionality to the table with their ability to interact with the immune system. Advances in Fc engineering mean that effector function can be ablated or enhanced,15,16 for example, for antibody-dependent cell-mediated cytotoxicity (ADCC), and this can have significant mechanistic impact, for example, in oncology settings where immune suppression or evasion plays a key role in disease. The effector mechanisms of antibodies can lead to ADCC, complement-mediated cytotoxicity (CDC), and antibody-dependent cellular phagocytosis (ADCP) of target cells. The Fc domain is also involved in interaction with the neonatal Fc receptor FcRn, which contributes to a long serum half-life of about 21 days for most IgG subclasses and unique pharmacokinetic (PK) characteristics compared with SMs. 17 This can be further manipulated to extend half-life 18 or incorporate pH-dependent antigen binding.16,19
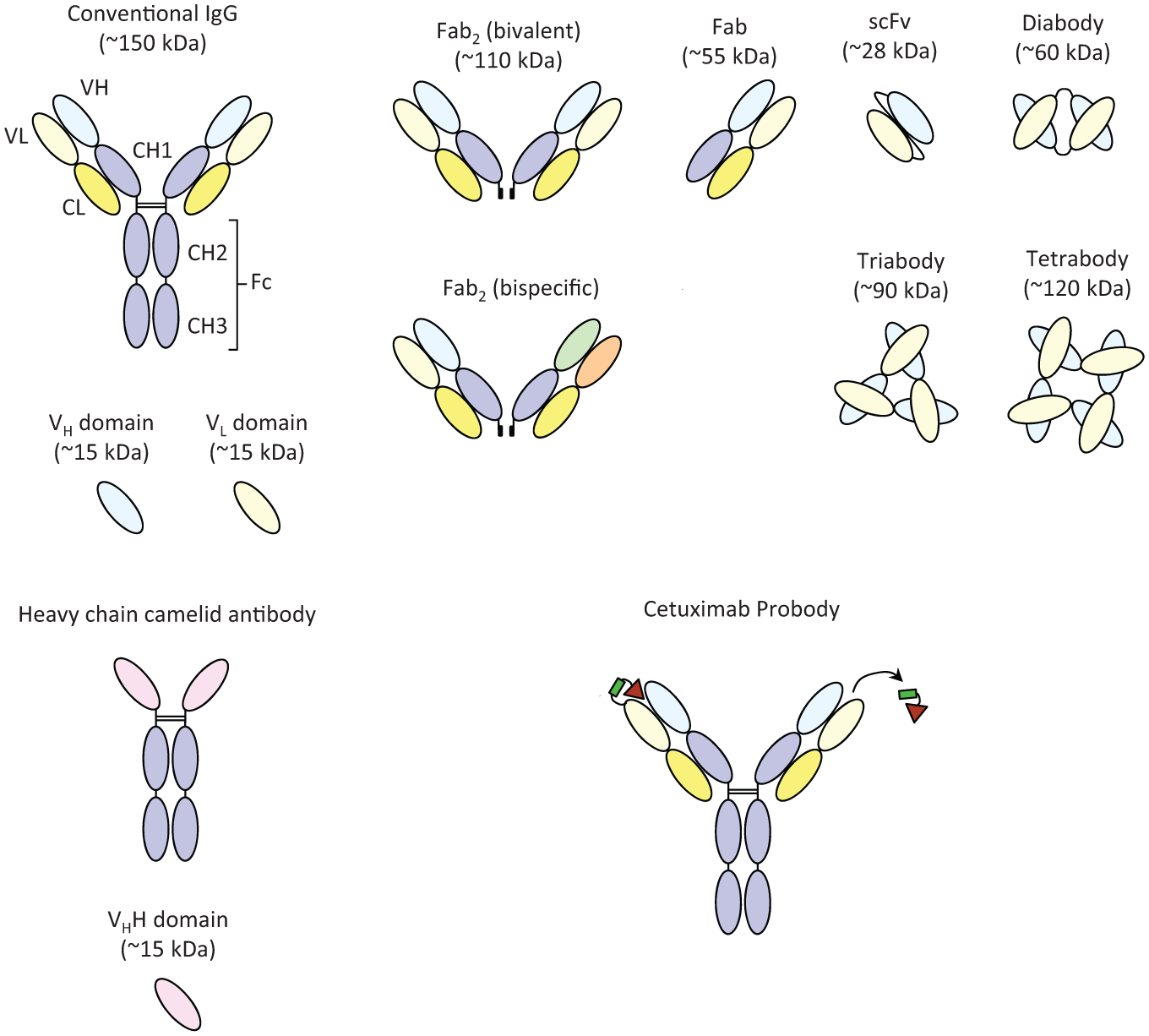
Schematic illustration of the structure of an intact classical IgG, a camelid heavy-chain IgG, and a range of antibody fragments, including an indication of approximate molecular size. Classical IgGs have two heavy and two light chains, which are further divided into variable (VH and VL) and constant domains (CH and CL). Constant regions determine the IgG subclass, and the heavy chain constant region is divided into CH1, CH2, and CH3 domains. The antigen binding site of an IgG is composed of six hypervariable complementarity determining regions (CDRs), three in the heavy chain variable domain (VH) and three in the light chain variable domain (VL). Single domains are the smallest antigen binding fragment. A variety of other antibody fragments are shown. Introduction of a flexible linker of at least 12 residues stabilizes the VH-VL dimer to form a scFv; reducing the linker to between three and 12 residues forms a bivalent dimer, and further reduction below three residues can force the molecule into trimers or tetramers. Camelid antibodies lack light chains and the CH1 domain. The Probody 65 is also shown. The left Fab arm represents the intact molecule, encompassing a masking peptide (red triangle), tethered by a substrate linker (green rectangle); the right Fab arm illustrates the activated Probody following protease cleavage of the masking peptide.
