
Abstract
Diamond-Blackfan anemia (DBA) is a bone marrow failure syndrome caused by mutations in ribosomal protein genes. Pathogenic mechanisms are poorly understood but involve severely reduced proliferation of erythroid precursors. Because current DBA therapies are ineffective and associated with severe side effects, disease-specific therapies are urgently needed. We hypothesized that druggable molecular pathways underlying the defect can be revealed through phenotypic small-molecule screens. Accordingly, a screening assay was developed using c-kit+ fetal liver erythroid progenitors from a doxycycline-inducible DBA mouse model. The addition of doxycycline to the culture medium induces the phenotype and reduces proliferation to <10% of normal, such that rescue of proliferation can be used as a simple readout for screening. Here, we describe the assay rationale and efforts toward validation of a microtiter plate–compatible assay and its application in a pilot screen of 3871 annotated compounds. Ten hits demonstrated concentration-dependent activity, and we report a brief follow-up of one of these compounds. In conclusion, we established a robust scalable assay for screening molecules that rescue erythropoiesis in DBA.
Introduction
Diamond-Blackfan anemia (DBA) is a pure red blood cell aplasia, characterized by severely decreased numbers of erythropoietin (Epo)–responsive red blood cell precursors in the bone marrow.1,2 DBA generally presents with severe anemia during infancy, often accompanied by skeletal abnormalities. Most DBA cases are caused by inherited mutations in ribosomal protein genes, among which ribosomal protein S19 (RPS19) is the most frequent, accounting for ≈25% of all cases.3–5 Patients always have one intact gene copy, resulting in haploinsufficiency of the affected ribosomal protein.
DBA patients are resistant to Epo therapy, which can be explained by the lack of Epo-responsive erythroid progenitors in the bone marrow. Blood transfusion is the first line of treatment; however, despite improved chelation therapies, iron toxicity caused by chronic blood transfusion therapy remains a major cause of death. Many DBA patients can instead be treated with corticosteroids.2,6 Although 80% of patients initially respond to 2 mg/kg prednisone, only 40% can be maintained on tolerated doses (≤0.5 mg/kg prednisone/d). Even at lower doses, corticosteroids cause side effects such as altered metabolism, osteoporosis, cataracts, and psychological symptoms of mood swings and insomnia. 2 The therapeutic effect of corticosteroids in DBA patients is not related to their anti-inflammatory properties but results from an activation of stress signals that stimulate progenitor cells in the bone marrow to produce more red blood cells.7,8 Hematopoietic stem cell transplantation is the only potentially curative therapy but is associated with significant morbidity and mortality. Because current therapies are ineffective and/or associated with adverse effects, there is an urgent need to develop novel therapeutic strategies specifically targeting the underlying molecular mechanisms.
Efforts to explain how ribosomal protein deficiency selectively affects erythroid progenitor cells have resulted in several suggestions for disease-specific therapies. Forced expression of RPS19 rescues the anemia in RPS19-deficient patient cells, whereas reduced RPS19 expression in normal cells induces the disease phenotype,9,10 suggesting gene therapy is a potential cure.4,11,12 Haploinsufficient levels of RPS19 arrest ribosome biogenesis, disrupt ribosomal RNA maturation (rRNA), and cause upregulation of genes downstream of p53. 13 Ribosomal protein deficiency results in insufficient globin protein synthesis and toxic accumulation of excess heme, contributing to erythroid progenitor cell death. 14 Disease-specific small-molecule therapy would need to improve ribosomal protein production or target the pathogenic consequences of ribosomal protein deficiency, leading to erythroid progenitor failure.
To develop and evaluate therapies for DBA, we generated a doxycycline-inducible mouse model for RPS19-deficient DBA. Upon induction, there is a downregulation of Rps19 expression that results in a proliferative block in erythroid development. This block in erythroid development is alleviated upon removal of p53. 15 Activation of p53 in the DBA mouse model is caused by ribosome biogenesis stress, leading to inhibition of Mdm2, the main negative regulator of p53, by the 5S ribonucleoprotein particle (RNP). Disruption of the 5S RNP-Mdm2 interaction rescues anemia in the DBA mouse. 16 Because this inducible DBA mouse model recapitulates key features of the human disease, it is an excellent tool for identifying and characterizing chemical compounds, genes, and molecular pathways to be used for DBA treatment. We therefore set out to develop a robust screening assay using erythroid progenitors from this inducible DBA model. Here, we present the development of this assay, a protocol scalable to screening of large libraries, results from a pilot screen, and examples of validated hits.
