
Abstract
Aberrant expression and/or activation of the MET receptor tyrosine kinase is characterized by genomic recombination, gene amplification, activating mutation, alternative exon-splicing, increased transcription, and their different combinations. These dysregulations serve as oncogenic determinants contributing to cancerous initiation, progression, malignancy, and stemness. Moreover, integration of the MET pathway into the cellular signaling network as an addiction mechanism for survival has made this receptor an attractive pharmaceutical target for oncological intervention. For the last 20 years, MET-targeting small-molecule kinase inhibitors (SMKIs), conventional therapeutic monoclonal antibodies (TMABs), and antibody-based biotherapeutics such as bispecific antibodies, antibody–drug conjugates (ADC), and dual-targeting ADCs have been under intensive investigation. Outcomes from preclinical studies and clinical trials are mixed with certain successes but also various setbacks. Due to the complex nature of MET dysregulation with multiple facets and underlying mechanisms, mechanism-based validation of MET-targeting therapeutics is crucial for the selection and validation of lead candidates for clinical trials. In this review, we discuss the importance of various types of mechanism-based pharmaceutical models in evaluation of different types of MET-targeting therapeutics. The advantages and disadvantages of these mechanism-based strategies for SMKIs, conventional TMABs, and antibody-based biotherapeutics are analyzed. The demand for establishing new strategies suitable for validating novel biotherapeutics is also discussed. The information summarized should provide a pharmaceutical guideline for selection and validation of MET-targeting therapeutics for clinical application in the future.
Keywords
Introduction
MET, a name abbreviated from the carcinogen N-Methyl N nitroso guanidine from previous studies that eventually led to the discovery of truncated MET fused with sequences from the translocate promoter region (TPR-MET), 1 belongs to a unique subfamily of receptor tyrosine kinases (RTKs) with distinct structural features and biological activities (Figure 1a). 2 The MET gene is located in chromosome 7 (7q31.2) with 21 exons encoding a 180 kDa protein. 3 The MET extracellular sequence contains several important domains, including a semaphorin (SEMA) domain followed by a plexin-semaphorin-integrin (PSI) domain, and four immunoglobulin-plexin-transcription (IPT) motifs (Figure 1b).1–3 The SEMA domain harbors a ligand-binding pocket responsible for interacting with hepatocyte growth factor (HGF) (Figure 1c) and is critical for receptor dimerization and subsequent phosphorylation.1–3 The PSI domain acts as a wedge between the SEMA domain and IPT motifs and facilitates the formation of a MET homodimer with interface formed by the SEMA domain from both the α-chain and β-chain.1–3 The MET intracellular sequence consists of a juxtamembrane (JM) domain, a tyrosine kinase (TK) domain, and a C-terminal multifunctional docking site.1–4 The JM domain contains several important amino acid residues including Y1003, which interacts with casitas B-lineage lymphoma (Cbl) and leads to ubiquitin-dependent MET degradation. 5 This process is a mechanism of a negative feedback loop, which controls the MET activation status.1,3,5 The TK domain, upon phosphorylation of Y1234 and Y1235, undergoes a conformational change resulting in increased TK activity,3,4 which leads to phosphorylation of two tyrosine residues, Y1339 and Y1356, in the docking site (Figure 1b).3,4 The docking site is responsible for recruiting adaptor molecules and transduction of different signals to activate multiple downstream signaling pathways (Figure 2).3,4

Schematic representation of structures of the MET gene, MET, and its ligand hepatocyte growth factor (HGF). (a) The MET gene is located in the 7p31 locus of chromosome 7. It contains 21 exons separated by 21 introns. The classical promoter contains two transcription factors including specificity protein 1 (SP1) and activating protein-2 (AP2)-binding elements and is responsible for the transcription of full-length MET with 1408 amino acids. (b) MET is first synthesized as a biologically inactive single-chain precursor (pro-MET). Proteolytic conversion is required to activate MET. Mature MET is composed of a 45 KDa α-chain and a 145 kDa β-chain linked by a disulfide bond. Structurally, the MET α-chain is an extracellular component containing a portion of the semaphorin (SEMA) domain. The extracellular sequence of the MET β-chain contains a large portion of the SEMA domain, followed by a plexin-semaphorin-integrin (PSI) domain, and 4 immunoglobulin-like plexin and transcription (IPT) motifs. The intracellular sequence harbors a short transmembrane (TM) segment followed by a juxtamembrane domain (JM), a tyrosine kinase (TK) domain, and a C-terminal tail. Regulatory tyrosine residues, Y1003 in the JM domain and Tyr1234 and Tyr1235 in the TK domain are indicated. Also, Tyr1349 and Tyr1356 in the MET C-terminal tail, which form the functional docking site, respectively, are marked. (c) HGF is first synthesized as a biologically inactive single-chain precursor known as pro-HGF. Proteolytic cleavage results in a biologically active two-chain form of mature HGF. The HGF α-chain contains a hairpin loop (HPL) followed by four kringle domains (K1 to K4). The HGF β-chain contains a serine protease-like domain with substation of amino acids in the active site. The high-affinity MET-binding site is in the HGF α-chain and the low-affinity MET-binding site is in the HGF β-chain.
