
Abstract
Highly sensitive, high-throughput assay technologies are required for the identification of antibody therapeutics. Multiplexed assay systems are particularly advantageous because they allow evaluation of several parameters within 1 well, increasing throughput and reducing hands-on laboratory time.
The mirrorball (TTP Labtech), using high-throughput fluorometric microvolume assay technology, offers simultaneous scanning with up to 3 lasers as well as laser scatter detection. This makes the mirrorball especially suitable for the development of highly sensitive and multiplexed assays.
We have developed bead- and cell-based binding assays that demonstrate how the multilaser capability of the mirrorball can be exploited to enhance assay sensitivity. In addition, using the multilaser simultaneous scanning capability, we have established multiplexed cytokine quantitation assays and antibody–cell binding assays.
Our results demonstrate the potential utility of this technology to improve the sensitivity and efficiency of biologics screening, resulting in streamlining of the lead antibody selection process.
Introduction
The discovery of new antibody therapeutics, using phage display and hybridoma approaches, typically requires screening tens of thousands of antibodies, or antibody fragments, to identify potential leads with the desired binding specificity and activity profile. The assays used for screening are ideally homogeneous, mix-and-read assays to enable higher throughput with reduced hands-on laboratory time. One way of increasing assay throughput is to multiplex assays (i.e., to generate multiple results from 1 well). This enables comparison of a sample in multiple assays within 1 well, reduced reagent and sample usage, saved time, reduced costs, and increased efficiency of screening.
Assay technologies traditionally used for antibody screening, such as enzyme-linked immunosorbent assay (ELISA) or flow cytometry, can be highly sensitive but require multiple wash steps, thereby reducing assay throughput. The introduction of high-throughput flow cytometry 1 has addressed some of the issues associated with this method; however, the development of homogeneous flow cytometry assays can still present a challenge due to high background fluorescence. Fluorometric microvolume assay technology (FMAT), described in 1999,2,3 presented researchers with a homogeneous, mix-and-read assay solution for measurement of binding to cells or beads. Numerous studies have described the use of FMAT for measuring the binding of antibodies to target antigens captured on beads or expressed on the surface of cells.4,5 The measurement of binding to cell surface proteins enables the screening of antigens in a native conformation that is particularly important for multispanning membrane targets. In addition, quantification of soluble proteins using FMAT has been described in single6,7 and multiplexed 8 assays.
FMAT uses a laser excitation source and photomultiplier tubes (PMTs) for detection of fluorescence emission within a fixed depth of focus. The electrical signal from the PMT is converted to a digital image of the cells or beads on the bottom of the plate. The analysis of this digital image allows for detection only of areas of concentrated fluorescence on cells or beads, with minimal background fluorescence from unbound detection reagent, resulting in a highly sensitive homogeneous assay format. Unlike flow cytometry and ELISA methods, binding to both adherent and nonadherent cells can be measured using FMAT. In addition, assays using this technology can be performed in 96-, 384-, and 1536-well microtiter plates, thus reducing reagent usage, cell number, and sample volume requirements.
The mirrorball (TTP Labtech, Melbourn, UK) has been developed using a similar principle and proprietary optics. A key advantage of the mirrorball is that it can be equipped with multiple excitation lasers, including 405 nm, 488 nm, or 640 nm excitation, in addition to a laser for scatter detection of beads. Simultaneous scanning of all lasers is a standard feature, significantly reducing read time and increasing flexibility while allowing rapid assessment of multiple binding events. The capability of the mirrorball to read multiple lasers simultaneously lends itself to the possibility of multiplexing assays. 9 In addition, the mirrorball allows whole well scanning; therefore, the read area is not restricted to a small portion of the total available area, which results in a reduced cell number requirement.
