
Abstract
Kinetic analysis of antibodies is crucial in both clone selection and characterization. Historically, antibodies in supernatants from hybridomas are selected based on a solid-phase enzyme-linked immunosorbent assay (ELISA) in which the antigen is immobilized on the assay plate. ELISA selects clones based on a combination of antibody concentration in the supernatant and affinity. The antibody concentration in the supernatant can vary significantly and is typically unknown. Using the ELISA method, clones that express high levels of a low-affinity antibody can give an equivalent signal as clones that express low levels of a high-affinity antibody. As a consequence, using the ELISA method, superior clones can be overshadowed by inferior clones. In this study, we have applied Bio-Layer Interferometry to screen hybridoma clones based on disassociation rates using the OctetRED 384 platform. Using the OctetRED platform, we were able to screen 2000 clones within 24 hours and select clones containing high-affinity antibodies for further expansion and subsequent characterization. Using this method, we were able to identify several clones producing high-affinity antibodies that were missed by ELISA.
Introduction
Therapeutic antibodies represent the fastest-growing class of new medicines being developed in the pharmaceutical industry. 1 The first human monoclonal antibodies (mAbs) were developed during the early 1980s. In the past 25 years, more than 30 antibody-based drugs have been approved for various indications.2,3 The current marketed antibody-based drugs have been approved to treat not only diseases affecting a large number of patients, such as cancer and inflammation, but also orphan or rare diseases such as nocturnal hemoglobinuria. 3 Significant advances in the genetic engineering of antibodies eliminated mouse-derived sequences and immunogenicity. Furthermore, technology advancements for antibody generation using transgenic mice and phage display have propelled antibody discovery efforts. These combined advancements rapidly increased the number of therapeutic antibodies entering clinical development and subsequent approval.
The majority of therapeutic antibodies currently available were derived from rodent immunizations that generate a panel of hybridomas.4,5 Although the hybridoma technology for the production of mAbs was first described in 1975, it still remains the most common technique used to generate antibodies for therapeutic applications. 5 The generation and screening of hybridomas from an immunized animal comprise a time-consuming process and sample only a fraction of the antibodies generated during the adaptive immune response. Generally, hydridomas are isolated into single cells by fluorescence-activated cell sorting (FACS) and transferred into multiwell assay plates. These hybridoma supernatants, typically 500–5000 clones, are screened by a single-point binding assay to detect antigen-positive antibodies. A fraction of positive clones are isolated and sequenced to identify unique antibodies, which are then expressed and purified in larger quantities for further characterization. Therefore, screening hybridoma supernatants for antigen-positive antibodies is a crucial step in the screening and selection cascade. To ensure that detailed characterization of antibody hits is manageable, only ~1% of the 500–5000 hybridoma supernatants are picked for further analysis, even though binding to antigen can be observed for more than 15% of the supernatants. Historically, antibodies in hybridoma supernatants are selected for further analysis based on a solid-phase enzyme-linked immunosorbent assay (ELISA) in which the antigen is immobilized on the assay plate. 6 The ELISA-based approach selects clones based on a binding signal and is dependent on the combination of concentration and affinity of the antibody in the supernatant. Both parameters can vary significantly and are typically unknown. Using the ELISA assay, clones that express high levels of a low-affinity antibody can give an equivalent signal to clones that express low levels of a high-affinity antibody. As a consequence, superior clones can be overshadowed by inferior clones. A better assay method for selecting high-affinity antibodies would require screening hybridoma supernatants kinetically and would allow clones to be ranked and selected based on binding, but, more importantly, off-rate. The off-rate kinetic measurement is a first-order rate constant, concentration independent, and therefore it can be determined in samples in which the antibody concentration is unknown. Ranking and selecting antibodies based on off-rate would ensure that a high-affinity antibody expressed at low levels is not overshadowed or missed during primary screening.
To screen hybridoma supernatants kinetically requires the use of a high-throughput, label-free, real-time biosensor platform. The OctetRED 384 instrument (Pall ForteBio, Menlo Park, CA) is a high-throughput, label-free platform that uses disposable fiber-optic biosensors to detect biomolecular interactions by bio-layer interferometry (BLI). BLI analyzes the interference pattern of a white light reflected from the biosensor tip. A change in the number of molecules bound to the biosensor tip causes a shift in the interference pattern that is measured in real time. 7 Traditional label-free platforms, such as the Biacore (GE Healthcare, Little Chalfont, UK), use surface plasmon resonance (SPR) coupled with a sophisticated microfluidics to flow analyte over ligand-coated sensors. 8 These traditional flow-based and SPR-based platforms have been previously used to kinetically screen hybridoma supernatants; however, the throughput is not very high (fewer than 800 samples a day).9,10 The OctetRED uses a non-flow-based dip-and-read platform that can measure 16 samples in parallel by immersing ligand-coated biosensor tips into sample wells of a 96- or 384-well microplate. The non-flow-based dip-and-read configuration enables the OctetRED to measure interaction kinetics in hybridoma supernatants in high throughput (>1000 samples a day).
In this study, we compared BLI to ELISA as a method to identify high-affinity antibodies by screening 2000 hybridoma supernatants. We found screening clones kinetically was an efficient method for identifying high-affinity antibodies. A detailed analysis of the screening data revealed that the off-rate parameter, and not binding signal, correlates most accurately with antibody affinity. We found that the highest-affinity antibody was identified by ranking the clones based on off-rate and would not have been identified using only the ELISA binding signal strength as the selection criterion.
Materials and Methods
Hybridoma Libraries
A Chinese hamster ovary (CHO) cell line that expresses a fragment of the proprietary target (MW >25 kDa) was purchased from ProSpec (Rehovot, Israel) and used as an immunogen to generate the target antibodies. Mice were immunized with the target protein immunogen with Ribi adjuvant using a PolyExpress protocol from Antibody Solutions (Sunnyvale, CA). Sera from mice were tested for binding to the target by ELISA, and B-cells from lymph nodes of mice showing high titers to the antigen were fused with mouse myeloma cells to generate hybridoma libraries. Hybridoma libraries that retained affinity for the antigen were subcloned to generate hybridoma clones.