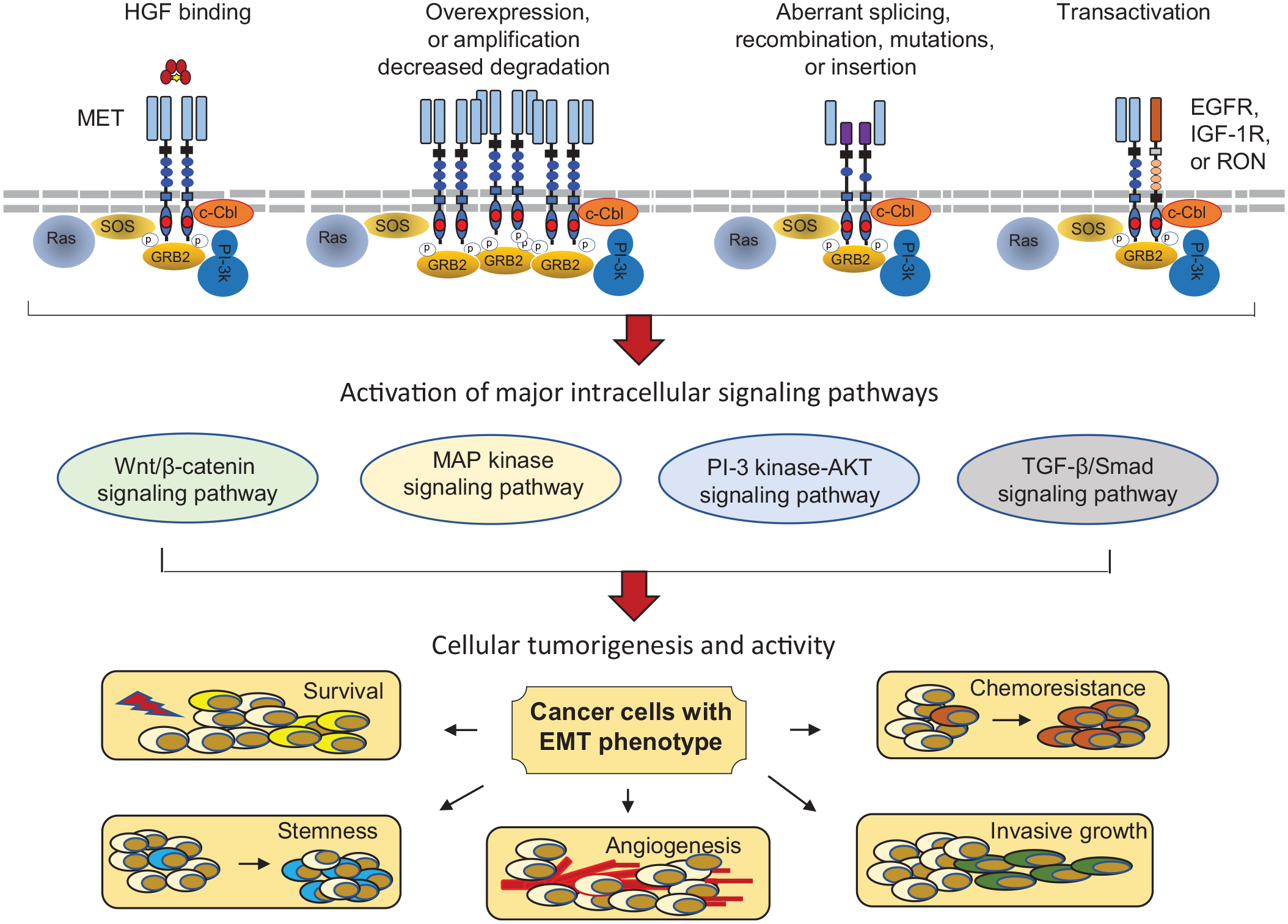
Dysregulated MET activation, signaling pathway, and tumorigenic consequence. Activation of MET in cancer cells, in general, is mediated through multiple mechanisms including ligand binding, activating mutation, receptor overexpression, aberrant splicing/alternative initiation, and transactivation through other receptor tyrosine kinases such as EGFR, IGF-1R, and RON. HGF-induced MET activation, a classical model, is functional through phosphorylation of several critical tyrosine residues and creates the C-terminal functional docking site, which recruits cytoplasmic molecules such as SOS and GRB2. The negative modulator c-CBL, a ubiquitin ligase, also binds the docking site and mediates MET endocytosis and degradation. Multiple signaling cascades, such as RAS/MAP kinase, PI3K/AKT, Wnt/β-catenin, and TGF-β/SMAD pathways, are activated upon MET phosphorylation in cancer cells, which creates a complex intracellular signaling network. The biological consequence is to induce cell proliferation with a malignant phenotype known as EMT, which leads to increased cellular survival, invasiveness, chemoresistance, and tumorigenic stemness.
Cancerous MET expression and activation are featured by genetic recombination, gene amplification, point mutation, alternative exon-splicing, increased transcription, increased protein accumulation, and their different combinations (Figure 3).6–13 The outcomes from these changes imply a complex picture of MET dysregulation, which provides the opportunity to target MET for cancer therapy. 14 Currently, therapeutics such as small-molecule kinase inhibitors (SMKIs) (Table 1),15–27 conventional therapeutic monoclonal antibodies (cTMABs),28–33 and antibody-based biotherapeutics targeting MET (Table 2) have been validated in preclinical studies and many of them have advanced into clinical trials.34–46 Significantly, four SMKIs, crizotinib, cabozantinib, tepotinib, and capmatinib, have been approved for clinical application (Table 1) (www.FDA.gov). Nevertheless, MET-targeting cTMABs, although some of them under clinical trials for almost 10 years, have made little progress. Up to now, none of the cTMABs or antibody-based biotherapeutics have been approved by the FDA. In addition, recent progress in MET-targeted therapy has led to the preclinical development of MET-specific chimeric antigen receptor (CAR) T cells and natural killer cells for the treatment of cancers overexpressing MET.47–49 Moreover, dual-functioning CAR T cells targeting both MET and programmed death-1 (PD-1) has also been described as a strategy for therapy of solid tumors. 50
Pharmaceutical features of MET-targeting small-molecule kinase inhibitors approved or currently in clinical trials.

Type I MET SMKIs target the ATP-binding pocket of the active form of the kinase domain in MET. Type II MET SMKIs interact with the ATP-biding pocket in the inactive form of the kinase domain in MET.
ABL1, ABL proto-oncogene 1, non-receptor tyrosine kinase; ALK, anaplastic lymphoma kinase; ATP, adenosine triphosphate; AXL, coming from the Greek word “anexelekto,” means uncontrolled; CDK, cyclin-dependent kinase; CRC, colorectal cancer; EGFR, epidermal growth factor receptor; FLT, FMS-like tyrosine kinase or fetal liver kinase; HCC, hepatocellular carcinoma; KIT, v-kit Hardy–Zuckerman 4 feline sarcoma viral oncogene homolog; MET, a name from carcinogen N-Methyl N nitroso guanidine; NSCLC, non-small cell lung cancer; p-MET, phosphorylated MET; RCC, renal cell carcinoma; RET, rearranged during transfection; RON, recepteur d’origine nantais; ROS1, v-ros UR2 sarcoma virus oncogene homolog 1 (avian); TEK, TIER2; TIER2, tyrosine kinase with immunoglobulin-like and EGF-like domains 2; TRKB, tropomyosin related kinase B; VEGFR-2, vascular endothelial growth factor receptor 2.
Biochemical and biological features of antibody-based biotherapeutics targeting MET and/or other signaling molecules in various stages of development * .