Antibodies display numerous mechanisms of action, and these include interactions at receptors (activation or inhibition), ligand or cytokine blockade by direct capture of ligand, effector mechanisms including ADCC/CDC, induction of apoptosis, internalization, and toxin or conjugate delivery. In some cases, mechanisms of antibody action may include multiple components. For full IgGs, valence and avidity can play a significant role in mechanism and function, potentially increasing potency, and crosslinking can, for example, drive receptor internalization. Antibodies have utility across all disease areas but have had most success to date in oncology, inflammation, infection, transplant rejection, and respiratory disease. Of those antibodies that have been approved, targets include CD20, IL2R, RSV, TNF, HER2, CD33, CD52, CD11a, EGFR, IgE, VEGF, integrins, IL1b, EpCAM, CD3, IL12/23, IL6R, IL6, RANK-L, BlyS, CTLS-4, CD30, IL-6, IL17a, and PD18 (www.antibodysociety.org). In oncology, targeting receptor tyrosine kinases and their ligands has been quite successful, with examples including cetuximab (Erbitux) and panitumumab (Vectibix) for EGFR, bevacizumab (Avastin) for VEGF, and trastuzumab (Herceptin) and pertuzumab (Perjeta) for HER2. However, in the case of EGFR mutations with constitutively active tyrosine kinase activity that occur commonly in cancer, there may be resistance to antibody therapy, 20 particularly where the receptor becomes ligand insensitive and constitutively dimerized. However, resistance can also develop to SM tyrosine kinase inhibitors. 21 Highly expressed antigens such as CD20 on hematopoietic cancers also have been shown to be druggable, for example, with rituximab (Rituxan), where the primary mechanism involves ADCC/CDC. Trastuzumab (Herceptin) 22 is another example of an antibody with an immune effector mechanism. There may also be an opportunity to modulate Fc effector function through protein or glycan engineering to further enhance ADCC.23,24 In addition, there are emerging classes of T-cell immune-modulating antibodies, including those targeting CTLA-4 such as ipilimumab and tremelimumab, those targeting PD1 such as nivolumab and pembrolizumab (lambrolizumab) or its ligand PD-L1, and CD137. 12 Such targets may be challenging to drug with SMs. In inflammation and autoimmune disease, targeting cytokines with antibodies has been very effective, for example, anti–tumor necrosis factor alpha (TNFα) antibody adalimumab (Humira) for rheumatoid arthritis and infliximab (Remicade) for Crohn disease.
Advances in antibody engineering have enabled the modular architecture of antibodies to be recombinantly manipulated to produce a wide range of fragments with varying properties (effector function, serum half-life, avidity). These include single-chain Fv (scFv), Fab, (Fab′)2, diabody, triabody, and single-domain antibody, the smallest antigen binding fragment8,25 ( Fig. 1 ). Single-domain antibodies or “nanobodies” are VHH fragments of heavy chain IgGs, occurring naturally in camelid species where light chains are absent.26,27 They are easily engineered and expressed and have reduced interaction with tissues expressing high levels of Fc receptors and thus low immunogenicity. Examples of domain antibodies in clinical trials include the anti-TNFα ART621 for immunologic disease and a bivalent nanobody ALX-0081 for coronary syndromes. 28
For antibody fragments, potential advantages include better penetration of tissues or tumors and the possibility to better bind less accessible epitopes such as enzyme active sites and viral targets. 29 However, they may have a reduced half-life due to lack of an Fc domain and cannot induce effector functions. Fragments have circulating half-lives measured in hours rather than days, and molecules below 50 to 60 kDa are subject to rapid renal clearance.25,30 However, a variety of half-life extension methods can be used to improve the PK of antibody fragments, including PEGylation and albumin conjugation. 31 Proportionally, there are fewer fragments than canonical antibodies so far in clinical study. 32 ScFv, although predominantly monomeric, can also form higher molecular weight species such as dimers and trimers. 5 The tendency to dimerize has been exploited to create diabodies 33 and tri- or tetrabodies34,35 by reducing the length of the scFv linker. Antibody fragments can also be used as building blocks to generate larger multivalent or multispecific molecules. 26
Bi- and multispecific antibodies present new therapeutic opportunities for intervening in disease where more than one target or pathway is involved. Many different formats of bispecific antibodies have been produced,36–38 using a variety of engineering techniques, including knob-into-hole technology for heavy chain heterodimerisation,39,40 and a number of such molecules are in trials. 37 These constructs can offer increased avidity and efficacy by engaging different epitopes on the same antigen (e.g., two epitopes on IGF-1R 41 ) or the opportunity to target different antigens (e.g., IGF-1R and EGFR 42 ), including the ability to retarget immune effector functions. Bispecific T-cell engagers (BiTEs) combine Fv fragments of two different antibodies fused by a flexible linker that facilitates optimal antibody-antigen interaction, with the aim of linking T cells (via CD3) and tumor-associated antigens (TAAs) such as CD19 in the case of blinatumomab.43,44 Bispecifics have also found utility for CNS targeting, with one arm facilitating transport across the blood-brain barrier and the other providing therapeutic benefit. 45 Trifunctional antibodies such as catumaxomab have an ability to engage three different cell types, typically tumor, T cell, and accessory cells such as macrophages, natural killer (NK) cells, or other Fc receptor–expressing cells. 46 Antibodies therefore offer exciting potential to build a variety of bi- and multispecific molecules capable of modulating targets in new ways.