Antibody Selection and Purification
Selected monoclonal antibodies produced by individual hybridomas derived from the libraries were expanded into 10 mL cultures and purified by one-step affinity chromatography using a protein resin matrix. Antibody protein concentrations were determined by absorbance at 280 nm.
ELISA Assay
Nunc plates (Affymetrix, CA) were coated overnight with 1 µg/ml of antigen in 50 mM sodium borate buffer (pH 8.0) at 4 °C (100 µl/well). On the following day, the plates were washed 3 times with phosphate buffered saline with Tween 20 (PBST; 10 mM sodium phosphate, 140 mM sodium chloride, 0.05% Tween 20, pH 7.4) and blocked with bovine serum albumin (BSA) solution (5% BSA in 10 mM sodium phosphate, 140 mM sodium chloride, pH 7.4, 200 µl/well) for 1 h at ambient temperature. Plates were then washed 3 times with 300 µl of PBST, and hybridoma supernatant containing target antibody or 10 nM (highest concentration tested) purified antibody (100 µl) was added to the blocked plates and incubated at room temperature for 1 h. Plates were washed again, and secondary antibody was added: 100 µl of a 1:10,000 dilution of goat anti-mouse HRP (horseradish peroxidase) secondary antibody (Pierce, Rockford, IL). Secondary antibody was diluted in 0.5% BSA solution (0.5% BSA in 10 mM sodium phosphate, 140 mM sodium chloride, pH 7.4), followed by incubation at room temperature for 1 h. Plates were washed again and developed using 100 µl of 3,3′,5,5′-tetramethylbenzidine and allowing the reaction to take place for 2 min. The reaction was quenched with the addition of 100 µl of 1 M hydrochloric acid. Quantitation was carried out on a SpectraMax M5 in absorption mode at 450 nm.
Based on previous hybridoma screens, we have found that a high optical-density (OD) cutoff (≥1.5 OD units) for clone selection substantially reduced the number of false positives and downstream work required for antibody expression, purification, and characterization. Furthermore, historically we tentatively observed a better correlation between clones having a high OD value and high affinity with the resulting purified antibodies. As a result, the clones selected in the hybridoma screen in this study were scored as positive for containing antibody when they had an absorbance value of at least 1.5 OD units higher than that of mock supernatants (no antibody). For purified antibodies, dissociation constants were determined by plotting the absorbance values versus the concentration of antibody and fitting the data to Equation 1:
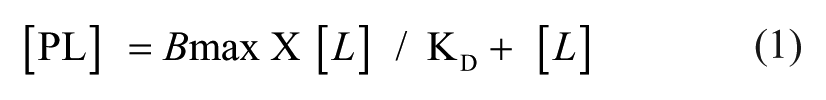
where PL is the bound concentration, which is proportional to the absorbance reading; L is the antibody concentration; Bmax is the maximal binding; and KD is the dissociation constant.
OctetRED-Based Screening
Samples of buffer were dispensed into polypropylene 96-well black flat-bottom plates (Greiner Bio-One, Frickenhausen, Germany) at a volume of 180–200 µl per well, and all measurements were performed at 30 °C with agitation at 1000 rpm. Anti-mouse immunoglobulin G (IgG) Fc (AMC)-coated biosensor tips (Pall ForteBio, Menlo Park, CA) were used to capture antibodies from hybridoma supernatants, and typical immobilization levels captured on the AMC sensors varied from 1 to 4 nm, which approximately translates to 3–15 µg/ml of captured antibody. Capture of antibody in hybridoma supernatants was done off-line from the OctetRED instrument. Preliminary assay development experiments using a set of purified control antibodies spiked into mock hybridoma supernatants revealed that a minimum of 0.5 µg/ml antibody had to be captured on the biosensor to generate a binding response that could be kinetically characterized and quantified. The same set of control antibodies were used to establish assay parameters for the kinetic screening of hybridoma supernatants, and this is described below.
Using a Hudson Plate Crane robotic arm (Hudson, NJ), a tray of 96 AMC biosensors were pre-wetted with assay buffer [10 mM sodium phosphate (pH 7.4), 140 mM sodium chloride, 0.05% Tween 20, and 0.01% BSA], then dipped into wells containing hybridoma supernatants and incubated for 1 h. The biosensors were then transferred to the OctetRED for kinetic screening of antigen binding. Using the OctetRED, biosensors were dipped in assay buffer containing wells for 1 min to remove any nonspecific protein or unbound antibody acquired from the hybridoma supernatants. Biosensors were then transferred into fresh assay buffer for 100 s to collect a baseline read. Kinetic measurements for antigen binding were performed by dipping the antibody-coated biosensors into wells containing a single concentration of antigen (8 nM) for 70 s, followed by a 180-s dissociation time by transferring the biosensors into buffer-containing wells. New AMC biosensors were used for each sample, and a total of 16 samples were kinetically characterized for antigen binding and disassociation in parallel. All sensorgrams were referenced for buffer effects and then fitted using the OctetRED user software (Pall ForteBio). The binding profile of each sample was summarized as an “nm shift” (the wavelength or spectral shift in nanometers), which represented the difference between the start and end of the kinetic cycle. Kinetic responses were fitted to a 1-site binding model to obtain values for association (kon), dissociation (koff) rate constants, and the equilibrium dissociation constant (KD). Curves that could not be reliably fitted with the software (R2 < 0.90), usually caused by heterogeneous binding, were excluded from further analysis.
Results
Label-Free Assay Configuration and Development
The antigen and antibodies used and identified during this study were directed toward a proprietary human antiviral target. Two assay configurations can be used on the OctetRED to characterize the binding interaction between an antibody and its antigen: (1) a format that directly captures the antigen onto the biosensor surface and monitors antibody binding in hybridoma supernatants, and (2) one that captures antibody in the hybridoma supernatant onto the biosensor and monitors antigen binding. We decided to develop and optimize the latter assay configuration for several reasons. First, capturing the antibody directly onto the biosensor from crude hybridoma supernatants eliminates the need to purify, concentrate, and quantitate the amount of antibody in each sample. Second, capturing minimizes avidity because the bivalent nature of antibodies can crosslink on the antigen surface, resulting in a misinterpretation of the dissociation rate constant and, thus, the affinity. 9 Ideally, by capturing the antibody at similar loading densities on the biosensor surface, we could further differentiate off-rate and subsequent affinity that could be masked by avidity. Third, the antigen is allowed to remain in solution with all epitopes available for antibody binding. Lastly, capturing antibody onto the biosensor surface allowed us to develop a simple but fast workflow for hybridoma screening (see the Kinetic Screening of Hybridoma Supernatants section).