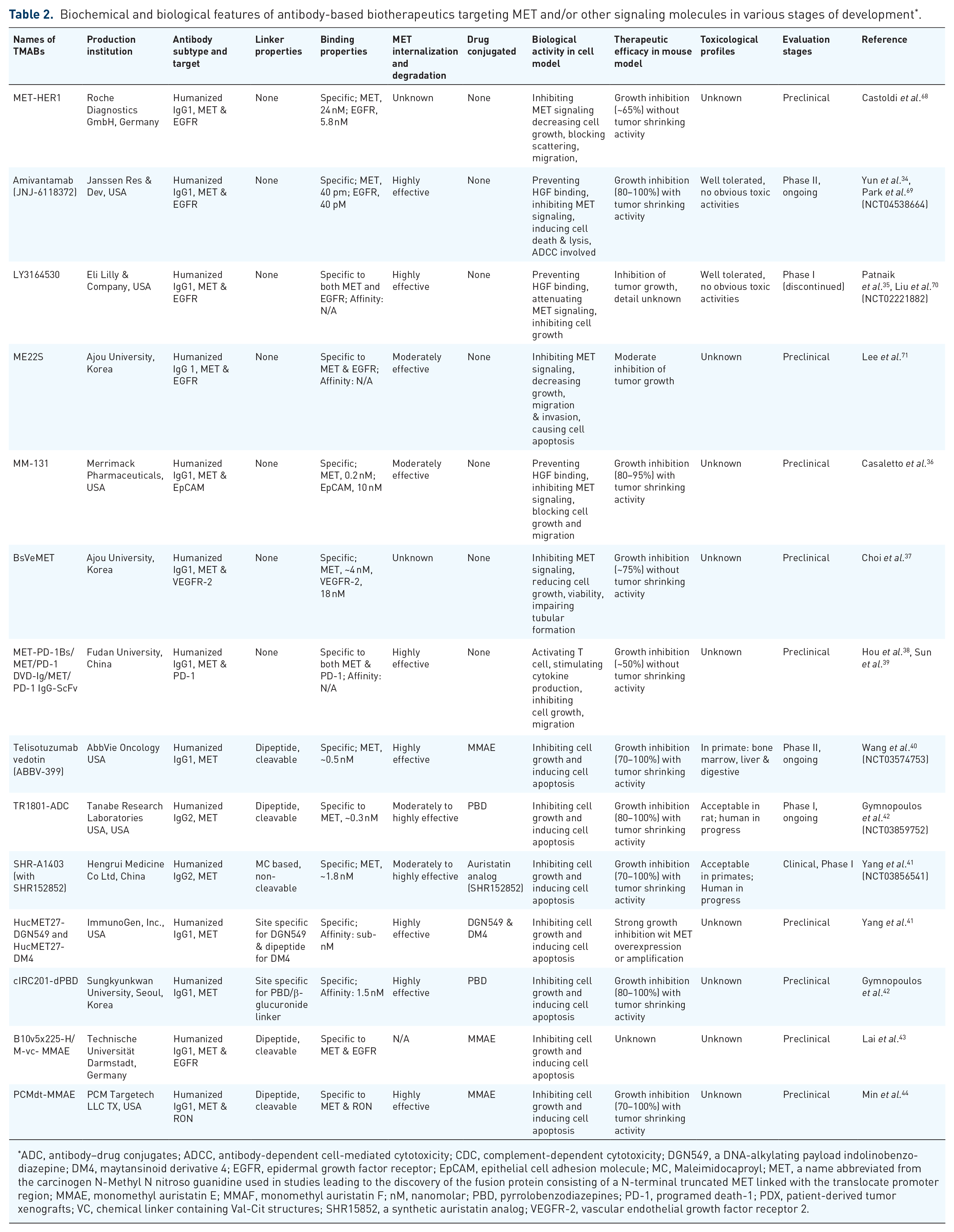
ADC, antibody–drug conjugates; ADCC, antibody-dependent cell-mediated cytotoxicity; CDC, complement-dependent cytotoxicity; DGN549, a DNA-alkylating payload indolinobenzo-diazepine; DM4, maytansinoid derivative 4; EGFR, epidermal growth factor receptor; EpCAM, epithelial cell adhesion molecule; MC, Maleimidocaproyl; MET, a name abbreviated from the carcinogen N-Methyl N nitroso guanidine used in studies leading to the discovery of the fusion protein consisting of a N-terminal truncated MET linked with the translocate promoter region; MMAE, monomethyl auristatin E; MMAF, monomethyl auristatin F; nM, nanomolar; PBD, pyrrolobenzodiazepines; PD-1, programed death-1; PDX, patient-derived tumor xenografts; VC, chemical linker containing Val-Cit structures; SHR15852, a synthetic auristatin analog; VEGFR-2, vascular endothelial growth factor receptor 2.

MET dysregulations observed in cancer cells from different tissues and therapeutics suitable for the targeted therapy. Different types of cancerous MET dysregulation are depicted in red oval circles. Various forms of therapeutics specific to MET that are suitable for targeting MET-expressing cancer cells are indicated in yellow boxes.
The communication presented here focuses on pathogenic mechanism-based validation of MET-targeting therapeutics for clinical trials. Based on complex mechanisms of MET dysregulation in different types of cancer, our objective is to summarize the latest development of strategies in pharmaceutical validation of MET-targeting therapeutics. Due to the page limitations, MET-targeting CAR T-cell therapy will not be discussed in this communication. As the first step in pharmaceutical development, mechanism-based validation serves as a key principle in selecting lead candidates for potential clinical trials. Considering the biological role of MET in tumorigenesis and its complex nature of dysregulation with various underlying mechanisms, the importance of a validation strategy used in the pharmaceutical development process should not be underestimated.
MET dysregulation and underlying mechanism
Aberrant MET expression and activation during tumorigenic progression have multiple facets with different underlying mechanisms (Figures 2 and 3).6–14 At present, the identified forms of MET dysregulation include DNA recombination/rearrangement,1,6,55,72,73 gene amplification,7,74–76 point mutation,8,76–79 alternative exon skipping,9,80–82 somatic insertion or deletion,80–82 increased transcription,10,83–85 impaired protein degradation,11,80–82 and abnormal protein accumulation.7,10,74–76,83–85 Several features of these abnormalities are worth mentioning. First, the form of MET dysregulation is different in different types of cancer.6–14 Second, the majority of MET abnormalities directly lead to MET activation but not overexpression.73–81 Third, mechanisms of MET signaling in regulating cellular tumorigenic phenotypes are complex.73–81 Fourth, MET signaling is integrated or addicted at variable levels by cancer cells for growth and survival.86,87 These pathogenic characteristics, acting either alone or in their different combinations, not only change the pattern of MET expression but also cause HGF-independent MET activation, leading to malignant progression.