Materials and Methods
Rps19-Deficient Mouse Model
We used a doxycycline-inducible Rps19-deficient mouse previously developed by us, carrying a reverse transactivator–dependent short hairpin RNA (shRNA) against Rps19 that develops robust anemia upon doxycycline induction. For all experiments, we used shRNA-D mice. 15 The hematopoietic phenotype in these mice is specific to Rps19 downregulation, as it can be cured by enforced expression of Rps19. 17 We refer to mice carrying one copy of the shRNA-D as D+ mice (milder DBA phenotype) and those carrying two copies as DD mice (severe anemia and bone marrow failure). The Lund University ethics committee for animals approved all experiments.
Mouse Transplantations and Harmine Administration
Eight- to 14-wk-old wild-type C57bl/6 mice were subjected to lethal irradiation (900 cGy) and transplanted with unfractionated bone marrow from Rps19-deficient DD or D+ mice. After 16 wk of transplantation, Rps19 deficiency was induced by feeding the mice with doxycycline-containing food pellets (200 mg/kg doxycycline; Bio-Serv, Flemington, NJ) or by adding doxycycline in the drinking water (2 mg/mL doxycycline; Sigma-Aldrich, St. Louis, MO) supplied with 10 mg/mL sucrose (Sigma-Aldrich). Harmine was dissolved in PEG:Tween (50:50) and administered intraperitoneally at a final concentration of 7 mg/kg every 48 h beginning 24 h after starting doxycycline. Peripheral blood was analyzed 15 d from the start of doxycycline induction.
Cell Culture
Mouse fetal liver cells
Fetal livers from heterozygote D/+ E14.5-15.5 embryos were dissected and enriched for c-kit (CD117)–positive cells using magnetic beads and LS columns (Miltenyi Biotech, Germany) according to the manufacturer’s instructions. The cells were cultured in serum-free expansion medium (SFEM; Stem Cell Technologies, Canada) with 2 U/mL Epo (LEO Pharma, Denmark), 100 ng/ mL murine stem cell factor (SCF; PeproTech, USA) with 100 nM dexamethasone (Sigma-Aldrich), with or without 0.5 µg/mL doxycycline (Sigma-Aldrich), and incubated at 37 °C.
DBA patient cells
Anonymized peripheral blood was collected from a DBA patient after obtaining informed consent. All procedures were followed according to the ethical guidelines of Lund University, Sweden, and the University of Freiburg, Germany. The sample was from a male patient carrying a mutation in Rps19. Mononuclear cells from normal and patient blood were enriched for the CD34+ fraction using magnetic separation columns (Miltenyi Biotech, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. Cells were expanded in a two-phase culture system differentiation. Cells were cultured in phase 1 medium (20 ng/mL TPO, 20 ng/mL FLT-3 ligand, 100 ng/mL SCF, 10 ng/mL interleukin-3 (IL3), and 1 nM dexamethasone in SFEM) for 7 d. The medium was then changed to phase 2 medium (30% serum, 50 ng/mL SCF, and 2 U/mL Epo in SFEM) for 3 d, after which the assay was set up. A total of 2500 cells were seeded per well of a 96-well plate (details below). Harmine was added at a concentration of 10 µM after 3 d of changing to phase 2 medium, and fresh additions of Harmine were made every other day thereafter. Five days after starting compound addition, cell viability was estimated using CellTiter-Glo reagent (Promega).
Cell Viability Assay
Murine fetal liver erythroid cells or patient-derived cells were cultured in white, clear-bottom, 96-well plates (BD, Ref. 353377). CellTiter-Glo luminescent cell viability reagent was used as described by Promega. Briefly, at the endpoint of the assay, 40 µL of the reagent was added to each well of the 96-well plate carrying cells. To mix contents and induce lysis, plates were placed on a shaker for 5 min. Subsequently, the plates were incubated at room temperature for 10 min to stabilize the luminescent signal. The signal was measured using a luminometer (Victor, 1 s per well). During optimization, different volumes, 40, 50, and 100 µL, of CellTiter-Glo reagent were tested.
High-Content Imaging
Fetal liver erythroid cells were plated on non–cell culture–coated 96-well plates. At the endpoint of the assay, DAPI was added to the wells. The Cellomics ArrayScan VTI HCS reader was used for automated imaging of the wells.
Kinase Selectivity Profiling
We used the DiscoverX KINOMEscan scanELECT platform for kinase profiling. Compounds were dissolved in DMSO, shipped to DiscoverX, and tested at 1 μM and 10 μM concentrations.