Before we conducted any screening activities, we had to characterize the selectivity of our antigen using tool antibodies on the OctetRED platform ( Fig. 1 ). Because the hybridoma supernatants are mouse derived, we used AMC biosensors to capture our control mouse monoclonal antibodies. We conducted 3 control experiments. First, we tested whether our antigen binds to the target mouse monoclonal antibody with an affinity comparable to reported values ( Fig. 1A ). Second, we determined specificity of the antigen binding to the blank AMC sensor or to sensor coated with a random or unrelated mouse monoclonal antibody ( Fig. 1B and 1C ). Finally, we wanted to make sure our positive control antibody does not bind to an unrelated antigen ( Fig. 1D ). The data shown in Figure 1 demonstrate that our antigen specifically binds to the target antibody, it does not bind to a naked AMC sensor or unrelated mouse monoclonal antibody, and our positive control antibody is specific for the target antigen.
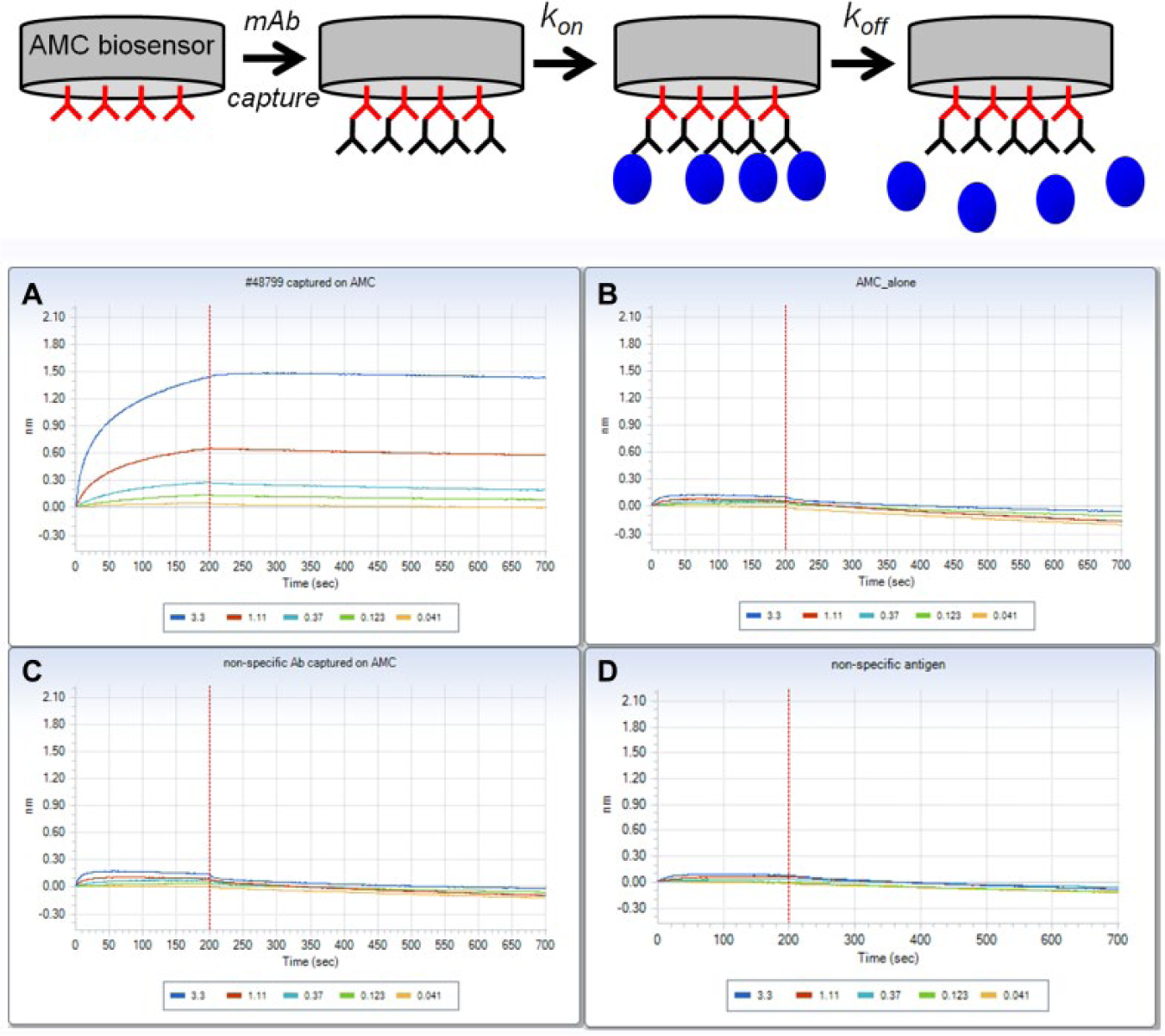
Assay development: determination of antigen specificity. At the top is a sequential schematic representation for the OctetRED 384 assay. AMC [anti-mouse immunoglobulin G (IgG) Fc] sensors are used to capture mouse antibodies and then dipped into wells containing antigen to measure the kinetics of association (kon). Rate of disassociation (koff) is determined by dipping sensors into buffer-containing wells. (
Kinetic Screening of Hybridoma Supernatants
To screen the 2000 hybridoma supernatants, we established a simple automated workflow that enabled us to complete the screen in less than 24 h ( Fig. 2A ). We configured a Hudson Plate Crane robotic arm that would feed and remove biosensor trays and assay plates from the OctetRED. Such trays contained 96 AMC biosensors coated with antibody from supernatants, and the assay plates contained buffer and antigen. Hybridoma supernatants were plated in 96-well plates and incubated with AMC biosensors off-line for 1 h to capture the antibody in the hybridoma supernatant. The antibodies were screened against a single antigen concentration to obtain kinetic and binding constants (kon, koff, and KD). Using the assay conditions described in the Materials and Methods section, we were able to screen an entire 96-well plate of supernatants in 45 min. Using the assay workflow described, it took us 15 h to screen 2000 hybridoma supernatants. The OctetRED workflow appears slower than the ELISA assay workflow, which took approximately 10 h to screen 2000 supernatants; however, an overnight incubation was required for the ELISA assay. Furthermore, the ELISA assay was significantly more labor intensive and involved multiple wash, incubation, and blocking steps in comparison to the OctetRED-based workflow, which required only sample plating and biosensor transfer steps.