MET fusion occurs when the MET extracellular sequence at a particular region is fused with different partner sequences under DNA recombination/rearrangement, resulting in various forms of MET fusion proteins such as CLIP2-MET, ST7-MET, CD47-MET, GPRC5C-MET, and others.1,6,55,72,73,86,87 TPR-MET was the first fusion protein identified under the action of certain carcinogens. 1 Mechanistically, MET fusion occurs through either intrachromosomal or interchromosomal rearrangements.1,6,47–50,72,86–89 Because the fusion partner sequences often contain a coiled-coil domain that facilitates protein dimerization, almost all of MET fusions exhibit ligand-independent MET activation.8,46–50,55,72,73,86–89 The frequency of MET fusions in cancer such as those from lung, gastric, hepatic, kidney, and pancreatic tissues is relatively low, ranging from 0.1 to 2%.86–89 The only exception is glioma, in which MET fusion has been found in ~12% cases.72,89 Gene amplification, occurring as polysomy and focal events, 86 exists with variable frequency in different types of cancer such as stomach and lung cancer.74–76 Tumors with identified frequencies of MET gene amplification include non-small cell lung cancers (NSCLCs, <1–5%), gastric cancers (<1–10%), colorectal cancer (CRC, 2–4%), and papillary renal cell carcinomas (3–135). 86 The outcome is often characterized by accumulation of a large amount of activated MET proteins.74–76 Somatic alterations in the MET gene serve as another pathological feature observed in several types of cancers, particularly in hereditary and sporadic papillary renal cell carcinomas.8,77–79 They are often manifested by insertion, deletion, and missense mutations with different frequencies in different domains of MET,8,77–79,90–92 which profoundly affect the structural and functional integrity of the SEMA, JM, and TK domains (Figures 2 and 3).10,51,78–80,90–92 The observed frequencies are ~15% in papillary renal cell carcinomas, ~7% in hepatocellular carcinomas (HCCs), and up to 14% in patients with head and neck cancers. 86 Alternative mRNA splicing, particularly exon-14 skipping, is currently a hot topic due to its clinical significance associated with oncogenesis.9,80–82 Exon-14 encodes the JM domain of MET, 3 which regulates the MET metabolic degradation through the Cbl-directed ubiquitin pathway.9,80–82 Alternative exon-14 skipping is caused by insertion/deletion in the acceptor or donor regions or by missense mutations in certain tyrosine residues including Y1003 (Figure 4). This results in the inability of the JM domain to interact with Cbl E3 ligase, 8,11,81–83 and ultimately leads to the accumulation of a large amount of MET protein with increased stability and elevated kinase activity.7,10,80–82 The frequency of MET exon-14 skipping occurs 3–4% of patients with NSCLCs. The alteration is further enriched in patients with sarcomatoid carcinomas (9–22%), an aggressive subtype of NSCLC. 86 The mechanism that causes cancerous MET overexpression is complex. Transcriptional upregulation appears to be the major cause.10,83–85 For instance, hypoxia-initiating factor (HIF)-1α is one of the triggering factors responsible for increased MET transcription. 83 Activation of signaling proteins such as reticular activating system (RAS) also upregulates MET expression through the transcriptional event. 85 Moreover, both gene amplification and exon-14 skipping are involved in abnormal accumulation of large amounts of MET protein.74–76,80–82 The documented MET overexpression in primary tumor samples determined by immunohistochemical staining include prostate cancer (~55%), gastric cancer (~65%), HCC (~50%), CRC (~55%), triple-negative breast cancer (TNBC, ~15%), and NSCLC (~50%). 86 Thus, various mechanisms are involved in cancerous MET overexpression.

Activating mutations in the different functional domains of MET. (a) Various mutations in the tyrosine kinase domain of MET. Point mutations in more than 16 amino acid residues in the kinase domain have been documented in different types of primary cancer samples. These mutations often result in a conformational change that facilitates the kinase domain to convert into an active mode with increased kinase activity. (b) Missense mutations in the exon 14 ubiquitination site. The JM domain is encoded by MET exon-4. The tyrosine residue Tyr1003 in the JM domain is responsible for the interaction with the ubiquitin E3 ligase, which promotes MET degradation, a negative feedback mechanism for controlling levels of MET activation. The mutation results in the inability of Tyr1003 to interact with ubiquitin E3 ligase, leading to an increase in stability of MET. (c) Alterations in the exon-14 splice site often results in exon-14 skipping, leading to formation of a MET slicing variant known as MET exon-14 skipping. The consequence is that this MET variant is resistant to ubiquitin-mediated protein degradation with increased stability and kinase activity. (d) Various mutations are documented in the SEMA domain of MET. Since the SEMA domain contains the MET-binding pocket; it is speculated that these mutations will affect the ability of HGF binding to MET with reduced affinity. However, pathological implication of these mutations in association with clinical oncological events currently are largely unknown.