Arming Antibodies: ADCs and Beyond
Antibodies can be used to selectively deliver cytotoxic drugs to targets such as tumor-associated antigens, allowing a more directed approach than systemic delivery of chemotherapeutics alone, with the potential to reduce toxicity and improve efficacy. Targeted delivery also potentially allows use of more potent cytotoxics than is possible with conventional chemotherapy. Antibody-drug conjugates (ADCs) also offer the potential to combine SM and LM modalities in a variety of creative ways. The expanding pipeline of ADCs47,48 commonly relies on the ability of target antigen to be internalized, together with the ADC, and correctly trafficked to release the payload. 16 Two breakthrough ADC therapeutics have been recently approved, brentuximab vedotin (Adcetris), which is an anti-CD30 chimeric IgG1 for hematological malignancies, and trastuzumab emtansine (Kadcyla), an anti-HER2 humanized IgG1 for solid tumors. In addition, a further 35 novel ADCs are currently in clinical testing. 49
Key considerations in an ADC strategy are good target antigen selection, type and stability of antibody-drug linker, potency of payload drug, conjugation method, and drug-to-antibody ratio.47,50–52 The targeted antigen should be expressed at high levels, ideally homogeneously, but more importantly with limited or no normal tissue expression in order to achieve an adequate therapeutic index. 47 Antigen shedding can increase the risk of toxicity. Receptor internalization and recycling have been studied for many receptor classes and may be accelerated by ligand binding. Some receptors internalize continuously such as the transferrin receptor, others may be accelerated by ligand or antibody binding such as EGFR, and some internalize poorly or not at all. 51 The impact of antibodies versus ligands on receptor internalization is less well characterized but likely depends significantly on antibody epitope. Antibodies with species cross-reactivity can help address on-target toxicity associated with normal tissue expression early in discovery, but often dose-limiting toxicity may be mediated off-target. Obvious issues are linker instability and systemic toxin release, which particularly affected some of the earlier ADC linkers.52,53 Much work has been done on linkers, and there is now appreciation that ADCs with higher drug loads can cause more toxicity, with increased clearance rates for more heavily modified molecules. 54 Site of conjugation is also important in this regard with site-specific methods reducing heterogeneity, increasing stability and improving PK.52,54
Cytotoxic drugs can be linked to the antibody with cleavable or noncleavable linkers. Depending on the payload, the former may have more capability for bystander effects, which can be useful for tumors with heterogeneously expressed antigens. Linkers have been designed to exploit intracellular conditions for drug release (e.g., acidic environment of endosomes and lysosomes, high cytosolic thiol concentrations, proteolytic enzymes in lysosomes). Linkers ideally should allow rapid release of drug inside cells but have good stability in plasma during circulation to limit toxicity.50,51 Optimal linker can depend on the antigen. ADCs with cleavable linkers were shown to be efficacious in vivo against seven B-cell targets (CD19, CD20, CD21, CD22, CD72, CD79b, CD180), but only CD22 and CD79b displayed efficacy with noncleavable linkers. 55 Conjugation methods are critical for effective ADCs. Away from the heterogeneous conjugates formed by nonspecific labeling of lysine and cysteine residues, site-specific conjugation can yield homogeneous ADCs with defined sites and drug stoichiometries. 52 Evidence suggests that the site of such conjugation modulates stability in vivo 56 and can improve the therapeutic index. 57 Engineering approaches to site-specific conjugation include substitutions or addition of accessible cysteines 57 or incorporation of nonnative amino acids.58,59 Most cytotoxic drug moieties are based on microtubule-targeting drugs (maytansinoids, auristatins, tubulysins) or DNA-damaging drugs (calicheamicins, pyrrolobenzodiazepines), and they tend to have greater potencies than conventional chemotherapeutics. Development of ADCs may therefore be challenging, with three separate components to consider50,51: antibody, linker, and payload. Data are limited on the relationship between antibody affinity and antitumor potency for ADCs, and in addition, the roles played by antigen density, internalization rates, and recycling need to be further understood in relation to ADC activity. 47
Other armed antibody approaches include antibody-directed enzyme prodrug therapy (ADEPT), antibody-cytokine fusion proteins or immunocytokines,60,61 immunotoxins, and radioimmunoconjugates.60,62–64 The possibility of activating drugs specifically when they are in the tumor microenvironment could be a neat strategy to reduce potential systemic toxicity of armed antibodies. Desnoyers et al. 65 describe an EGFR-directed Probody therapeutic that remains inert until activated by proteases upregulated in the tumor microenvironment. The Prodrug is a modified version of cetuximab where the antibody is coupled via a protease cleavable linker to a masking peptide that blocks EGFR antigen binding until the antibody is at its tumor target ( Fig. 1 ); it shows comparable efficacy to normal cetuximab but decreased toxicity at high doses. Engineered immunoliposomes can also provide an alternative drug delivery strategy to ADCs, with high drug capacity of up to 150,000 molecules per liposome and potential avidity effect resulting from multivalent display of 5 to 100 antibody fragments, which can crosslink cell surface receptors to drive internalization. 66 Nielsen et al. 67 identified Erb2 scFvs capable of triggering receptor-mediated endocytosis, which were then coupled to sterically stabilized liposomes containing doxorubicin. These scFv-targeted nanoparticles were avidly internalized by Erb2-expressing tumor cells and had antitumor activity in vivo. Finally, a variety of other SM drugs could theoretically be coupled to antibodies for more targeted and potentially cell type–specific delivery.
Challenging Targets and Diverse Mechanisms
Protein-Protein Interactions
Protein-protein interactions (PPIs) constitute a large and important class of potential therapeutic targets. Antibodies have had some notable success, specifically with regard to disrupting cell surface or extracellular ligand and receptor interactions, providing high target specificity, affinity, and isoform-specific selectivity with few off-target liabilities. However, there is also a rich landscape of potential intracellular PPI targets that are currently not accessible to LMs due to issues with penetration of cellular membranes. Such intracellular PPI targets may offer novel mechanisms to affect specific signaling pathways downstream of receptor activation.68,69 This could allow more subtle and specific intervention in signaling cascades and downstream cellular function than can be achieved at the level of the receptor with standard agonists or antagonists. Despite their clear potential for intracellular penetration, PPIs have proved to be challenging targets for SMs due to the large size and the flat geometry of the protein interaction interface, often with a lack of well-defined binding pockets. 70 Some progress has been facilitated by the identification of discrete binding hot spots on protein interaction surfaces,68,70 which have been well characterized for the IgG Fc domain, co-crystallized with three protein ligands and a phage-optimized peptide. 71 In addition, SM libraries tend to be highly biased toward classical drug target classes, so further exploration of chemical space may be required to identify new molecular templates. Use of in silico methods, structure-based design, and fragment-based discovery 69 may allow identification of other SMs such as those reported to inhibit interactions between BCL/BAK in regulation of apoptosis, 70 MDM2 and p53, 70 or those differentially inhibiting G protein βγ subunit regulation of selected downstream effectors. 72
Antibodies with their larger footprint may generally be more amenable to targeting these PPI interfaces, but intracellular delivery currently remains a significant hurdle. Advances in the engineering of smaller antibody fragments 25 may help make such targets more tractable, including sites that might be less accessible to conventional antibodies such as enzyme active sites and viral pockets.