Using the mirrorball, we have evaluated a selection of typical assays that may be used for identification of antibody therapeutics, including receptor–ligand binding assays (to determine the inhibitory activity of an antibody) and receptor–antibody binding assays (e.g., to identify binding antibodies for purposes such as the development of antibody–drug conjugates). Assay sensitivity is a critical factor in these assays, particularly for the screening of phage display selection outputs where binders may have low affinity for the target. We have investigated ways to maximize assay sensitivity using the flexible, multilaser capabilities of the mirrorball system. In addition, we have assessed the multiplexing capability of the mirrorball for receptor–antibody binding assays and for simultaneous quantification of multiple cytokines within a single sample. These assays would enable the rapid assessment of antibody cross-reactivity or specificity for the target of interest in a single well and increase the amount of information gained from testing of a single sample.
Materials and Methods
A mirrorball system equipped with 488 nm and 640 nm excitation lasers was used for assay development. Four channels are available for detection of emitted fluorescence—FL1 (488–540 nm), FL2 (560–610 nm), FL3 (650–690 nm), and FL4 (700–780 nm).
Antibody Labeling
Antihuman insulin monoclonal antibody (IN05) (ab46707; Abcam, Cambridge, UK) was labeled with AlexaFluor647 according to the kit manufacturer’s instructions (A20186; Invitrogen, Carlsbad, CA). Anti-interleukin-6 (anti-IL-6) (MAB208; R&D Systems, Minneapolis, MN), anti-tumor necrosis factor alpha (anti-TNF-α) (MAB610; R&D Systems), and anti-interleukin-8 (anti-IL-8) (MAB208; R&D Systems) monoclonal antibodies were labeled with DyLight488 using labeling kits according to the manufacturer’s instructions (53025; Thermo Scientific, Waltham, MA; or 322-0010; Innova Biosciences, Cambridge, UK).
Preparation of Cell Supernatants from THP-1 and Peripheral Blood Mononuclear Cells
THP-1 cells were plated into 12-well plates at a density of 1×106 cells per well in RPMI-1640 medium (61870; Gibco, Carlsbad, CA) containing 10% fetal bovine serum (19106C; SAFC, St. Louis, MO). Cells were differentiated by the addition of 0.1 µg/mL Phorbol 12-myristate 13-acetate (P1585; Sigma-Aldrich, St. Louis, MO) for 48 h, washed once with Dulbecco’s phosphate buffered saline (PBS) (14190; Gibco), then stimulated with 1 µg/mL lipopolysaccharide (LPS) (LSM33-02; InvivoGen, San Diego, CA) for 24 h at 37 °C, 5% CO2. Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized whole human blood. The blood (20 mL) was layered onto 10 mL of Ficoll-Paque PLUS (17-1440-02; GE Healthcare, Little Chalfont, UK) and centrifuged at 400 g for 25 min. The buffy coat was gently removed, washed twice with PBS, and resuspended at 1×106/mL in RPMI-1640 medium containing 10% FBS and 1× penicillin–streptomycin. PBMCs (1×107) were stimulated with 1 µg/mL LPS (tlrl-eblps; InvivoGen) for 24 h at 37 °C, 5% CO2. Cells were removed by centrifugation. All supernatants were stored at −80 °C until use.
Bead-Based Binding Assays
Insulin–biotin (I5636; Sigma-Aldrich) at various concentrations was incubated with antihuman insulin AlexaFluor647 antibody at 0.4 nM and streptavidin-coated 6.7 µm polystyrene beads (SVP-60-5; Spherotech, Lake Forest, IL) (5000 per well). Incubation was performed in a mirrorball assay plate, a black-walled, clear-bottom, nonbinding surface, 384-well microtiter plate (3655; Corning, Corning, NY) for 5 h at room temperature in the dark followed by 18 h at 4 °C. All reagents were prepared in bead assay buffer comprising PBS containing 0.1% bovine serum albumin (BSA) (A9576; Sigma-Aldrich) and 0.1% Tween20 (P2287; Sigma-Aldrich).