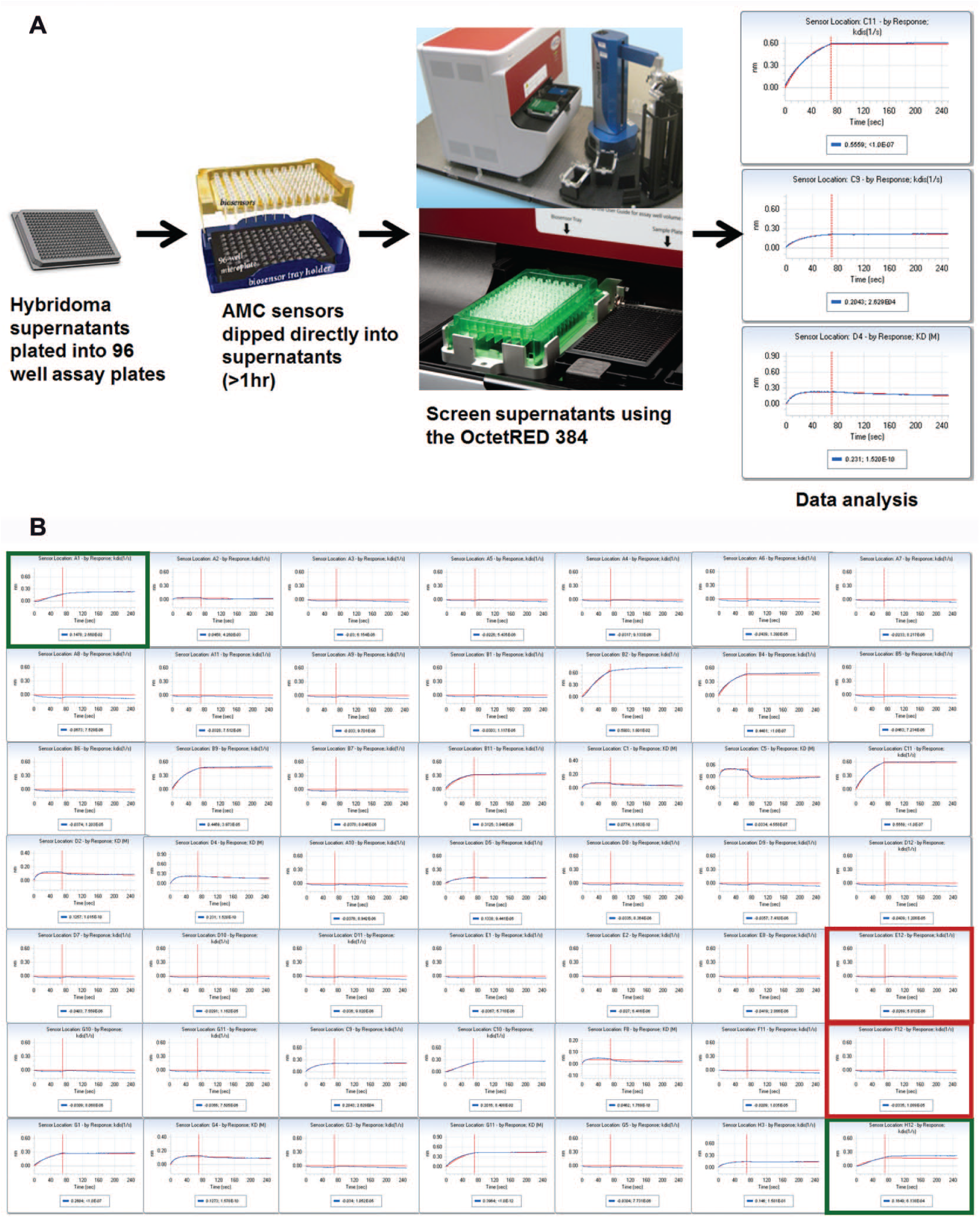
Hybridoma screen setup and execution. (
Figure 2B shows hybridoma screening data analyzed by BLI from an assay plate using a single antigen concentration. For each assay plate screened, we included negative and positive control wells. Negative control wells were mock supernatant (no antibody) that represented nonbinding wells. Positive binding controls were assay wells containing either polyclonal cells or fusion sera. The 2000 hybridoma supernatants were derived from 2 hybridoma libraries, each resulting in 1000 supernatants. The 2000 hybridoma supernatants were plated into 22 96-well assay plates. For each assay plate, all positive control wells showed a binding response, and all negative control wells scored as nonbinders for both ELISA- and OctetRED-based screens (data not shown). For the ELISA assay, clones were scored as positive for containing antibody when they had an absorbance value ≥1.5 OD units higher than that of intraplate mock supernatants (no antibody) containing wells. For the OctetRED, we selected clones based on off-rate and a binding signal at least 0.1 nm higher than that of intraplate mock supernatants (no antibody) wells. Although in this exercise we only focused on selecting antibodies with the slowest off-rates, we could have picked antibodies with a fast off-rate to increase the probability of selecting antibodies with unique epitopes and functional activity.