Intracellular Delivery
Intracellular targets represent a major hurdle for biologics compared with SMs, and the issue has been challenging to address, with many approaches still at an early stage of validation. Delivery of LMs into the cytoplasm is challenging, and for antibodies, intracellular expression of correctly folded protein is difficult due to the reducing cytoplasmic environment. One approach used to enhance intracellular delivery is by coupling the “cargo” to peptides that can translocate across cell membranes. These cell-penetrating peptides or protein transduction domains (PTDs), such as Tat, SynB, and penetratin, are cationic, interact with lipid membranes, and have been used to deliver peptides, oligonucleotides, and proteins, as well as enhance the uptake of liposomes.73–75 For Tat, the mechanism appears to be lipid raft–dependent, receptor-independent macropinocytosis, and although the macropinosomes do not seem to traffic to lysosomes, the release of PTD-conjugated cargo to the cytosol is limited. Endosomal escape may potentially be enhanced by pH, using noncovalent cleavable linkers, 74 or by developing new agents based on knowledge of methods used by viruses and bacteria for endosomal escape. 76 One application of cell-penetrating peptides has been to develop intracellularly targeted allosteric modulators of G protein–coupled receptors (GPCRs) using peptides derived from the third intracellular loop. 77 These lipidated peptides or “pepducins” allosterically activate or block GPCR function, with the lipid moiety facilitating rapid translocation across the membrane and anchoring the peptide on the cytosolic face. 78 A range of pepducins have now been produced from segments of cytoplasmic loops 1, 2, or 3, or the C-terminal tail, with a variety of lipid tethers and linker regions.78,79 Stapled peptides have also received attention, although there is some controversy in the literature about cell permeability. 80 A second technique to enhance cellular delivery, similar to cationization-enhanced adsorptive-mediated transcytosis across the blood-brain barrier (BBB), is to increase protein net positive charge such as that described for +36 GFP-mediated small interfering RNA (siRNA) delivery.81,82 Of particular interest is a subset of lupus autoantibodies reacting against host DNA, which can penetrate living cells, even localizing in the nucleus. A particular lupus anti–double-stranded DNA (dsDNA) antibody 3E10, which unusually is not harmful to cells, was isolated from a mouse model of systemic lupus erythematosus and penetrated cells via an equilibrative nucleoside salvage transporter, which is ubiquitous on human cells. 3E10 has been shown to transport a variety of LMs, including antibodies, into the cell nucleus.83,84
A potential route for success with functional intracellular antibodies, or “intrabodies,” has come with the engineering of smaller antibody fragments. In situ expression of intracellular mAb fragments could provide an attractive alternative to systemic administration, and advances have been made in both selection technologies and in framework designs to enable correct folding. 25 ScFv can be problematic as intrabodies because disulfide bonds cannot form in the reducing environment of the cell, potentially resulting in incorrect folding, insolubility, and instability.82,85
Intracellular antibody capture (IAC) technology enables direct selection of antibodies that can fold correctly and bind their target in the reducing environment of the cell cytoplasm. The method involves a modified two-hybrid system where antibody libraries are screened directly in yeast using an antibody-antigen interaction assay. For example, when a prey protein (Ab fragment fused to VP16 transcriptional activation domain) interacts with a bait protein (target fused to a LexA-DNA binding domain), the association results in transactivation of the His3 gene, which enables yeast cells to grow in the absence of histidine. 86 Tanaka et al. 85 generated diverse single-domain VH intrabody libraries and used IAC to identify antibodies that inhibited RAS-dependent oncogenic transformation of NIH3T3 cells. Visintin et al. 87 engineered a library of intracellular scFv antibodies and used the genetic selection strategy whereby PPI inhibition constitutes a selective advantage for yeast cells. On antibody disruption of the target PPI, a tetracycline repressor that suppresses HIS3 gene expression is not expressed, and yeast cells survive in the absence of histidine. Intrabodies inhibiting interaction of GABARAP with the GABAA γ2 subunit and interfering with the p65 dimerization domain, resulting in NF-κB transcriptional activation, were identified. While first-generation IAC employed an initial phage display selection step to generate a sublibrary of antibodies in a yeast prey vector, which was then screened in yeast with a target bait protein, second- and third-generation IAC methods employ direct screening of libraries in yeast. 86
However, the key issue for intrabodies remains therapeutic delivery. While delivery of proteins themselves is difficult, nucleic acid expression vectors are being explored for direct expression in target cells.82,86 Options include viral delivery, antibody-coupled vector delivery via antigen internalization, and liposome nanoparticles using antibodies for cell surface antigen targeting, with encapsulated single domains or vectors able to express them.