MET therapeutics with different mechanisms of action
Small-molecule kinase inhibitors
SMKIs discussed here are structurally designed and chemically synthesized small molecules that are specific to a unique kinase domain of MET and other proteins with similar kinase structure. The use of SMKIs has several pharmaceutical advantages and has been clinically proven to be effective. The principle of using SMKIs for cancer therapy is based on cellular oncogenic signaling addiction/dependence.15–27 Currently, chemical design and large-scale synthesis of SMKIs are not a technical challenge due to the use of computer-aided structural analysis and synthetic chemistry platforms. The use of these advanced technologies, in general, ensure to generate MET-specific SMKIs with variable targeting specificity. Besides four SMKIs specific to MET, including crizotinib, cabozantinib, tepotinib, and capmatinib, that have already been approved by the FDA (Table 1), additional SMKIs such as AMG-337, bozitinib (APL-101), glesatinib (MGCD265), Golvatinib (E7050), merestinib (LY2801653), savolitinib, Sar125844, and others appear to be promising in clinical trials (Table 1).15–27 Mechanistically, SMKIs are the choice for inhibiting both cell-surface and intracellular MET protein that displays both an inactive and active status in the TK domain. An inhibitory effect is achieved by SMKIs binding to the critical region in the TK domain, either competing with adenosine triphosphate (ATP) for binding to the ATP-binding pockets in the TK domain or by preventing the conversion of the TK domain from an inactive conformation into an active mode.15–27 Moreover, the therapeutic activity of SMKIs is independent of HGF-mediated MET activation regardless of the presence or absence of HGF in the tumor microenvironment or via a cancer cell autocrine-producing fashion. The major disadvantage of SMKIs is that their anticancer action is heavily dependent on the strength of MET signaling integrated into the cellular signaling network and the addictive levels acquired by cancer cells for growth and survival.15–27 In the preclinical studies, mechanism-based validation appears to be able to objectively determine the effectiveness of individual MET-targeting SMKIs. Nevertheless, in clinical trials and practice, the status of MET signaling addiction by cancer is difficult to assess. Although immunohistochemical (IHC) staining, fluorescence in situ hybridization (FISH), and next-generation sequencing (NGS) have been used as biomarkers for patient selection,13,86,87 these methods are unable to determine the addictive status of cancer cells to MET signaling.
Therapeutic monoclonal antibodies
Therapeutic monoclonal antibodies (mAbs) described here are defined as natural or recombinant mAbs specific to MET (cTMABs) or to both MET and other signaling proteins (bispecific mAbs) without drug, cytotoxin, or radioisotope conjugation. Both cTMABs and bispecific antibodies have been evaluated as MET-targeting biotherapeutics. Representative cTMABs are ARGX-111, emibetuzumab, onartuzumab, SAIT301, telisotuzumab, and Sym015, which have been in different phases of clinical trials.28–33 Anti-HGF TMABs ficlatuzumab and rilotumumab are also under clinical trials.93,94 However, none of the therapeutic mAbs specific to MET or HGF have currently been approved by the FDA.
The objective of using cTMABs is to suppress HGF-dependent and -independent MET activation, resulting in inhibition of cell proliferation, induction of cellular apoptosis, and regulation of host immune activity.28–33,93–95 In this sense, the induction of these activities is a biological criterium for the selection of MET-targeting cTMABs for clinical application. However, the mechanisms of action by these cTMABs rely on the levels of cellular addiction to MET signaling. Preclinical studies have demonstrated that anti-MET cTMABs have therapeutic activities against different types of cancer. Nevertheless, the observed efficacies vary significantly among individual TMABs tested.28–33,93–95 Moreover, outcomes from clinical studies at different phases are disappointing.93–100 Currently, conventional anti-MET TMABs, although under clinical trials for almost 10 years, have not been approved for clinical application, mainly due to the lack of therapeutic efficacy but not pharmacokinetic or toxicological issues.93–100
Five MET-based bispecific antibodies targeting partner proteins, including EGFR, VEGFR-2, epithelial cell adhesion molecule (EpCAM), and programmed cell death (PD)-1, have been preclinically evaluated (Table 2).34–39 The rationale to select these partner targets is either to achieve a coordinated inhibition of two signaling pathways or to regulate the immune response by targeting immunocheckpoint molecules to enhance anticancer activity.34–39 Inhibition of two signaling pathways has clinical relevance for treatment of tumors that develop resistance to chemotherapeutics or kinase inhibitors. Similarly, restoration of T-cell activity by targeting PD-1 is an approach in the format of a bispecific antibody.38,39,101,102 Currently, only two bispecific antibodies, amivantamab and LY3164530 (both targeting MET and EGFR), have entered into clinical trials (Table 2).34,35,80,95 Amivantamab is effective in NSCLC patients with EGFR exon-20 insertional mutation, which has led the FDA to grant it the Breakthrough Therapy Designation status (www.FDA.gov). Interestingly, the role of amivantamab in targeting MET is not mentioned in this group of NSCLC patients. LY3164530 has been terminated in clinical trials due to toxicity. 35
Single and dual-targeting antibody–drug conjugates
Antibody–drug conjugates (ADCs) are a class of targeted biotherapeutics consisting of a target-specific mAb, a versatile chemical linker, and a highly potent cytotoxic payload.103,104 The combination of antibody-based antigen specificity with payload cytotoxic potency results in an increased therapeutic index, favorable pharmacokinetic profile, and acceptable toxicological activity.40–46 Up to now, the FDA has approved nine ADCs, including gemtuzumab ozogamicin, brentuximab vedotin, trastuzumab deruxtecan, sacituzumab govitecan and others, for oncological application (www.FDA.gov). These ADCs target HER2, CD22, CD30, Trop-2, and others for treatment of various types of cancer. Currently, all MET-targeting ADCs are still under clinical trials without any approval by the FDA. The major mechanisms of action by ADCs are mediated by antibody-directed delivery of a cytotoxic payload for cancer cell killing. Other activities exerted by antibodies, such as antibody-dependent cell-mediated cytotoxicity, are also involved in cancer cell killing.40–46 Currently, five single targeting ADCs specific to MET, namely ABBV-399 (telisotuzumab vedotin), SHR-A1403, TR1801-ADC, HucMet27-based ADC, and cIRCR201-dPBD have been preclinically validated (Table 2).40–44 The obtained results indicate that these MET-targeting ADCs are highly effective against cancer cellular models and patient-derived xenografts (PDXs) that harbor different forms of MET dysregulation. These forms of dysregulation include overexpression, amplification, exon-14 skipping, and activation mutation regardless of the level of MET signaling status involving cancer cell addiction.40–44 Two MET-based dual-targeting ADCs, including B10v5x225-H/M-vc-MMAE (targeting both MET and EGFR) and PCMdt-MMAE (targeting both MET and RON) have been preclinically studied (Table 2).45,46 B10v5x225-H/M-vc-MMAE is a dual-targeting ADC specific to both MET and EGFR. 45 Preclinical studies indicate that B10v5x225-H/M-vc-MMAE coordinately binds to both MET and EGFR, blocks ligand-induced MET and EGFR activation, and induces both receptors to internalize. These activities in vitro result in inhibition of MET/EGFR-mediated tumorigenic signals and cytotoxicity of various types of cancer cells. 45 PCMdt-MMAE is a MET and RON dual-targeting ADC developed by PCM TargeTech in Texas. 46 RON belongs to the MET family, important in epithelial tumorigenesis, and is a validated drug target. 105 Results from both in vitro and in vivo studies have demonstrated that PCMdt-MMAE is highly effective against the growth of xenograft tumors mediated by various types of cancers that express different levels of MET, RON, or both receptors with a favorable pharmacokinetic profile. 46 Currently, PCMdt-MMAE is ready for government-regulatory approval and transition into clinical development.