Real-Time qPCR
For real-time (RT) qPCR, 60,000 c-kit+ fetal liver cells were seeded in each well of a 96-well plate in the presence or absence of doxycycline. Harmine, dissolved in DMSO, was added at a concentration of 10 µM. Control wells received only DMSO. Twenty-four hours after adding Harmine, cells were frozen with 300 µL RLT buffer. RNA extraction was done using RNeasy micro kit (Qiagen, Venlo, the Netherlands) according to manufacturer’s instructions. One hundred microliter reactions were set up using SYBR Green PCR buffer, in which the following primers were used: actin,
F:5′-ATGGTGGGAATGGGTCAGAA-3′,
R:5′CCATGTCGTCCCAGTTGGTAA-3′ and Rps19,
F:5′-GCAGAGGCTCTAAGAGTGTGG-3′,
F:5′-GCAGAGGCTCTAAGAGTGTGG-3′,
R:5′-CCAGGTCTCTCTGTCCCTGA-3′.
When using the Taqman (Life Technologies, Carlsbad, CA), the following primers were used: p21 (00355782_m1) and Bax (00180269_m1).
Reactions were run and analyzed using 7900 HT fast RT-PCR (Applied Biosystems, Foster City, CA).
Statistical Analysis
Comparisons of data in the in vivo experiments were made by an unpaired Student’s t test (Graphpad Prism version 6). Statistical significance was defined as p < 0.05.
Results
Assay Development and Optimization
Choice of disease-relevant cells and phenotype: rescued proliferation of Rps19-deficient primary erythroid progenitor cells
The first step in assay development was to determine which cell type to use for screening. We realized it is not feasible to use primary DBA patient cells as their limited availability severely limits the number of compounds that can be evaluated. Immortalized cell lines 18 are also not suitable because the normal regulation of cell proliferation is disturbed. Hence, primary c-kit+ E14.5-15.5 fetal liver erythroid progenitor cells from our mouse model of DBA with doxycycline-inducible expression of rps19-shRNA were used. 15 Using these cells, a DBA-like erythroid phenotype can be induced by adding doxycycline to the culture medium, which reduces erythroid proliferation to <10% of normal. We considered rescue of this proliferation defect a simple and relevant readout for large-scale compound screening.
Assay design and development
Next, we evaluated different methods for determining effects on cell proliferation in a 96-well microtiter plate format. We tested the high-content imaging platform of Cellomics to analyze whether healthy cells could be quantitated. Because we used primary cells on noncoated plastic surfaces, the cells tended to roll and settle on the periphery of the wells. The cells were not uniformly distributed within a well, and selection of representative fields was not possible ( Suppl. Fig. S1 ). Moreover, staining and imaging were time-consuming. Hence, we looked for options with a simpler one-step protocol, short incubation times, and robust readouts. We tested two reagents, the PrestoBlue cell viability reagent and the CellTiter-Glo luminescent cell viability reagent, both of which are compatible with large-scale assays, and readouts can be made on a compatible microplate reader. Our cultures showed reduced proliferation and increased cell death upon doxycycline addition. Within 48 h of doxycycline addition, treated wells were left with very few cells (see below for multiple examples). The CellTiter-Glo reagent, which determines viable cells based on quantitation of present adenosine triphosphate, an indication of metabolically active cells, generated data with superior linear correlation. Next, we evaluated different volumes of the CellTiter-Glo reagent per well, with each well already containing 100 µL of culture medium. Forty microliters of CellTiter-Glo per well resulted in the best signal-to-background ratio in the luminescence readings ( Suppl. Fig. S2 ).
The luminescence readout of CellTiter-Glo from cultures of uninduced cells was set to represent an activity value of 100% rescue on an arbitrary rescue scale, and the readout from doxycycline-induced controls was set to represent 0%. This allowed reliable normalization between plates and gave toxic chemicals a negative value, meaning less cells were observed in these wells compared to wells with doxycycline induced control cells. Thus compounds rescuing proliferation received a positive value, with a rescue score of 100% indicating complete rescue.
To enable large-scale screening, the assay was further optimized for the following:
Cell numbers per well
Media composition
Culture duration
Doxycycline concentration
Timing of induction with doxycycline
Timing of addition of test chemicals
To objectively quantify how changes in different parameters improved variability in both induced and uninduced cells, we calculated the Z′ factor as described by Zhang et al. 19 The Z′ factor takes into consideration both the range and variability of data to calculate the suitability of an assay for high-throughput screening. It is represented as

where SD represents the standard deviation. The larger the value, the higher the data quality, and a Z′-factor greater than 0.5 is considered robust.