GPCR Targets
At first glance, certain target classes such as GPCRs (prototypical structure shown in Fig. 2a ) seem to have been well addressed by SMs. However, a significant number of GPCRs have proved intractable to drug discovery, particularly away from the family A receptors targeted by many existing drugs, and in addition, there are often challenges in obtaining receptor selectivity with SMs, such as seen for the monoamine receptor family. 88
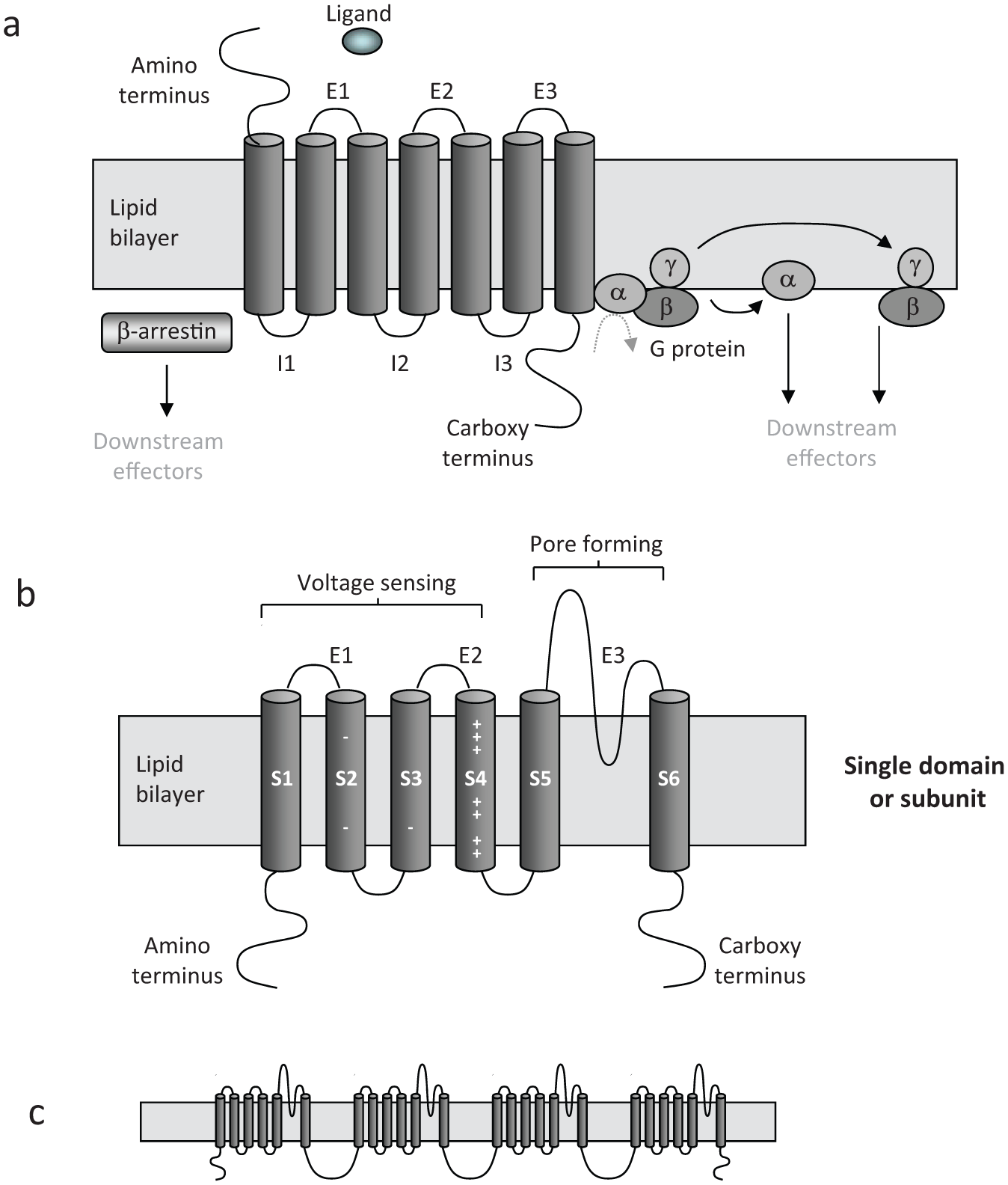
Schematic representation of key structural features of complex receptors. (
Most sequence diversity resides in the extracellular N-terminus and loop regions, domains likely to be targeted by antibodies, lending support to the idea that antibodies may be useful GPCR therapeutics, particularly with their potential for high selectivity, affinity, and extended serum half-life. However, despite the fact that a range of GPCR- and GPCR ligand-targeting antibodies are now in various stages of clinical and preclinical development,88,89 it has not been easy or straightforward to target these receptors with LMs, and substantial effort has been invested in this pursuit. In addition, certain GPCRs may be more intractable to antibody therapeutics, especially those with limited extracellular domains or small, buried, and lipophilic binding pockets, where SMs may have better penetration.
As with other membrane-spanning proteins, there have been technical challenges in raising functional antibodies against GPCRs, predominantly arising from issues with antigen reagents. The ideal is pure, stable, homogeneous antigen in specific native conformations, which is challenging outside of a membrane context. Low cellular receptor expression levels may also be a limiting factor for cell surface selections or immunization purposes, and the extent to which the extracellular domains are exposed may also influence the success with a given target. 89 A variety of antigen strategies for antibody isolation have been used, including isolated peptides, constrained peptides, overexpressing cell lines, cell membranes, DNA immunization, liposomes, purified protein, and StaR GPCRs, engineered with a small number of point mutations to increase thermostability and drive the receptor into a specific conformational state. 88 Cell selections generally require a high copy number of receptors and ideally a deselection stage on untransfected parental cells. 90 Examples where cell-based panning has been successfully used are the isolation by subtractive selection of scFvs specific to the activated conformation of integrin αIIbβ3 91 and OncoMed’s use of a hybrid cell panning/recombinant protein strategy to isolate different anti-MET antibodies that synergistically inhibit HGF binding to MET. 92 The use of cells in selections potentially also allows selection of clones with specific phenotypic characteristics such as the ability to internalize, affect proliferation, and recognize novel cell type–specific (e.g., tumor) antigen forms. 90
Orthosteric versus Allosteric Mechanisms
The issue of receptor subtype selectivity is one factor that has driven the pursuit of SM allosteric modulators,93–95 since classically targeted orthosteric sites tend to be more highly conserved across GPCR subfamilies. Identification of SM drugs with allosteric mechanisms of action came about primarily with the increasing use of functional screening assays,96,97 but it is becoming increasingly evident that antibodies can also exhibit such modulatory effects.
Other factors that have focused attention on allosteric modulators include the ability to more subtly modulate physiological systems, the potential to elicit probe-dependent effects, and separate control of affinity and efficacy.95,96 With regard to the latter, there is an interesting report of a CXCR1 (class A GPCR) antibody that binds the receptor N-terminal region and can block interleukin 8 (IL-8)–mediated functional responses but has no detectable effects on ligand binding. 98
Allosteric mechanisms affect many target types besides GPCRs, such as ligand-gated ion channels (e.g., GABAA 99 ; NMDA 94 ), receptor tyrosine kinases (RTKs) (e.g., insulin receptor 100 ), and enzymes. They generally involve modulation of an orthosteric ligand, although some allosterics can induce direct effects in their own right, such as observations of antibody-induced internalization of mGluR7 class C receptors 101 and direct activation of the insulin receptor by an allosteric antibody. 100 Thus, it is of interest to note, in addition to SM modulators, the growing number of reports of antibodies with allosteric mechanisms, both at GPCRs (e.g., allosteric inhibitor of glucagon class B GPCR 102 ; allosteric inverse agonist at class A adenosine receptor A2A 103 ) and at other target classes (e.g., insulin receptor 100 ; allosteric inhibitors of IGF-1R 104 ; allosterics at TrkB BDNF receptor 105 ; allosteric inhibition of BACE1 aspartic protease 106 ; allosteric inhibitor of αV integrins 107 ). Autoantibodies, which can influence receptor activity, have been described for a number of GPCRs, often targeting the second, most variable extracellular loop,108–110 and further allosteric antibodies binding this region have been developed (e.g., partial agonist mAbs and inverse agonist scFvs at β2-adrenergic and muscarinic M2 class A receptors111,112).