Mechanism-based evaluation of MET-targeted therapeutics
Tremendous efforts have been made during the last 20 years to optimize mechanism-based validation strategies for MET-targeting SMKIs and cTMABs.15–33 Pharmaceutical innovation resulting in novel biotherapeutics also pushes for the development of new strategies to meet validation demands. The principle of a mechanism-based validation strategy depends on the type of MET-targeting therapeutics being tested. Practically, the therapeutic efficacy of SMKIs, cTMABs, and bispecific antibodies highly rely on the addictive status of cellular models to MET signaling for growth and survival.15–39 In contrast, the activity of ADC-based biotherapeutics is associated with levels of MET expression and sensitivity of cancer cells to cytotoxic payloads attached to the mAb.40–46 Thus, logical selection of a proper mechanism-based drug validation strategy is the first step required for drug evaluation.
Increased MET expression as a validation mechanism
Quantitative MET analysis has made this model highly attractive for initial drug screening. For instance, 49 gastric cancer cell lines with integrated genomic profiling have been analyzed to establish a pattern of MET expression as a reference. 106 Moreover, MET amplification, HGF production, and expression of other oncogenic kinases such as RAS, EGFR, HER2, and PI-3 kinase have been matched in many individual cell lines. 106 The use of this 49-cell-based model is highly valuable for validating various types of MET-targeting therapeutics, particularly ADCs, which depends on the level of MET expression and their subsequent internalization for delivering cytotoxic payloads. As indicated in a previous study, the ADC-mediated responsiveness in vitro is proportionally correlated with levels of cancerous MET expression. 40 A similar correlation trend has also been observed in animal studies, in which the effectiveness of MET-targeting ADCs is positively correlated with xenograft tumors expressing different levels of MET expression.40–44 Moreover, the use of advanced drug-linker technologies and the selection of highly potent payloads have dramatically lowered the threshold of MET expression required for an ADC to exert significant cytotoxicity.42,44 These observations have potential implication in clinical trials for selecting patient populations showing variable levels of MET expression.
Levels of MET expression as a validation marker has limitations. Increased MET expression is only a phenotypic appearance, which reflects only alterations by a particular genetic or cellular pathway. However, these aberrations, alone or in combination, contribute to increased MET expression.6–14,74–76,83–85 Importantly, levels of MET expression, including overexpression, are not equivalent to a MET-dependent or addictive status by cancer cells.86,87 Nevertheless, overexpression indeed results in MET phosphorylation with activation of downstream signaling pathways, which leads to increased cellular activities such as malignant phenotypes.72,55,73–85 However, the detection of MET signaling activation by no means implies that cancer cells are addicted to MET for growth and survival.86,87 Clinical studies show that increased MET expression is not directly associated with the efficacy of MET-targeted therapy using either SMKIs or cTMABs.86,87 The lack of signaling addiction or low levels of MET signaling addiction is the major reason for the inefficacy of MET-targeted therapeutics regardless the level of MET expression. Thus, MET overexpression is not a reliable biomarker and performs poorly for predicting clinical benefits for MET-targeting SMKIs and conventional TMABs.15–33,86,87
MET amplification as a validation mechanism
Validation of therapeutics for MET-amplified tumors is an essential pharmaceutical step. Amplification is a distinctive feature of MET dysregulation and often shows increased signaling activation with advanced oncogenesis.74–76 Currently, more than 20 cancer cell lines harboring variable degrees of amplification (Table 3) have been used to evaluate the effectiveness of MET-targeting therapeutics.15–33 This evaluation has helped identify those, such as AMG-337, that are highly effective against tumor models caused by MET-amplified cancer cells. 19 The cellular MET amplification model is also suitable for analysis of MET-targeting cTMABs, bispecific antibodies, ADCs, and dual-targeting ADCs. This is mainly due to MET overexpression by MET-amplified cancer cells. In this sense, the pharmaceutical principle of applying the MET-amplified validation strategy is highly similar to that showing MET overexpression as described above. Regardless, results from using both MET-amplified cell lines and PDXs in testing the efficacy of MET-targeting TMAB Sym015, and ADCs TR1801, ABBV-399, and SHR-A1403 have proven that this model is highly reliable.33,42–46
Biochemical features of different types of cancer cell lines with MET dysregulation and their responsiveness to MET-targeting therapeutics * .

More than 40 cancer cell lines originating from different tissue/organs are summarized here. Five normal human epithelial cell types are included for comparison. Variable levels of MET expression in various types of cancer cell lines are determined by Western blotting and cell-surface immunofluorescence analysis and artificially categorized as: (++++), overexpression with more than 100,000 MET molecules per cell; (+++), high expression with MET molecules from 99,000 to 50,000 per cell; (++), moderate expression with MET molecules from 49,000 to 10,000 per cell; and (+), low expression with MET molecules from 9000 to 1000 per cell. The MET activation status is determined by detecting phosphorylated MET in Western blot analysis. Amplification of the MET gene was analyzed by MET gene copy numbers from individual cell lines using FISH or quantitative real-time PCR. Results shown here are indicated as A): MET/CEP7 ratio, B): GCN (log2) ratio, or C): GCNs. Cellular sensitivity to individual SMKIs or TMABs are categorized according to levels of cell growth inhibition as: highly sensitive: 80 to 100%; moderately sensitive: 50–79%; low sensitivity: 10–49%; and insensitive: less than 10% of growth inhibition. The effectiveness of targeting ADCs in killing/inhibiting cancer cells in vitro and in blocking xenograft tumor growth are leveled according to the levels of cell death: highly effective: >80–100% death; moderately effective: 50 to 79% death; lowly effective: 10 to 49% death; and insensitive: <10% death. Cellular addiction to MET signaling for growth and survival is determined by individual SMKIs or TMABs and indicated by levels of growth inhibition as: highly addictive, >80–100%; moderately addictive; 50 to 79%; lowly addictive, <50%; and unknown, no information is available.