Bead-Based Cytokine Quantitation Assays
Streptavidin-coated fluorescent polystyrene beads (PAK-5067-5A; Spherotech) were incubated for 1 h at room temperature with 0.3 nM biotinylated polyclonal antibodies against IL-6 (BAF206; R&D Systems), IL-8 (BAF208; R&D Systems), or TNF-α (BAF210; R&D Systems) in bead assay buffer. Beads were washed 3 times with PBS by centrifugation and resuspended in bead assay buffer. For quantification assays, cell supernatants were diluted to ensure that concentrations of cytokine were within the dynamic range of the assay and incubated for 3–4 h with beads (1000 per well) and 1 nM DyLight488-labeled antibody in a mirrorball assay plate. Standard curves for each cytokine were generated using a range of concentrations of purified IL-6 (206-IL; R&D Systems), IL-8 (208-IL; R&D Systems), or TNF-α (210-TA; R&D Systems). Data were analyzed using a 4-parameter curve fit, and cytokine concentrations were interpolated from the standard curve.
Quantikine ELISA kits for IL-6 (D6050; R&D Systems), IL-8 (D8000C; R&D Systems), and TNF-α (DTA00C; R&D Systems) were carried out according to the manufacturer’s instructions.
Cell Staining
Vybrant cell dye (V22889; Invitrogen) was prepared in DMSO (D2650; Sigma-Aldrich) and added to cells prepared at 1×106 cells per mL in Dulbecco’s modified eagle medium (DMEM) (41966; Gibco). For cells with no stain, the equivalent volume of DMSO was added. Cells were incubated for 30 min at 37 °C, 5% CO2, before being washed twice by centrifugation. Cells were resuspended in cell assay buffer comprised of Hank’s Balanced Salt Solution (HBSS) (H8264; Sigma-Aldrich) with 0.1% BSA and 0.01% sodium azide (S8032; Sigma-Aldrich).
Cell-Based Binding Assays
Receptor–Ligand Binding Assay
Human embryonic kidney (HEK 293) cells, stably transfected with the complement receptors C5aR1 or C5aR2, and stained with 10 nM DiO Vybrant cell dye (1000 cells per well), were incubated with C5a peptide, custom synthesized with an AlexaFluor647 fluorophore incorporated (Almac, Craigavon, UK), at various concentrations. All reagents were prepared in cell assay buffer. Incubation was performed in a mirrorball assay plate for 18 h at room temperature in the dark.
Receptor–Antibody Binding Assay
Chinese hamster ovary (CHO) cells stably transfected with human formyl peptide receptor (FPR) or cynomolgus FPR were stained with 10 nM DiO cell dye or 60 nM Dil cell dye. Cells (1000 per well) were incubated with various concentrations of monoclonal antibodies raised against FPR (MedImmune, Gaithersburg, MD) and antihuman Fc AlexaFluor488 antibody (A11013; Invitrogen) at 2 nM. All reagents were prepared in cell assay buffer. Incubation was performed in a mirrorball assay plate for 18 h at room temperature in the dark.
Results and Discussion
Effect of Independent Definition of Beads and Cells on Assay Sensitivity
Increased assay sensitivity may be an advantage for the identification of low-affinity binding antibodies and for reducing reagent usage, and it may extend the application of the mirrorball to cell lines with lower target expression. When using this technology, assay sensitivity can be affected by how the beads or cells are defined. For example, if defined by a fluorescent binding partner, the bead or cell count will reduce as the concentration of the binding partner is reduced. This may negatively affect the assay sensitivity at low concentrations. If the beads or cells are defined independently of the binding, for example using the scatter laser for beads or a fluorescent label in the bead or cell membrane, a consistent count is obtained, thus potentially increasing sensitivity. The multilaser capability of the mirrorball enables this independent bead or cell definition whilst simultaneously recording binding to the bead or cell.
To investigate this, binding-dependent and -independent methods of bead definition on the mirrorball were compared using a high-throughput, bead-based, antibody-binding assay. Biotinylated insulin was captured on the surface of a streptavidin-coated polystyrene bead, and binding of an AlexaFluor647-labeled anti-insulin antibody measured. The presence of the bead was determined by using either the fluorescent antibody binding or the independent scatter laser ( Fig. 1a ). The triggering function on the mirrorball was used to differentiate these 2 methods. 10 For independent bead definition, the trigger was set in the scatter channel; a fluorescent binding signal is only recorded when the presence of a bead is first detected (a signal greater than 4 standard deviations higher than background). For binding-dependent bead definition, the trigger was set in the FL3 channel; all fluorescence is recorded.