After assay development, we arrived at the conditions as provided in Table 1 , resulting in a Z′ factor of 0.7. Compounds were added to the plates 24 h after cell seeding and doxycycline addition, to allow time for induction and to avoid small-molecule interference with induction of the proliferation-perturbed phenotype. A schematic outline of the final assay logistics is shown in Figure 1A .
Conditions in the Primary Screening Assay.

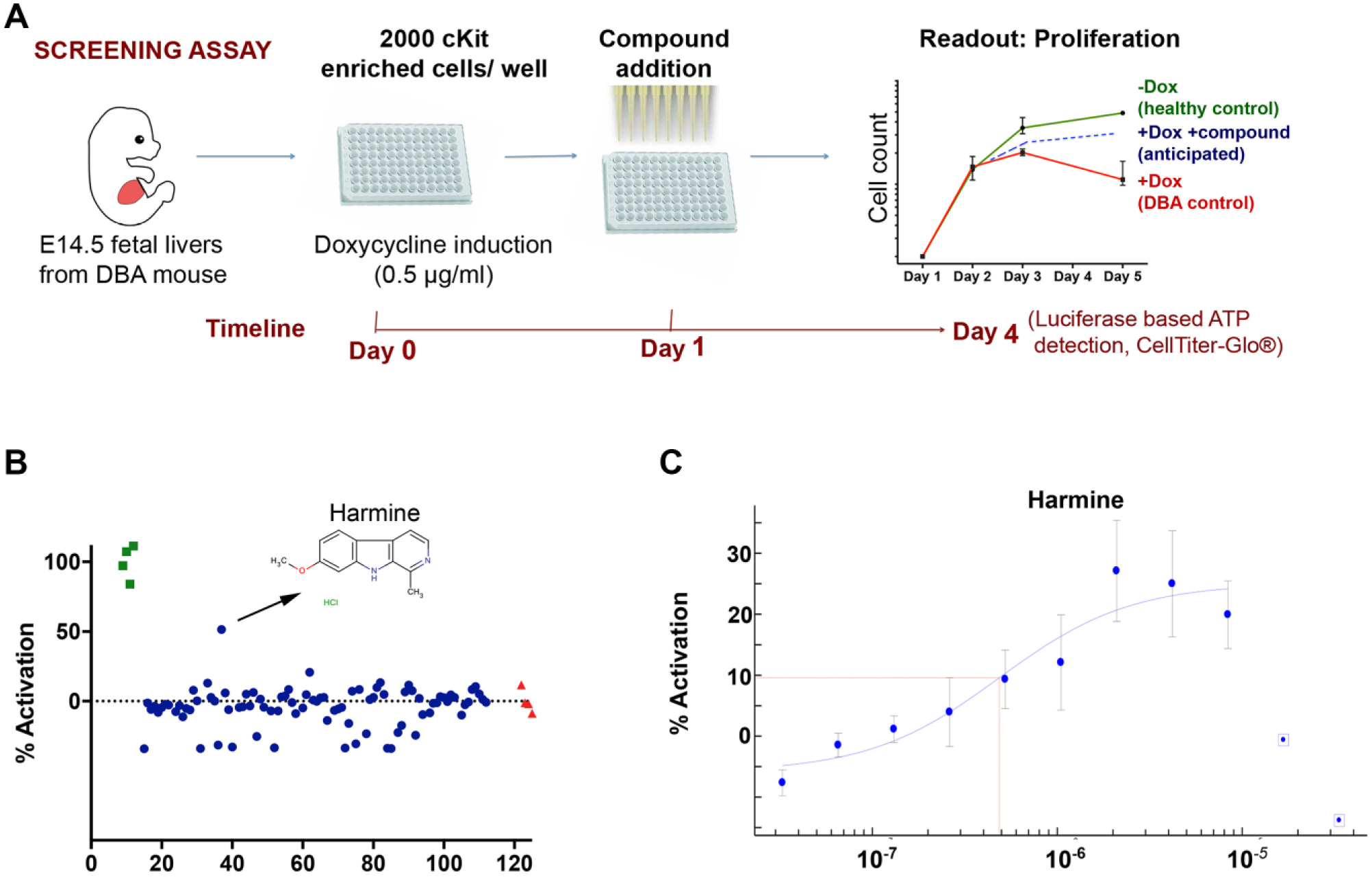
Primary screen identifies Harmine as a potential hit. (
IL3, a cytokine known to have a positive effect on the proliferation of erythroid progenitors, was used as the positive pharmacologic control throughout assay development and later screening efforts. We validated the assay response by testing and quantifying the impact of IL3, which consistently gave a positive rescue score of up to 25% in our assay ( Suppl. Fig. S3P ).