Efficacy Profiles and Biased Signaling
Depending on the target biology, there is likely to be a requirement for molecules with defined efficacy profiles such as agonist or inverse agonist antibodies. A variety of SMs, previously classified as GPCR antagonists, have subsequently been shown to possess partial agonist or inverse agonist activity in some sensitive functional systems. In addition, the recognition that receptors can exhibit constitutive activity in the absence of agonist 108 has reinforced the potential therapeutic value of inverse agonists. Examples of GPCR antibodies reported to have inverse agonist activity include those for the thyrotropin receptor 113 and glucagon receptor. 114 This again highlights a need for functional cell-based screening, particularly given the known complexities of biased GPCR signaling in response to different conformations of the receptor.89,115,116 There is interest in molecules capable of recognizing and stabilizing agonist, antagonist, or inverse agonist conformational states (reviewed in Webb et al. 89 ; generation of conformation-specific antisera 117 ; conformationally selective single domain intrabodies 118 ). Being able to influence biased agonism—namely, the ability of a given ligand to preferentially activate one signaling pathway over another—offers the potential to selectively affect different signaling pathways and potentially provide superior therapeutics, without a requirement for intracellular delivery. Reported examples where biologics have exhibited this phenomenon of GPCR-biased signaling are agonistic anti–β1-adrenergic monoclonal antibodies, which were unable to signal via β-arrestin in contrast to natural ligand isoprenaline—the potency of isoprenaline was reduced despite antibodies enhancing its binding affinity 109 —and an anti-mGluR7 antibody that inhibited ortho- and allosteric agonists through promoting receptor internalization via a G protein–independent pathway. 101 In contrast to SMs, which tend to be very receptor centric, antibodies offer additional potential to target the ligands that elicit biased signaling.
Traditional SM pharmacology, aided by availability of new structural data, has described an increasingly complex and elegant world of drug-receptor interaction over the years, from concepts of agonism and inverse agonism, through allosteric modulation and alternative signaling pathways, to biased agonism and conformation-specific states. Some of the above examples clearly place antibodies on a level playing field with SMs in terms of their ability to interact with their targets in a variety of complex ways beyond the traditional neutralizing antibody (e.g., pan-TGFβ antibody 119 ; anti-TNFαs infliximab and adalimumab 120 ; anti-VEGF Avastin 13 ).
In addition to GPCRs, the feasibility of generating antibodies with defined efficacy profiles is also evident for other target classes, such as agonists at the Trk receptor family of RTKs 105 and cytokine receptors, including the TNF receptor superfamily. Agonist anti-CD40 antibodies have been produced, which may have utility in cancer immunotherapy,121,122 and Chodorge et al. 123 describe Fas receptor agonists where the relationship between affinity and agonist potency was systematically investigated to reveal that bivalent agonists may have a potency ceiling that is not surmountable via conventional affinity maturation. Finally, hetero- and homodimerization have been described for a number of receptor classes, including GPCRs, 124 and modulation of these interactions may offer novel ways of intervening in disease. Some of the antibodies directed against receptor tyrosine kinases for cancer 125 affect receptor dimerization mechanisms; pertuzumab is an anti-HER2 inhibitor blocking ligand-induced HER2-HER3 heterodimerization, and cetuximab blocks dimerization of EGFR.
Ion Channel Targets
Voltage- and ligand-gated ion channels constitute another class of challenging multimembrane spanning targets for antibodies, where highly dynamic protein conformational changes govern function. In addition, for voltage-gated channels, resting, activated, and inactivated states are controlled by membrane potential, and crystal structures and functional studies are helping reveal how membrane potential–induced conformational changes are propagated to the pore domain.126,127 Reviews of potential ion channel targets in relation to existing approved (SM) drugs suggest that there is significant untapped potential for targeting ion channels. 128 SMs and peptide toxins have been widely explored for ion channels but often lack target selectivity and therefore have potential side effect issues. Thus, antibodies are attractive candidates for subtype-specific ion channel modulation, although there are few candidates in clinical development,129,130 reflecting the difficulty of generating functional antibodies to these targets. An example of the subtype specificity that might be achievable is a polyclonal antibody showing selective inhibition of a splice variant of the NaV 1.5 voltage-gated sodium channel expressed in cancer cells, which differs from the main form by only seven amino acids. 131 The main pore-forming α subunit of voltage-gated sodium channels contains four homologous domains (D1–D4), each with six transmembrane segments (S1–S6) ( Fig. 2b , c ). These seven divergent residues are located in the extracellular loop between S3 and S4 of D1. Endogenous physiological evidence that functional antibodies to ion channels were possible came from studies of a variety of autoimmune diseases where autoantibodies are produced. 132 For example, antiserum from patients with Lambert-Eaton syndrome has been shown to inhibit Ca2+ channel function. 133 As for GPCRs, targeting the right epitope in the correct conformational state is key to achieving functional antibodies. Wyatt et al. 134 describe a depolarization-sensitive antibody targeting a region of the voltage-dependent Ca2+ channel α1D subunit, C-terminal to the pore-forming SS1 to SS2 loop in domain IV, which is only exposed when cells are depolarized. The antibody attenuated the Ca2+ channel current and reduced the ability of the 1,4-dihydropyridine agonist S-(–)-Bay K 8644 to enhance this current.
Strategies to isolate ion channel antibodies therefore need to use antigen displayed in native conformations. Ways in which this might be achieved include exploiting cells endogenously or recombinantly expressing the channel of interest and targeting the exposed extracellular domains. A deselection step on the same cell background without target can be incorporated during in vitro antibody selections from recombinant antibody libraries or at the primary screening stage to filter out nontarget binders. For a hybridoma approach, cell immunizations can be conducted with the target expressed in different cell backgrounds, or potentially alternating with recombinant protein, to reduce the number of antibodies binding the cell surface nonspecifically. One issue with ion channels is that heterologous expression of target at high levels can often impair cell health. An additional challenge is how to control conformational states of the ion channel, particularly given that exposure of certain epitopes might vary with channel state. 134 It may be possible to control the depolarization status of cultured cells in vitro by using chemically defined medium, 134 or it may prove possible to introduce mutations into the channel to control conformational state, similar to the StaR approach for GPCRs. 88 Other approaches include raising antibodies to synthetic peptides representing partial sequences of extracellular domains (examples below) or using alternative ways of displaying the target protein. Latter techniques include the use of magnetic proteoliposomes as described by Mirzabekov et al. 135 and expression of high levels of membrane protein targets in functional form in the membrane of the yeast Pichia pastoris. 136
One strategy for generating functional ion channel antibodies has been to target the third extracellular (E3) loop of the channel ( Fig. 2b ) (successes and practicalities reviewed in Naylor and Beech 137 ). Xu et al. 138 applied E3 targeting to the transient receptor potential (TRP) Ca2+ channel member TRPC5 and the voltage-gated sodium channel NaV 1.5 to generate target-specific polyclonal inhibitors, while Yang et al. 139 used a 14–amino acid E3 peptide from the external end of the human Kv1.3 pore region to generate a polyclonal antibody by immunization. A variety of functional polyclonal antibodies have been described, 137 although one study found that polyclonals raised against E3 (Glu600 to Pro623) of TRPV1 allosterically blocked function, 140 but monoclonal antibodies were ineffective. The latter were raised in a separate immunization using Thr598 to Cys636, and lack of effect could be explained by targeting single versus multiple epitopes but possibly also by clone prioritization based on cell binding rather than function. However, functional ion channel monoclonals have been reported for both voltage- and ligand-gated channels. An E3-targeted antibody blocked human Eag1 potassium channels and exhibited antitumor effects in vitro and in vivo 141 ; an inhibitory NaV 1.7 antibody raised against the S3 to S4 loop at the tip of the voltage-sensor paddle, which moves in response to membrane potential changes and is key in channel gating, suppressed inflammation and neuropathic pain in mice 142 ; a monoclonal raised against the adenosine triphosphate (ATP)–gated P2X7 ion channel by transfected cell immunizations reduced currents and inhibited IL-1β release 143 ; and Lee et al. 144 identified monoclonal antagonists of TRPA1. The potential to use other extracellular regions is likely to vary on a target-by-target basis, 138 dependent on the size of the exposed domain, sequence diversity across channel subtypes, posttranslational modification (E1 and E2 are commonly glycosylated), and what sort of conformational changes occur in the domain on channel activation.