Using the ELISA assay, we selected 130 clones for follow-up; and, using the OctetRED assay, we selected 155 clones. All the clones selected using the ELISA method were also selected by the OctetRED method, leaving 25 clones found only by the OctetRED platform. Comparison of the binding signal for the selected clones between the two assay formats is shown in Figure 3A . The absorbance signal for the 130 clones selected by ELISA correlates well with the OctetRED binding signal (R2 = 0.71). Clones that had an absorbance value of less than 1.5 OD but appeared to have a slow-off rate were selected by the OctetRED. A subset of these clones specifically selected by the OctetRED assay also contained several high-expressing antibodies that did not show a large binding signal in the ELISA and therefore were not selected ( Fig. 3A ). A possible explanation could be related to assay configuration and capture of antigen in one assay format versus the other. A comparison of the OctetRED binding signal on-rate (kon) and off-rate (koff) to affinity (KD) is shown in Figure 3B , 3C , and 3D , respectively. We observe no correlation between either binding signal or on-rate with affinity (R2 = <0.03). However, we observe a strong correlation between antibody off-rate and affinity (R2 = 0.82). We find that antibodies with a slow-off rate had a high affinity, whereas antibodies with a faster off-rate generally had lower affinity ( Fig. 3D ). More importantly, the highest-affinity antibody-containing clone was identified by ranking the clones based on off-rate and would not have been identified using either the on-rate or ELISA/OctetRED binding signal strength as the selection criterion.
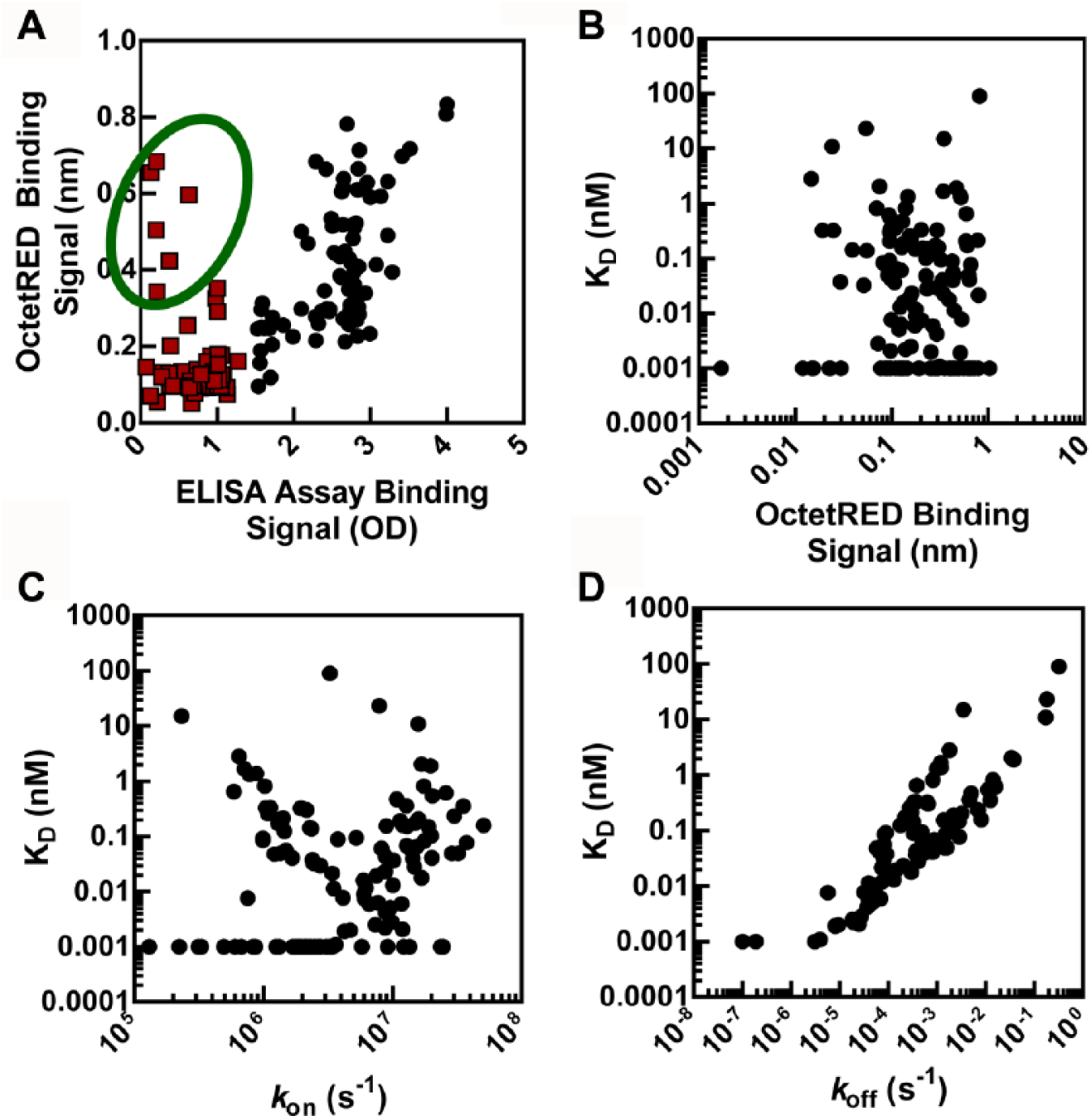
Correlation plots of the hits selected from the hybridoma screen. (
Hit Conformation and Correlation with ELISA
The 155 clones selected during the primary screen using the OctetRED platform were expanded, and the resulting antibodies were purified. These purified antibodies were retested for binding on the OctetRED at multiple antigen concentrations ( Fig. 4 ). All purified antibodies were confirmed for binding to the antigen, and this translated to a 100% conformation rate. The very high conformation rate indicates that the process of expanding the selected antibody-containing clones and subsequent purification was very efficient. The single antigen concentration KD determined in the primary screen by the OctetRED showed an excellent correlation with the KD values determined using multiple antigen concentrations (R2 = 0.99). For some antibodies, however, the OctetRED affinities determined in hybridoma supernatants using a single antigen concentration were slightly overestimated (5–10-fold more potent) than the values obtained with the corresponding purified antibodies using multiple antigen concentrations ( Fig. 4C ).