Small-molecule screening
Following validation of the screening assay, we chose to screen 12 different small-molecule libraries carrying a total of 3871 compounds (details in Suppl. Table SI ). The screening campaign was conducted over several different occasions and entailed isolation of primary cells for each run. The reproducibility in terms of response to doxycycline and the IL3 pharmacological control was excellent, despite the use of primary cells. All library compounds were dissolved in DMSO and were screened in parallel at two concentrations of 1 µM and 10 µM, respectively. This was done as we suspected that compounds with a beneficial impact on cell proliferation could also be associated with cell toxicity at higher concentrations because of the promiscuous and nonselective nature of small molecules.
While many compounds showed apparent toxicity (i.e., cell numbers below that of untreated controls as exemplified in Figure 1B ), we identified 15 hits that demonstrated the desired proproliferative effect ( Table 2 ). Ten of these 15 compounds were shown to give a concentration-dependent effect in follow-up experiments, where they increased proliferation of Rps19-deficient erythroid progenitors more than 4- to 8-fold ( Suppl. Fig. S3A–J ). Many of the top hits, including Harmine, D4476, and iodotubercidin, had activation scores of greater than 30% in our screening assay ( Fig. 1C ; Suppl. Fig. 3A, C, F ).
Hits Identified in the Primary Screen.

Hit Characterization and Screen Follow-Up
Investigation of compound structures as well as previously reported activities quickly revealed many of the confirmed hits as kinase inhibitors. Given the promiscuous nature of these, we wanted to understand which of these were responsible for the beneficial proproliferative effects in the progenitor cells. For this purpose, we chose the DiscoverX KINOMEscan scanELECT platform for testing the in vitro specificity of these compounds on the following 18 kinases, selected based on published activities and implications in disease-related pathways. These included CDK11, multiple casein kinase 1 variants (A1, A1L, D, E, G1, G2, and G3), DAPK1, three DYRK isoforms (1A, 1B, and 2), GCN2, HIPK1, p38-alpha, PIM1, RSK4, and TGFBR1. Of the kinases tested, both Harmine and iodotubercidin strongly inhibited DYRK1A whereas D4476 did not ( Table 3 ). We decided to follow-up on Harmine with in vivo experiments using the DBA mouse model and in vitro experiments using primary cells from a DBA patient.