CNS Targets and the BBB
The brain constitutes one of the least accessible organs in the body. A major challenge for therapeutic delivery of antibodies in the CNS is the inability of LMs to cross the BBB. 145 The barrier is formed by complex tight junctions between endothelial cells of brain capillaries, which have low endocytic activity, and this prevents the passage of polar, lipid-insoluble, or large molecules >600 Da.146,147 Although some small, lipophilic drugs can penetrate by passive diffusion, up to 98% of SMs and practically all LMs are restricted. 148 Some solute carriers and active transport mechanisms exist on the barrier, 147 which permit nutrient access and efflux of metabolites. In addition, receptor-mediated transport systems allow BBB transport of selected endogenous LMs,147,148 and these include the transferrin, insulin, insulin-like growth factor, and leptin receptors.
Strategies to enhance brain drug delivery above levels achievable by passive diffusion include modifying the drug itself or coupling it to a vector for receptor-mediated or adsorption-mediated transcytosis.146,148 Invasive techniques such as direct physical administration or BBB disruption by osmotic, chemical, or mechanical means are also possible but carry obvious risks. Pharmacological modification of drug can include lipidization, reducing the number of polar groups, and using carriers such as nanoparticles or liposomes, while more physiological methods exploit existing BBB carriers and receptors. The latter method has been used to deliver L-DOPA for Parkinson disease and therapy for brain tumors.
Basic proteins have an enhanced ability to enter the CNS via adsorptive-mediated transcytosis where charge mediates a nonspecific binding to anionic sites on the apical side of the endothelial cell membrane, followed by endocytosis.145,147 Thus, cationization of antibodies or of protein vectors such as albumin may enhance brain delivery, 145 although primary sorting endosomes need to be routed away from the degradative acidic lysosomal compartment, which tends to occur more than in peripheral endothelia. 147 Use of nanoparticles, including liposomes,145,149 is another approach, where drug moieties can be encapsulated or adsorbed onto the surface. Gaillard et al. 150 used pegylated, doxorubicin-loaded liposomes with an additional glutathione coating to enhance brain delivery for glioblastoma therapy in vivo in mice, reporting better delivery and efficacy than with nonglutathione liposomes. Glutathione occurs naturally at high levels in brain tissue, and transporters for the antioxidant substance are highly expressed on the BBB. Gao et al. 151 reported enhanced brain delivery of vasoactive intestinal peptide in vivo via an intranasal route using nanoparticles conjugated with wheat germ agglutinin.
Hijacking receptor-mediated transcytosis forms the basis of the “molecular Trojan horse” strategy,145,148 where antibodies against receptors that undergo transcytosis across the BBB can be used to deliver drugs or therapeutic peptides into the brain. HIRmAb is a molecular Trojan horse that targets the human insulin receptor and has been used to deliver proteins into the CNS. For use in rodents, the murine OX26 antibody targets the rat transferrin receptor, and a rat antibody, 8D3, targets the mouse transferrin receptor. 148 Phage display has been used to identify other molecules that can cross the BBB, including novel human brain endothelium-specific single-domain antibodies, FC5 and FC4. 152 FC5 uptake was saturable in vitro, suggesting a receptor-mediated process, colocalized with clathrin, and was targeted to early endosomes, bypassing late endosomes/lysosomes. 153 CNS delivery of proteins and antisense molecules has been reported using this Trojan horse strategy, for example a SA-OX26 delivery vector targeting the transferrin receptor to deliver bFGF in rats, which evoked a time-dependent neuroprotection, 154 and a human immunodeficiency virus (HIV) antisense drug delivered in a similar manner in animal models. 155
For antibodies, additional transport mechanisms exist for rapid removal from the brain, presumably to prevent engagement of Fc domains with effectors that promote a proinflammatory response. 156 Boado et al. 157 exploited this by fusing anti-amyloid scFv antibodies, able to disaggregate Abeta fibrils, to the carboxyl terminus of the heavy chain of HIRmAb for brain delivery in Rhesus monkeys, with the Fc domain of HIRmAb able to bind FcRn and effect brain efflux. Bispecific antibodies, with one arm providing the therapeutic efficacy and the other facilitating transport, appear ideally suited to address issues of antibody brain penetration, 45 although biodistribution studies suggested that much of the high-affinity antitransferrin receptor antibody remained trapped in brain capillaries. In an elegant study, Yu et al. 158 reported that reengineering the construct by reducing affinity for the transferrin receptor while maintaining high affinity for the enzyme BACE1 enhanced release into mouse brain and the observed efficacy of BACE1 inhibition. Once the brain compartment is accessed, considerations are similar to SMs—namely, attaining suitable overall levels, ensuring appropriate availability and receptor occupancy at the target, ability for cell type–specific targeting (neuron, astrocyte, etc.). One advantage of the normally limited CNS penetration of antibodies is that this can provide therapeutic benefit with respect to side effect profiles when peripheral antigens are targeted, in contrast to SMs.