ADC, antibody–drug conjugate; CEP7, chromosome enumerating probe against chromosome 7; FISH, fluorescence in situ hybridization; GCN, gene copy number; PCR, polymerase chain reaction; TMAB, therapeutic monoclonal antibody; SMKI, small-molecule kinase inhibitor.
The limitation of the MET-amplified validation model is the extremely low frequency of MET amplification in clinical samples.74–76,86,87 In this sense, the use of MET amplification as the biomarker for patient selection is a challenge. It requires to have an advanced laboratory with sophisticated technologies for performing FISH, NGS, and other methods, resulting in an increase in clinical cost and expenditures. In addition, cancer cells with MET amplification are not always responsive to SMKIs or conventional TMABs. As described above, certain proteins with oncogenic mutations in the MET signaling pathway with disruptive cascades can support cancer cell growth and survival independent of the presence of MET-targeting SMKIs or cTMABs.21,107
MET exon-14 skipping as a validation mechanism
The use of MET exon-14 skipping as a validation approach has gained special attention due to exciting results from MET-targeted clinical trials of NSCLCs.22,108,109 Oncogenic evidence has shown that MET exon-14 skipping acts as a vital oncogenic driver,108,109 but its frequency is low with minor occurrence in lung (~4%), stomach (~7), and colorectal (~5%) cancers.108,109 These observations suggest that cancer patients with MET exon-14 skipping is a particular population suitable for MET-targeted therapy.
Currently, the cellular models that truly reflect the oncogenic effect of MET exon-14 skipping are still lacking. Only two cell lines, Hs746T and NCI-H596, have MET exon-14 skipping (Table 3). However, Hs746T cells are accompanied with MET overexpression and gene amplification.108,109 In contrast, levels of MET expressed by H596 cells are relatively low (Table 3). Thus, precaution must be taken in interpretation of results from using these two cell lines. Establishment of a mouse model expressing mouse MET exon-15 deletion (equivalent to human MET exon-14 skipping) through a molecular approach has been reported resulting in the formation of mouse lung adenoma, but not adenocarcinoma. 110 The use of this animal model has shown that crizotinib is able to stabilize tumor progression but the efficiency is relatively low. 110 Two PDX models with confirmed MET exon-14 skipping, namely LU2503 and LU5381, are available from Crown Bioscience (www.crownbiscience,com). They have been tested for their responsiveness to MET-targeting SMKIs, such as glesatinib, 22 and to cTMABs including Sym015. 35 Their pharmaceutical values are confirmed from results showing the responsiveness of both models to the action of MET-targeting SMKIs and conventional TMABs.22,33
MET mutation as a validation mechanism
The strategy using single or multiple MET mutation(s) as a model to validate MET-targeting therapeutics has not been reported in detail. Only a subset of MET point mutations found in papillary renal cell carcinoma, such as V1092I, H1094R, and others, have been tested with an enzymatic assay for the action of several SMKIs.22,25,51,111 As shown in Figure 4, numerous MET mutations in the different domains of MET have been identified. Results from preclinical studies have confirmed the role of MET mutations in tumorigenesis.86,87 Nevertheless, it is probably not practical to test the responsiveness of individual mutations to determine efficacies of MET-targeting therapeutics. The lack of available cell lines is probably due to the overwhelming numbers of MET mutations discovered in different regions of the MET sequence. With the growing interest in development of novel MET-targeting therapeutics, it is hoped that a strategy will be developed to validate MET-targeting therapeutics using models harboring individual mutations in the critical region of MET sequences/domains.
PDXs with defined MET dysregulation as a validation strategy
The use of PDXs with different MET dysregulations has been a favored choice for the last several years.22,29,32,33,41,42,51 The underlying reasons are obvious, owing to pathogenic features of PDXs highly resembling those from primary tumors. Currently, MET-based PDX models derived from lung, gastric, CRC, and head & neck cancers with MET overexpression, amplification and exon-14 skipping have been established.22,29,32,33,41,42,51 SMKIs, cTMABs, and ADCs have all been tested in PDX models with acceptable therapeutic responsiveness.22,29,32,33,41,42,51 For instance, glesatinib at a therapeutic dose of 60 mg/kg is highly effective against PDX LU2503 and LU5381 models with MET exon-14 skipping. 22 Similarly, TR1801-ADC, a second-generation MET-targeting ADC at a single-dose injection of 0.125 to 1 mg/kg, has been validated in PDX models derived from stomach, CRC, and head & neck cancer samples with demonstrated therapeutic activity. 42 Thus, PDX models are an exciting addition to the list of currently used validation strategies and should have pharmaceutical advantages in conjunction with traditional models for objectively evaluating MET-targeting therapeutics.
Additional MET alterations as a validation mechanism
Development of novel MET-targeting therapeutics, such as bispecific antibodies and dual-targeting ADCs, demands a proper strategy for validation. A MET-based bispecific antibody has a co-targeting antigen-binding arm that regulates the partner signaling pathway or T-cell activity, respectively.34–39 Validation of these agents requires selection of proper cellular models to determine anticancer activities of both antigen-binding arms. Several models including PDX-derived ex vivo 3D spheroids have been developed to evaluate the efficacy of MET-targeting therapeutics such as TR1801-ADC.34–46 However, comprehensive analyses at mechanistic levels of these models in terms of the strength of signaling integration, levels of addictive status, biological responsiveness, and activity coordination have not been studied in detail. For instance, efficacies of three MET-based bispecific antibodies targeting PD-1, as evaluated in several cellular models, are not impressive in terms of tumor growth inhibition and levels of T-cell activation.38,39 Thus, the complexity in mechanism of action and tumorigenic feature included in the models must be considered to objectively evaluate the efficacy of these novel MET-targeting therapeutics.
Pharmaceutical criteria for mechanism-based drug validation
Utilizing a mechanism-based validation strategy has significantly contributed to the progress and success in the development of MET-targeting therapeutics. Approval of four SMKIs by the FDA is an example. Nevertheless, strategies used to validate the efficacy of MET-targeting cTMABs appear to have some issues. Results from preclinical studies seem to be promising; however, outcomes from clinical trials, which have been conducted for almost 10 years, are disappointing.28–33 This raises serious concerns about the reliability of these strategies for validating MET-targeting cTMABs. Thus, it is time to evaluate current approaches in order to identify deficiencies that cause unobjective conclusions, and to avoid mistakes of moving these unjustified MET-targeting TMABs into clinical trials. The following is a summary of criteria to be considered when a mechanism-based validation strategy needs to be applied.