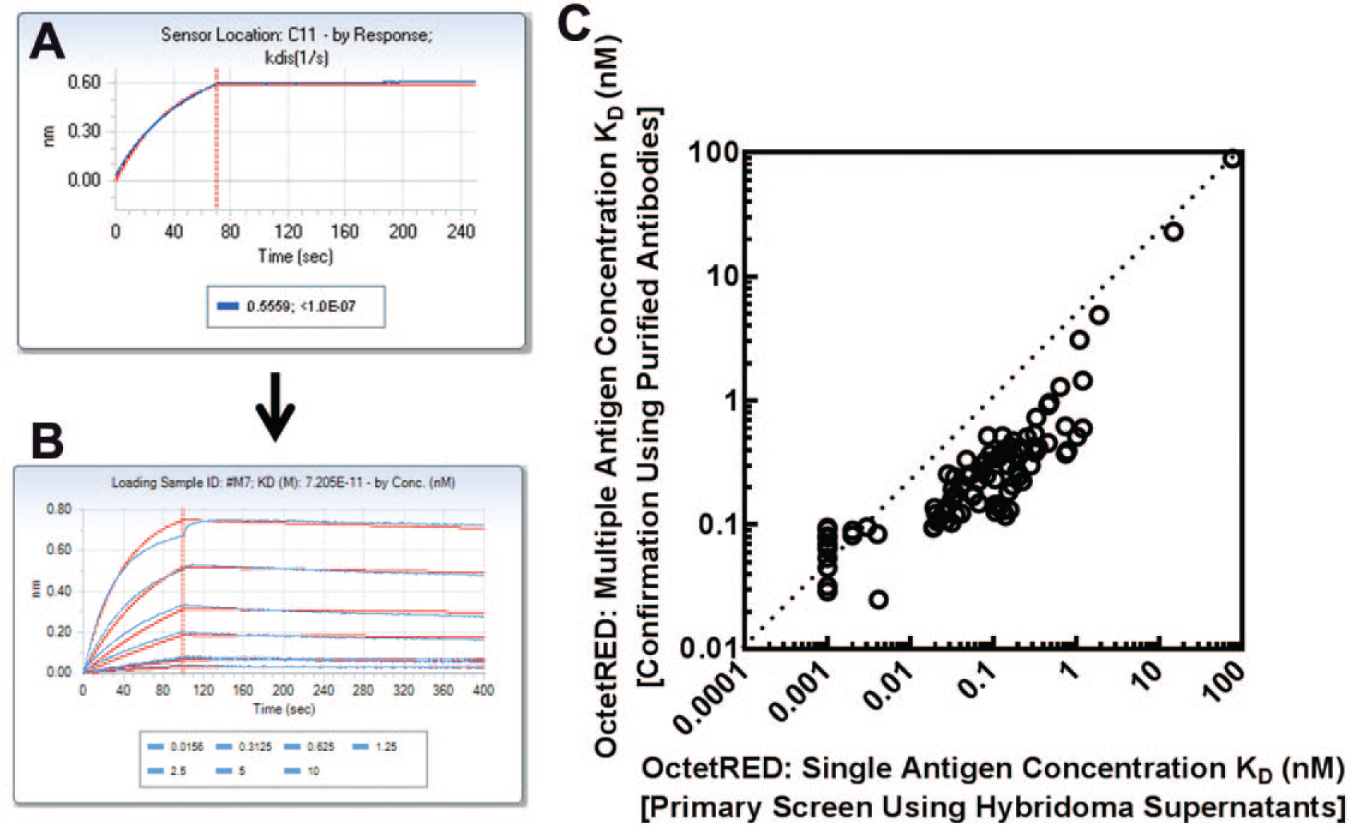
Hit confirmation using purified antibodies. (
We then compared the KD values for the purified antibodies identified in both the ELISA and OctetRED platforms. Figure 5 shows a correlation plot comparing the KD values determined using multiple antigen concentrations for 130 purified antibodies between ELISA and OctetRED. We observe a good correlation between the 2 platforms; however, the ELISA platform is unable to differentiate among antibodies with a KD value less than 100 pM. This suggests that the OctetRED platform is a more sensitive method compared to ELISA for measuring the affinity (KD) of potent antibodies.
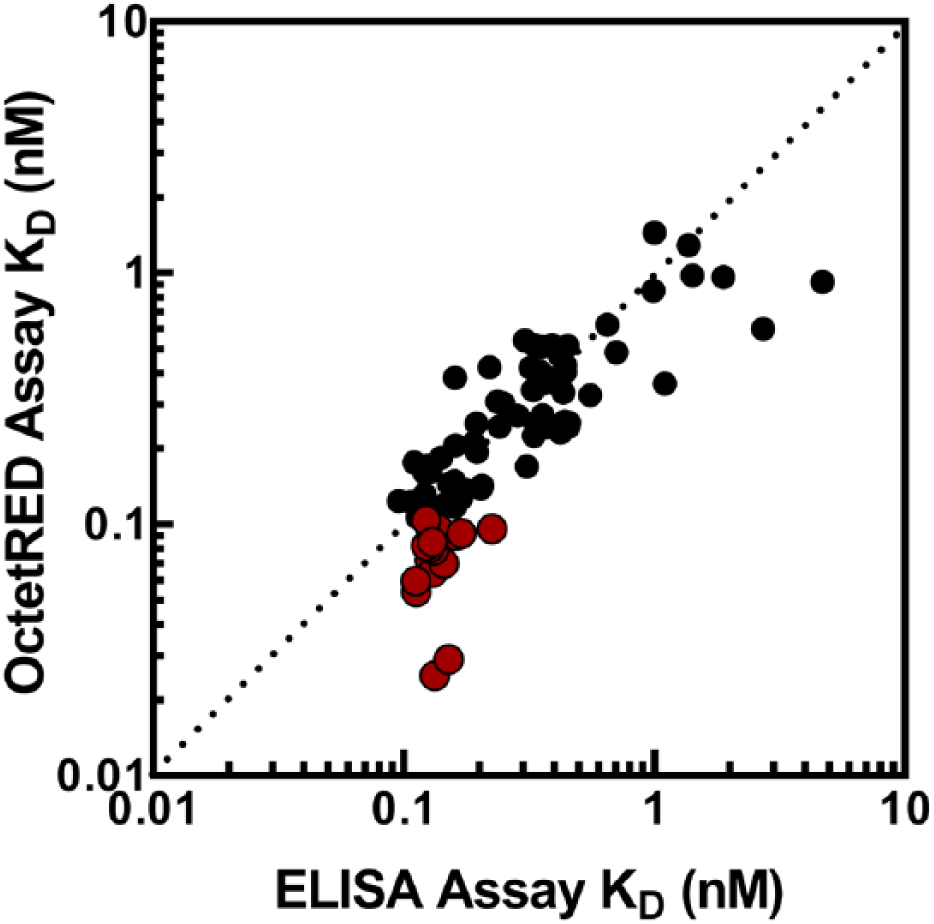
Correlation plot comparing the KD values for purified antibodies in an enzyme-linked immunosorbent assay (ELISA) versus OctetRED assay. Data points in red are antibodies with a KD value less than 100 pM, which cannot be resolved using ELISA.