Results From KINOMEscan ScanELECT. a
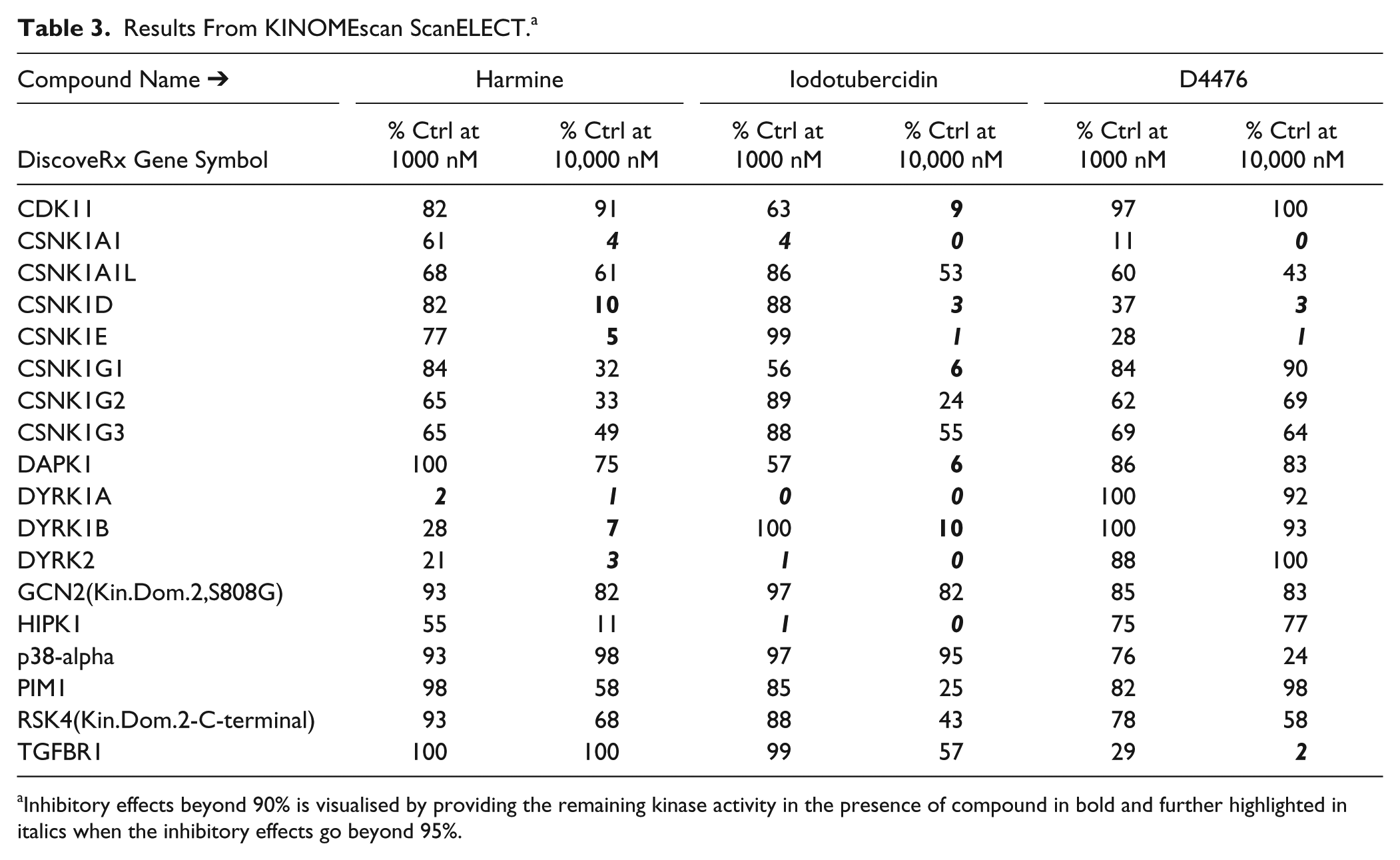
Inhibitory effects beyond 90% is visualised by providing the remaining kinase activity in the presence of compound in bold and further highlighted in italics when the inhibitory effects go beyond 95%.
Rps-19–deficient erythroid cells exhibit a dramatic upregulation of Trp53 downstream genes, including p21 and Bax. 15 When we tested whether Harmine had an effect on this upregulation, using RT-qPCR, we found that Harmine lowered the levels of p21 and Bax, whereas an Rps19-deficient state was maintained in the sample we tested ( Fig. 2A ).
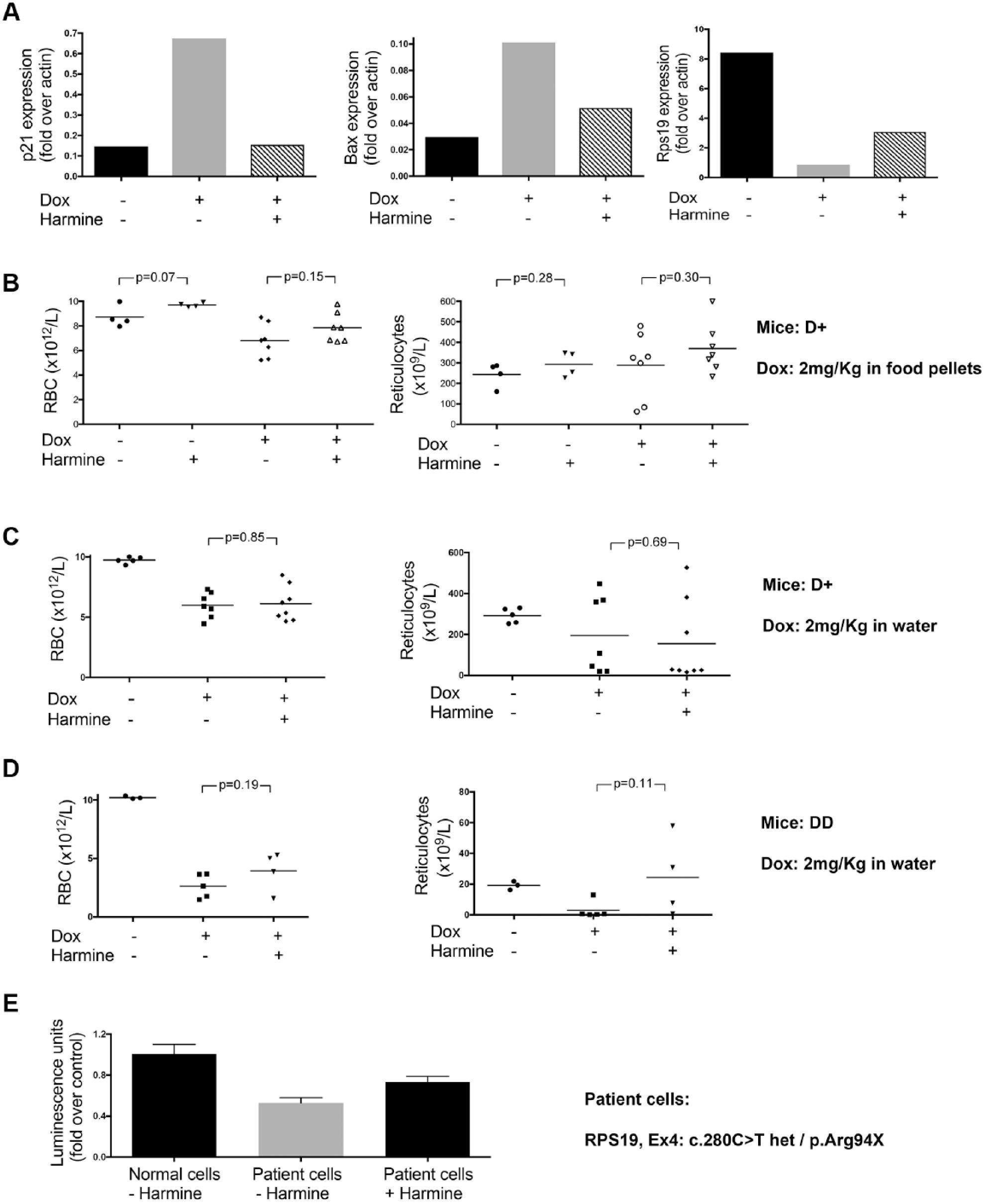
Harmine partially rescues the defect in Diamond-Blackfan anemia. (
When starting with in vivo animal experiments, we first ensured that Harmine did not have an effect on normal erythroid cells by administering Harmine to mice with normal levels of Rps19 (
CD34-positive cells differentiate in vitro to erythroid progenitors when exposed to Epo. Similar cells from DBA patients show an impaired differentiation.
21
We tested Harmine in vitro on erythroid cells obtained from an RPS19-deficient DBA patient. The impairment in proliferation in that sample was partially corrected by the addition of Harmine (
Discussion
Although academic hypothesis-driven research on DBA has been performed for more than 50 y, no disease-specific therapy is available, and our understanding of the underlying pathogenesis remains incomplete. The increasing availability of small-molecule libraries and screening capabilities for academic researchers today provides a new hope to overcome this challenge. Here, we describe a robust scalable method for phenotypic screening of any small-molecule library for discovery of new chemical probes and potential molecular targets for DBA therapy. The combination of a well-defined and disease-relevant cell type and phenotype is the single most important consideration in designing a screen. 