Target Landscape Summary
While the above examples illustrate strong progress with antibodies for challenging targets, it is clear there is significant further untapped potential. There are many GPCRs not effectively targeted by SMs; ion channel antibodies are still in their infancy with respect to development of clinical drug candidates; ion channels can be further exploited as targets in new disease areas such as cancer and respiratory disease; there is a huge array of new targets in the intracellular environment with the potential to more subtly and specifically affect downstream biology; and finally, there are new opportunities to affect the increasing burden of CNS diseases such as Alzheimer disease and neurodegenerative conditions. Achievements to date suggest antibodies can modulate their targets in a variety of complex ways.
Screening Challenges for the Next Generation of Antibody Therapeutics
Many of the above examples highlight a need for earlier use of functional cell-based assays during screening. This has already happened in the SM world, where a move away from classic binding and ligand inhibition assays to front-line cell-based screens has resulted in identification of molecules with diverse function. Implementation of cell-based screens has been facilitated by advances in assay technology, improved sensitivity and dynamic range, immortalized cell lines engineered for high target expression and high signal, and advances in automation and miniaturization.159–161 While similar suites of cell assays are available for both SM and LM discovery, high-throughput screening (HTS) sample type and process differences impose some practical constraints in antibody primary screening.
In contrast to SM discovery, protein libraries are often initially screened from extracts or supernatants containing protein of variable and undetermined concentration, rather than using purified proteins. These include hybridoma supernatants 162 and crude bacterial lysates or extracts.163,164 Compared with screening chemical libraries, there is an initial antigen selection step, in vitro or in vivo, resulting in highly focused, relatively low-throughput screening campaigns, typically around 10,000 to 20,000 scFv antibodies during lead isolation. While later-stage titration and pharmacological characterization is done using purified proteins, primary HTS is often conducted on crude preps. Cell assay-based HTS can be hampered by assay tolerance to crude bacterial preparations, bacterial by-products and osmotic shock agents, or components in hybridoma supernatant medium, including serum and growth factors. In addition, early lead isolation scFv antibodies can be low affinity, and expression levels can be low or extremely variable,164,165 and these factors all affect observed cellular responses. Some engineered assays may be more tolerant, for example, responses proximal to the receptor may be less affected, although it may be possible to use downstream assays such as reporter genes, depending on the target, its signaling cascades, the cell background, and the sample type being screened. 166
Purifying antibodies or reformatting to IgG for functional testing has often been a bottleneck prior to functional cell-based assay evaluation.7,47 For example, in vitro cytotoxicity is generally one of the first-line assays for ADCs but requires reformatting of antibody fragments to IgG for drug conjugation. In some cases, screening assays have proved challenging even for SMs, exemplified by ion channel targets. 128 Electrophysiology is the gold standard, allowing detailed analysis of channel conductance, but methods are slow and low throughput. Higher-throughput ion channel screening technologies have been developed, including fluorescence and luminescence methods, and advances made with higher-throughput automated electrophysiology platforms such as QPatch, IonWorks, and PatchXpress.128,167
Ways of approaching such issues could include reformatting antibody libraries, IgG display on mammalian cells, 168 and screening in IgG product format. Empirically, cell culture medium components from cells secreting IgG are still likely to cause issues in some assays. Mammalian display libraries may also often be of reduced library size, which could potentially limit hit diversity. There is also potential for yeast display libraries, and companies such as Adimab (Hanover, NH) have capitalized on this sort of technology with a platform allowing dual-mode surface display and secretion of full-length antibodies with human-like N-glycans in P. pastoris. 169 Other options include high-throughput purification techniques or use of deep sequencing to interrogate selection outputs and guide antibody lead selection, while achieving good diversity.170–172 Advances in assay technology may provide some solutions such as the two-hybrid IAC system described above for direct screening of antibody libraries in yeast for functional PPI inhibition.86,87 Another interesting approach was described by Melidoni et al., 173 in which antibody gene populations were expressed in embryonic stem cells using a targeting vector and, following expression and secretion, changes in differentiation of individual antibody-expressing ES clones were monitored. This in-cell expression and reporting system was validated by generating blocking antibodies to FGF4. Zhang et al. 174 devised a system to directly select functional thrombopoietin receptor agonists using combinatorial antibody lentiviral libraries to infect eukaryotic cells expressing a receptor and a fluorescent reporter construct. The method thus overcomes the need for a two-step process of selection for binding followed by functional characterization. Yea et al. 175 also used lentiviral transduction to infect TF-1 cells with an unbiased scFv-Fc antibody library and measured colony formation in the resulting infected cells. Targets for the recovered antibodies were shown by mass spectroscopy to be integrins and ion channels, with the former antibody able to induce differentiation of CD34+ stem cells into dendritic cells.
Making It More Relevant
A hot topic in SM discovery is the increasing effort to use more physiological and disease-relevant systems, including primary cells, stem cells, and 3D cultures.176,177 Some of these assays have been facilitated by advances in assay technologies such as high-content screening,178–180 where it becomes possible to measure more complex cellular behavior and phenotypes as well as multiplexing a variety of signaling readouts. The ability to track single-cell responses in complex cultures has also been enabled. However, some authors have suggested there is greater potential to enhance the information content from such platforms. 181
Away from a target-based approach, phenotypic screening approaches have also received attention, both as a mechanism of identifying molecules eliciting a relevant physiological outcome and as a means of potentially identifying new targets. However, target deconvolution can present a substantial challenge. 182 Examples of the use of phenotypic methods in antibody drug discovery include combining phenotypic and proteomic approaches to identify membrane targets in triple-negative breast cancer cells 183 and using a phenotypic approach to identify antibodies to Pseudomonas aeruginosa infections. 184 The ability to transduce mammalian cells with antibody libraries, providing a direct link between antibody genotype and cell phenotype, can facilitate direct phenotypic selections and screening.173,175
Conclusions
In conclusion, antibodies and antibody fragments have added significantly to the portfolio of available therapeutics and are likely to continue to do so with the many advances being made. There is significant further potential for targeting complex multimembrane-spanning receptors and protein-protein interactions with selective antibody drugs, and there is a rich landscape of additional potential targets, including intracellular ones and those that lie behind the BBB. There are a number of challenges in driving some of this progress, but the unique and multifunctional characteristics of antibodies have significantly added to the potential for the treatment of disease.
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article outside of employment with Medimmune.