Discussion
Therapeutic antibodies represent the fastest-growing class of new medicines. Generation of antibodies that meet specific criteria is increasingly important for therapeutic applications. In many well-studied cases, increased antibody affinity has been shown to correspond to improved biological efficacy. 11 Although an antibody’s efficacy can depend on several factors such as stability and pharmacokinetics, affinity of the antibody for its target will always be a key parameter for candidate selection. Furthermore, a high-affinity (potent) antibody would enable lower dosing, possibly reduce off-target toxicities, and lead to a lower cost of therapy. 12 Novel approaches that can accurately identify high-affinity antibodies early during the discovery process would therefore be extremely useful.
There are many methods and platforms available for screening antibodies. These include flow cytometry, ELISA, electrochemiluminescence using the Meso Scale Discovery (MSD; Rockville, MD) platform, and, more recently, homogeneous time-resolved fluorescence (HTRF)-based detection. 13 The optimal screening method should be high-throughput, simple, cost-efficient, and sensitive to accurately detect high-affinity antibodies. Flow cytometry methods are very costly and not high-throughput; however, they do enable multiplexing by the use of microsphere beads that are distinguished by either size 14 or intrinsic fluorescence.15,16 The ELISA method is the most common method used for hybridoma screening. ELISA is high-throughput and relatively inexpensive, but the signal intensity is mainly influenced by the antibody concentration and scarcely reflects specific affinity. Another concern with the ELISA method is the overreporting of false positives when using concentrated supernatants and low sensitivity for supernatants containing low levels of antibody. Diluting the supernatant, reducing the contact time between the antigen and antibody in the hybridoma supernatant, or increasing the number of washes could potentially reduce the number of false positives; however, it has also been shown that screening a hybridoma library using an ELISA with antibody immobilization has a lower ratio of false positives than an ELISA with antigen immobilization. 17 The MSD platform is similar to ELISA with respect to throughput, but in contrast to ELISA, it allows for multiplexing and has a larger dynamic range.18,19 The MSD’s electrochemiluminescence detection requires a special reader and expensive carbon electrode plates. This substantially increases the cost to conduct a hybridoma screen compared to an ELISA, and as a result the MSD platform has been used rarely for screening of hybridoma supernatants. Furthermore, both MSD and ELISA are technically complex because they can involve multiple reagent addition and washing steps and are laborious. More recently, Rieger and coworkers devised an advanced screening method that makes use of antibody microarrays generated by contact printing of hybridoma supernatants on glass chips coated with capture antibodies. 20 This assay method is based on the specific binding of an analyte–HRP conjugate, and binding is detected using a chemiluminescence readout system. Compared to the ELISA assay, this microarray-based technique requires nanoliter volumes of supernatant and is highly automated, resulting in a significant reduction in the required workload during a screening campaign. The use of HTRF technology has also been successfully used to screen hybridoma supernatants to identify high-affinity antibodies. 21 The HTRF assay method uses fewer components than ELISA and therefore eliminates multiple reagent additions, overnight incubation, and washing steps, making the screening assay homogeneous and very high throughput. However, HTRF technology is not very sensitive, has a small dynamic range, and is severely prone to assay hook effects, which can result in a large number of false negatives when screening concentrated supernatants. 22
A common feature for all of these assay formats is that they all score antibodies based on binding signal strength and do not provide accurate affinities or dissociation rate constants. For some methods, by performing the assay at multiple antigen or antibody concentrations, an affinity constant can be determined, but this would require multiple serial dilutions for each sample, which makes these methods more labor intensive and expensive if screening a large number of antibodies (>1000). Screening antibodies kinetically offers several advantages over common historical platforms like ELISA because the binding interaction can be monitored in real time, during which rates for association (kon) and dissociation (koff) can be analyzed accurately. This allows detection and characterization of low- and high-affinity antibodies that could often be missed using alternative readouts such as ELISA. Furthermore, kinetic screening of hybridoma supernatants would be less prone to reporting false positives or negatives compared to endpoint binding assays because the contact time between the antibody and antigen is very short and each interaction is monitored in real time by an association and dissociation phase. The kinetic size exclusion assay (KinExA) has been used as a highly sensitive and accurate method to identify potent antibodies from hybridoma supernatants. 23 The KinExA uses a small column of beads that are coated with antigen. The hybridoma supernatant is passed through the solid phase, and the amount of antibody captured is detected by fluorescence with a labeled anti-species secondary antibody at a certain flow rate. In practice, the KinExA has been shown to be 10–1000-fold more sensitive than ELISA and is thought to be the most accurate method to determine the affinity of extremely tight binding antibodies.24,25 However, the assay method and instrumentation used for detection are semiautomated and not very high throughput. Furthermore, determination of kon and koff is not straightforward because samples need to be monitored as they approach equilibrium, and this involves multiple injections of each sample. As a result, screening of hybridoma supernatants is not measured kinetically on the KinExA. 23 SPR methods are better suited to measure the kinetics of antibody–antigen interaction. 26 SPR-based biosensors are able to detect, in real time, the kinetics of association and dissociation as proteins absorb and desorb from the sensor surface in the absence of labeled fluorophores (label-free). This is achieved through measurement of the change in the refractive index of the buffer near the sensor, with the change in refractive index being proportional to the protein mass bound to the surface. The fully automated Biacore SPR instruments have been used to screen hybridoma supernatants and reliably identified potent antibodies.9,10 The highest-throughput instrument, which contains 4 independent flow systems, each containing 5 detection spots that can be monitored simultaneously, is capable of screening 800 samples a day at a single antigen concentration. 10 Furthermore, the biosensor surface could be recycled by a simple regeneration step within the assay protocol, giving rise to the possibility of screening hundreds of hybridoma samples using the same sensor surface. This would dramatically reduce antigen consumption and biosensor cost. In contrast to the Biacore SPR instruments, the Biacore Flexchip SPR instrument uses a microarray approach to provide kinetic constants for up to 400 different interactions in a single experiment. 27 The Flexchip system uses a slide composed of an optical grating that is coated with a thin layer of gold onto which proteins are immobilized into a single microarray. The application of an antibody microarray system in which SPR technology is used for signal detection can dramatically increase the throughput compared to Biacore instruments and was successfully used to screen hybridoma supernatants. 28