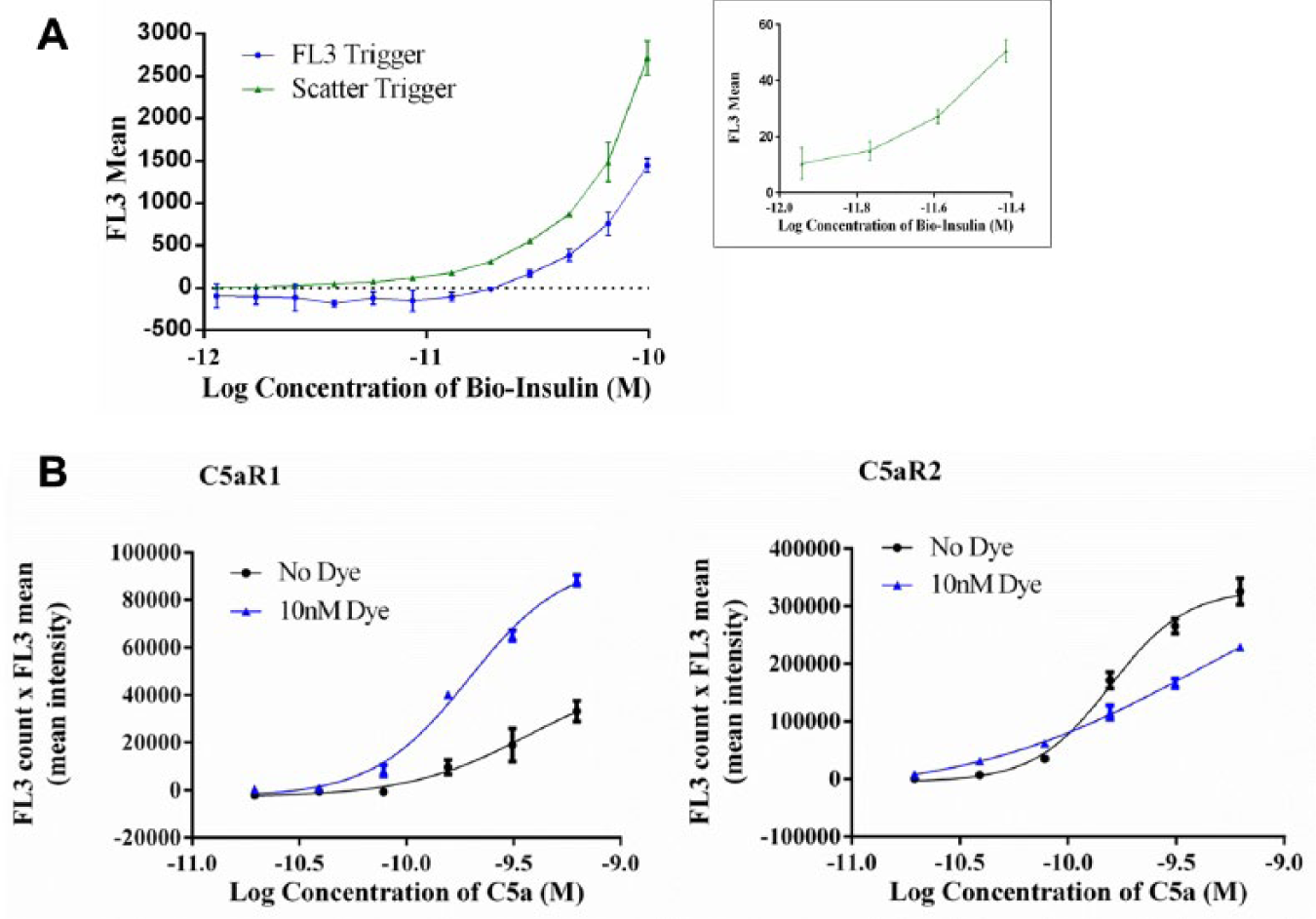
Effect of independent definition of beads or cells on assay sensitivity. (
The results demonstrate that if the binding of the fluorescent antibody is used to define the bead population, the lower limit of sensitivity (the lowest concentration of biotinylated insulin required to give a signal that is 3 standard deviations higher than the background) is 29 pM (n = 3). If scatter is used to independently define the bead population, the lower limit of sensitivity is 2 pM (n = 3). Therefore, a 15-fold increase in assay sensitivity was observed when the beads were independently defined using the scatter laser.