22 A medically relevant disease phenotype in DBA is the failure of erythroid progenitor proliferation. This defect is therefore a suitable basis for a phenotypic screen aimed at identifying tool compounds and starting points for DBA studies and therapy.
Limited availability and disease heterogeneity prevent the use of primary DBA patient cells for large screens. Considering the relevant alternatives, we first considered that the phenotype is relatively restricted to erythroid progenitor cells (i.e., it was necessary to use equivalent cells for the screen). We further considered that the p53 inactivation is known to rescue the antiproliferative phenotype. Thus, the use of immortalized cell lines may miss modulators of pathways that have been disturbed to allow immortalization. With these considerations, primary erythroid progenitors from a DBA mouse model were the next best choice of cell type for the screen.
Because DBA causes depletion of erythroid progenitors, it was important to use an inducible mouse model in which unperturbed progenitors can be isolated before induction of the DBA phenotype in the assay. 15 This was necessary to support the required sourcing of cells for the herein described screening campaigns. The most easily obtained source of pure erythroid progenitors is the mouse fetal liver. 23 Each timed pregnancy generates 5 to 10 fetuses, each containing >1 million of these progenitors. We used 2000 cells per well, and testing 100,000 different conditions in a screen, for example, will require 40 pregnant females. Although the assay can likely be downscaled to 384 or even 1536-well format, it was outside the scope of this work. Our assay has proven to be very robust, and more than 50% of hits were later validated in vitro. Although the pilot screen presented in this article was performed in house, we have transferred the assay to a second screening location at Karolinska Institutet for screens of larger libraries. For these experiments, we transport freshly isolated c-kit+ fetal liver cells that are plated at the remote screening location with maintained assay robustness.