There are a number of limitations to SPR and other kinetic-based technologies (KinExA). For example, assay configuration and design are extremely important to avoid misinterpretation of reaction data. Historically, hybridoma supernatants are injected over the biosensor immobilized with antigen. A major limitation to this configuration is the possibility of the bivalent antibody crosslinking to the antigen surface, resulting in avidity and leading to a misinterpretation of the dissociation rate constant and subsequent affinity. Also, when using this assay configuration, the antibody concentration in the supernatant needs to be determined to calculate the association rate constant for the binding interaction, and this is rarely done. Lastly, all SPR- and KinExA-based instruments require sophisticated microfluidics to inject samples over the biosensor or solid phase surface. These fluidics are very sensitive to sample composition, and they can fail or be clogged frequently due to improper sample preparation or the accumulation of particulates in the sample or buffers.
In this report, we compared results from screening 2000 hybridoma supernatants using an OctetRED 384 instrument and by a traditional ELISA assay. The OctetRED instrument, which uses BLI, is a high-throughput, label-free platform that allowed us to screen 2000 supernatants kinetically within 15 h. Unlike traditional SPR instruments, the OctetRED uses a plate-based dip-and-read configuration, does not require the use of microfluidics, and therefore is less sensitive to sample and buffer composition compared to traditional flow-based SPR instruments.7,8 Screening results showed that all antibodies selected by ELISA were also selected using the OctetRED platform. However, we identified several antibodies using the OctetRED that were missed in ELISA. Some of these antibodies were missed by ELISA due to low expression but selected by OctetRED because they had a slow-off rate. When these antibodies were purified and retested at a higher concentration (10 nM), binding was observed in ELISA (data not shown). There were several high-expressing antibodies that did not show a binding signal in ELISA. In the OctetRED assay, the antigen is in solution with all binding sites available for antibody binding. In ELISA, the antigen is directly immobilized to the assay plate, and we postulate that certain antibody-binding sites could be masked, resulting in clones being scored as nonbinders.
A detailed analysis of the antibodies selected from the hybridoma screen revealed that binding signal alone does not correlate with affinity ( Fig. 3 ). Importantly, using the OctetRED, we are able to select antibodies using multiple parameters such as binding signal, kon, koff, and KD. In the ELISA assay, antibodies are only selected on a binding signal that is predominantly dependent on the concentration of the antibody in the hybridoma supernatant. Our data also showed that binding signal and kon did not accurately select antibodies with the highest affinity. The only parameter that correlated with affinity (KD) was off-rate ( Fig. 3D ), and a slow-off rate resulted in a high-affinity antibody. This observation is consistent with a recently published data set showing that ranking antibodies based on off-rate is an efficient method of selecting high-affinity antibodies. 29 Lastly, comparison of the affinity (KD) data for the purified antibodies identified in both the ELISA and OctetRED platforms showed that ELISA was not capable of accurately determining the KD value of an antibody less than 100 pM ( Fig. 5 ). Being able to identify the most potent antibodies early during antibody generation is extremely valuable because it significantly reduces the number of antibodies required for in-depth characterization, saving time and money. Kinetically screening antibodies using the OctetRED enables accurate and rapid identification of high-affinity antibodies early in the antibody selection process.
In summary, we have compared a traditional assay against a novel assay technology to screen monoclonal antibodies. We developed a workflow for high-throughput screening of antibody containing hybridoma clones. In this study, we used the OctetRed 384 system as our kinetic screening platform. The OctetRED 384 allowed us to screen 16 supernatants in parallel because it contained 16 channels. In a typical screening experiment, each 96-well plate containing hybridoma supernatants was screened in 45 min. The most recent Octet system, Octet HTX, offers higher throughput because it can monitor up to 96 biosensors simultaneously, and this would allow us to screen each 96-well plate in less than 8 min. To reduce the cost of screening on the Octet systems, the biosensor tips could be regenerated and reused. Depending on the sample and the coated surface on the biosensor tip, we have regenerated the sensor surface and reused it up to 20 times. Although regeneration of biosensor tips was not included in the workflow described in this study, it could easily be incorporated, but the time required to screen each assay plate would increase. Lastly, in this study, we have demonstrated that screening antibodies kinetically is an efficient method for identifying high-affinity antibodies. The off-rate kinetic parameter correlates most accurately with antibody affinity and should be used to rank antibodies during a primary screen.
Footnotes
Acknowledgements
We thank Dharmaraj Samuel and Leanna Lagpacan for antibody purification. We also thank David Koditek, Sarah Wise, Annapurna Sapre, Jayshali Lad, and Leah Lad for encouragement and helpful discussions during the preparation of the manuscript.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors, except Terrence Hui, are full-time Gilead Sciences employees.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: All work reported in the publication was sponsored by Gilead Sciences.