It is vital to select a mechanism-based validation strategy that suits the purpose of a particular therapeutic to be tested. MET dysregulation occurs predominantly in certain types of tumors such as those from stomach, lung, kidney, and liver.51,55,72–92 The majority of validation programs have pre-determined objectives favoring particular types of cancer. Dependent on the nature of drug candidates, some studies screen drug efficacy by employing a large number of cancer cell lines in order to find defined MET-targeting activity. For instance, AMG-337, a type I, ATP-competitive, and highly MET-selective SMKI, has been profiled against a diverse panel of 260 cancer cell lines. 19 Only two cell lines, SNU-5 and Hs746T with MET amplification, have shown sensitivity to AM-337. 19 Studies then focused on cellular models with MET amplification for further validation. 19 In contrast, other studies have utilized an approach of focusing on a unique MET abnormality. An example is glesatinib, a unique type II MET SMKI, which is evaluated in lung cancer models harboring MET exon-14 skipping and mutation-associated resistance to type I MET SMKIs. 22 Such a focused strategy increases the potential for selecting a lead candidate moving into clinical trials. Thus, selection of a mechanism-based validation strategy must be considered in a balanced way.
Understanding the mechanism of MET dysregulation helps in selecting a proper validation strategy. The mechanism of action exhibited by individual MET-targeting therapeutics is fundamentally different. For instance, type I and II SMKIs act at different regions in the TK domain of MET with different structure conformations (active versus inactive).15–27 As described above, the TK domain of MET can be activated under various conditions and manifested through single or multiple events.55,72–91 In this sense, cellular models featured by HGF-dependent and independent MET activation have to be carefully selected before different types of MET-targeting SMKIs are applied. Similarly, different MET-targeting TMABs that bind to different regions in the MET extracellular sequences result in different biochemical impacts, such as preventing HGF binding, inducing MET internalization/degradation, attenuating MET signaling, or enhancing immune regulatory activity.28–46 All these activities must be considered when a validation strategy is selected.
The status of cellular MET signaling integration/addiction in individual cellular models is a factor determining the success of a validation strategy. The therapeutic efficacy of MET-targeting SMKIs, cTMABs, and bispecific antibodies is highly dependent on the level of addictiveness of the cancer cell to MET or partner protein signaling for growth and survival.28–46 In preclinical studies, many MET-targeting SMKIs and TMABs display only moderate inhibitory effects on cellular models showing limited levels of addiction. Clearly, these “positive results” are not sufficient to be reflected in clinical trials. In contrast, only those showing the strongest anticancer activity with complete growth inhibition in cellular models with full MET signaling addiction have the chance to achieve an objective response in cancer patients.28–46 Thus, studies validating SMKIs, cTMABs, and bispecific antibodies should select cellular models that exhibit full MET-signaling addictive status.
Consideration of acquired drug resistance is another strategy for validation of MET-targeting therapeutics. Aberrant MET expression and signaling have been established as a compensation mechanism during the treatment of cancer with SMKIs targeting EGFR and other signaling proteins. 101 The compensated MET pathway significantly contributes to the acquired drug resistance in various types of cancer undergoing chemo and targeted therapy. 101 In this sense, targeted inhibition of MET signaling using SMKIs or antibody-based biotherapeutics has clinical relevance. The use of MET-targeting SMKIs for treatment of tumors resistant to EGFR inhibitors is currently a recommended clinical practice. Demonstration of the effectiveness of antibody-based biotherapeutics to these drug-resistant tumors is also an objective in the validation procedures, and is highly anticipated in many MET-targeting clinical trials. Clinically, different types of cancer with variable levels of drug-resistant phenotypes have different drug sensitivity and/or treatment profiles. In this sense, the use of drug resistance as a biological criterium to validate the effect of MET-targeting therapeutics should be highly recommended.
Last but not least is the strategy of using MET-targeting therapeutics to target cancer stem cells to achieve a therapeutic objective. Aberrant MET expression and activation contribute to cancer stemness in certain types of cancer.112–115 For instance, increased MET expression in cancer stem cells from CRC and glioblastoma contributes to malignant phenotypes and behaviors,112–115 which has therapeutic value. Thus, the use of MET-targeting ADCs that have mechanisms of action independent of signaling addiction is an attractive approach to eradicate cancer stem cells as a therapeutic objective. ADCs targeting other RTKs, such as RON and leucine-rich repeat-containing G protein-coupled receptor 5 (LGR5), are examples for eradicating cancer stem cells.116,117 Thus, the same strategy should be applied to determine the effectiveness of MET-targeting ADCs to kill cancer stem cells. The outcome will help us not only dissect the pathogenic role of MET in oncogenesis, but also broaden our understanding about the underlying mechanism of MET-targeting therapeutics in clinical application.
Conclusion
Pathogenic mechanism-based evaluation of different types of MET-targeting therapeutics is critical to select and validate lead candidates for clinical trials and approval for patient application. Technological innovation resulting in novel therapeutics also requires appropriate new models to meet the pharmaceutical demand. During the last 20 years, the achievement in dissecting oncological MET dysregulation and its underlying mechanism has significantly improved the quality of mechanism-based validation by using well-defined models with characterized biochemical and biological features. These models not only try to mimic the clinical complexity of MET-driven tumorigenesis, but also serves as a pharmaceutical tool for drug screening and evaluation. At present, novel MET-targeting biotherapeutics, such as bispecific antibodies, ADCs, and dual-targeting ADCs, have emerged as new players in MET-targeted cancer therapy.36–50,72 The mechanisms of action by these biotherapeutics are different from previously established SMKIs and cTMABs. Thus, development and optimization of novel mechanism-based drug validation strategies is an urgent need, which will greatly facilitate the clinical approval of MET-targeting therapeutics for oncological application.
Footnotes
Acknowledgements
We greatly thank Ms. R. Hudson (TTUHSC School of Pharmacy in Amarillo, TX) for editing and proofreading the manuscript.
Author contributions
HPY, XMT, and MHW discussed the necessity for writing this manuscript. MHW wrote the original draft. HPY and XMT reviewed the draft with detailed comments. MHW made the revision. All authors read and approved the final manuscript for submission.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by funds from National Natural Sciences Foundation of China grant #81872883 (HPY), the Major Project of Zhejiang Provincial Sciences and Technology Department #2019C03038 (XMT), and by Amarillo Area Foundation for Cancer Biology Research (MHW).