Confirmed hits from screening described in this study include palmitoylethanolamide, known to target peroxisome proliferator-activated receptor alpha (PPARα) and cannabinoid-like G-coupled receptors GPR55 and GPR119; anandamides that are an endogenous ligand of cannabinoid receptors; the ROCK inhibitor Fasudil; casein kinase inhibitor D4476; and Harmine, an inhibitor of monoamine oxidase A (MAOA) and dual-specificity tyrosine phosphorylation-regulated kinases (DYRKs). Because the underlying molecular mechanisms preventing progenitor proliferation in ribosomal protein-deficient erythroid progenitors is incompletely understood, the targets of hits identified in this pilot screen including PPARα, casein kinases, and DYRKs may help advance this research.
When considering which of the confirmed hits to prioritize for further characterization, we looked at the observed efficacy of the molecules and also at factors such as availability, known mechanism of action, and proven in vivo potency. For these reasons, we first focused on Harmine, which has a well-characterized target profile and has been used in several in vivo studies. Harmine is a tricyclic β-carboline alkaloid that has two major pharmacological properties, as an MAOA inhibitor and as an inhibitor of DYRKs.
24
Various plants containing Harmine have long been used in traditional folk medicine around the world,
25
and Harmine is, for example, the main ingredient in a sacred hallucinogenic drink called “ayahuasca.” Because another validated hit, iodotubercidin, also targets DYRKs, we hypothesized that DYRKS are the main therapeutic targets. Inhibition of MAOA instead accounts for the tremors observed after Harmine administration, which deterred us from further in vivo studies. Interestingly, Harmine has been shown to induce pancreatic β-cell proliferation and increase islet mass in vivo
26
and neural progenitors
27
through inhibition of DYRK1A. It is possible that the same mechanisms that promote proliferation of these otherwise poorly proliferating cell types also contribute to the effect seen in DBA. Inhibition of DYRK1A by Harmine has been shown to increase S-phase entry,
28
where increased proliferation could involve reduced Ser-15 (mouse Ser-18) phosphorylation of p53 and the observed subsequent reduced induction of p21 expression (
Harmine is also known to target DYRK3, a kinase with expression restricted to erythroid progenitor cells and testes. In response to hemolytic anemia, DYRK3 KO mice increase reticulocyte production significantly faster than wild-type mice, suggesting DYRK3 is a negative regulator of stress erythropoiesis that potentially can be targeted for treating Epo-resistant anemia such as DBA. 30 A phase I clinical trial evaluating a DYRK3 inhibitor for anemia has been completed, but results are not known (ClinicalTrials.gov identifier: NCT00443170). Several other groups are currently performing small-molecule screen in cell lines, zebra fish, and induced pluripotent stem cells 31 to identify lead compounds for DBA therapy. Cross-evaluation of Harmine and other hits from these parallel initiatives in different models can help accelerate our understanding of potential mechanisms worthy of full-scale drug discovery efforts.
In summary, we present a phenotypic screening setup that allows screening of large chemical libraries and efficient downstream in vitro and in vivo validation. From a pilot screen of 3871 annotated molecules, more than 50% of hits could be validated in vitro. We further evaluated a DYRK inhibitor in the DBA mouse model and on patient erythroid cells cultured in vitro. Identified hits may provide insight into DBA pathology and can be used as tool compounds for DBA research. Larger screens are ongoing to identify promising lead compounds and drug targets for disease-specific DBA therapy.
Supplemental Material
Supplemental_Material_for_PhenotypicscreeningassayidentifiesmodulatorsofDBA_by_Siva_et_al – Supplemental material for A Phenotypic Screening Assay Identifies Modulators of Diamond Blackfan Anemia
Supplemental material, Supplemental_Material_for_PhenotypicscreeningassayidentifiesmodulatorsofDBA_by_Siva_et_al for A Phenotypic Screening Assay Identifies Modulators of Diamond Blackfan Anemia by Kavitha Siva, Fredrik Ek, Jun Chen, Abdul Ghani Alattar, Kristmundur Sigmundsson, Roger Olsson, Marcin Wlodarski, Thomas Lundbäck and Johan Flygare in SLAS Discovery
Footnotes
Acknowledgements
We thank Anna Hammarberg for help with Cellomics imaging. This work was supported by grants to J.F. from the Ragnar Söderberg Foundation, DBA Foundation, Captain Courageous Foundation, DBA Canada, Swedish Research Council, Swedish Foundation for Strategic Research, Åke Wiberg’s Foundation, and a Marie Curie Integration Grant. Chemical Biology Consortium Sweden was funded by the Swedish Research Council and Karolinska Institutet.
Supplemental material is available online with this article.
Authors’ Note
Marcin Wlodarski is currently affiliated with the Department of Hematology, St. Jude Children’s Research Hospital, Memphis, TN USA.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.