The principles of independent definition, as demonstrated by the bead-binding assay, were applied to a high-throughput receptor–ligand binding assay using cell lines overexpressing C5aR1 or C5aR2. Binding of AlexaFluor647-labeled complement C5a to the cells was measured in the FL3 channel using the 640 nm laser for excitation. A fluorescent lipophilic membrane stain, DiO, which has an emission measured in the FL1 channel following 488 nm laser excitation, was used for independent definition of the cells. A comparison of cell definition using the binding of AlexaFluor647-labeled C5a and independent object definition using the DiO fluorescent cell stain was performed ( Fig. 1b ). For independent cell definition, the trigger was set in the FL1 channel; a fluorescent binding signal is recorded only when the presence of a dyed cell is first detected (a signal greater than 4 standard deviations higher than background). For binding-dependent cell definition, the trigger was set in the FL3 channel; all fluorescence is recorded.
Independent definition of the C5aR1 cells with the DiO cell stain resulted in an 8-fold improvement in sensitivity, with a lower limit of sensitivity of 20 pM (n = 2) compared to 160 pM (n = 2) when cells were defined using the fluorescently labeled C5a. A twofold improvement in the sensitivity was achieved for the C5aR2-expressing cells, with a lower limit of 20 pM (n = 2) using an independent DiO-mediated definition compared to 40 pM (n = 2) using the fluorescently labeled C5a. In this case, a lower maximum signal is observed in the presence of the cell stain, due to an increase in background requiring more stringent gating. When the fluorescent signal from the ligand binding is high, such as for the C5aR2, the independent definition of cells may not have such an impact on the sensitivity of the detection. When the fluorescent signal from ligand binding is low, such as for the C5aR1, the definition of the cells is compromised, and thus independent definition of cells improves assay sensitivity. A cell-based receptor–ligand binding assay with increased sensitivity may allow use of cell lines or primary cells in which a low fluorescent assay signal is encountered. In the case of the C5aR1 binding assay, independent definition of the cells using a fluorescent membrane dye enabled the development of an assay that otherwise would not be viable due to the low fluorescent assay signal. Independent definition has the potential to be advantageous for other binding assays with low assay signals; however, this would need to be determined empirically.
A Multiplexed Cytokine Quantitation Assay
We have shown that when using the multilaser capability of the mirrorball, independent definition of cells may increase assay sensitivity. This capability can also be exploited for multiplexing, allowing the generation of more information about a sample from 1 well, saving time and reagents. These types of assays have been described previously, 8 in which a single laser was used for excitation of 2 fluorophores with different emission wavelengths, detected via 2 PMTs. The multiple lasers available in the mirrorball, however, open up the possibility of increased multiplexing options.
Multiplexing is particularly advantageous for high-throughput measurement of cytokines that are released from a cell population on stimulation. Cytokine quantitation is commonly performed using ELISA methods, which involve multiple steps and can be time-consuming; therefore, a homogeneous mix-and-read assay is preferable. In addition, cells may release multiple cytokines in response to stimuli. Therefore, having the ability to quantify multiple cytokines in the same assay would not only provide valuable information but also reduce time and sample requirements. The key challenge for the successful development of a multiplexed assay is the differentiation of binding; quantification of each cytokine must be specific, with no interference from any other cytokine.
The use of beads of different sizes has been suggested as a suitable method for multiplexing assays on plate-based cytometers. 8 This method was evaluated on the mirrorball using the scatter laser to identify streptavidin-coated bead populations ranging from 3 to 17.9 µm in size. A number of parameters were measured, including depth, width, volume, area, and perimeter ( Fig. 2a for representative perimeter and area data). The bead populations displayed a large amount of overlap, and therefore the ability to multiplex is restricted to a combination of 2 bead sizes. As an alternative, we therefore examined the use of fluorescence, instead of size, to identify bead populations.
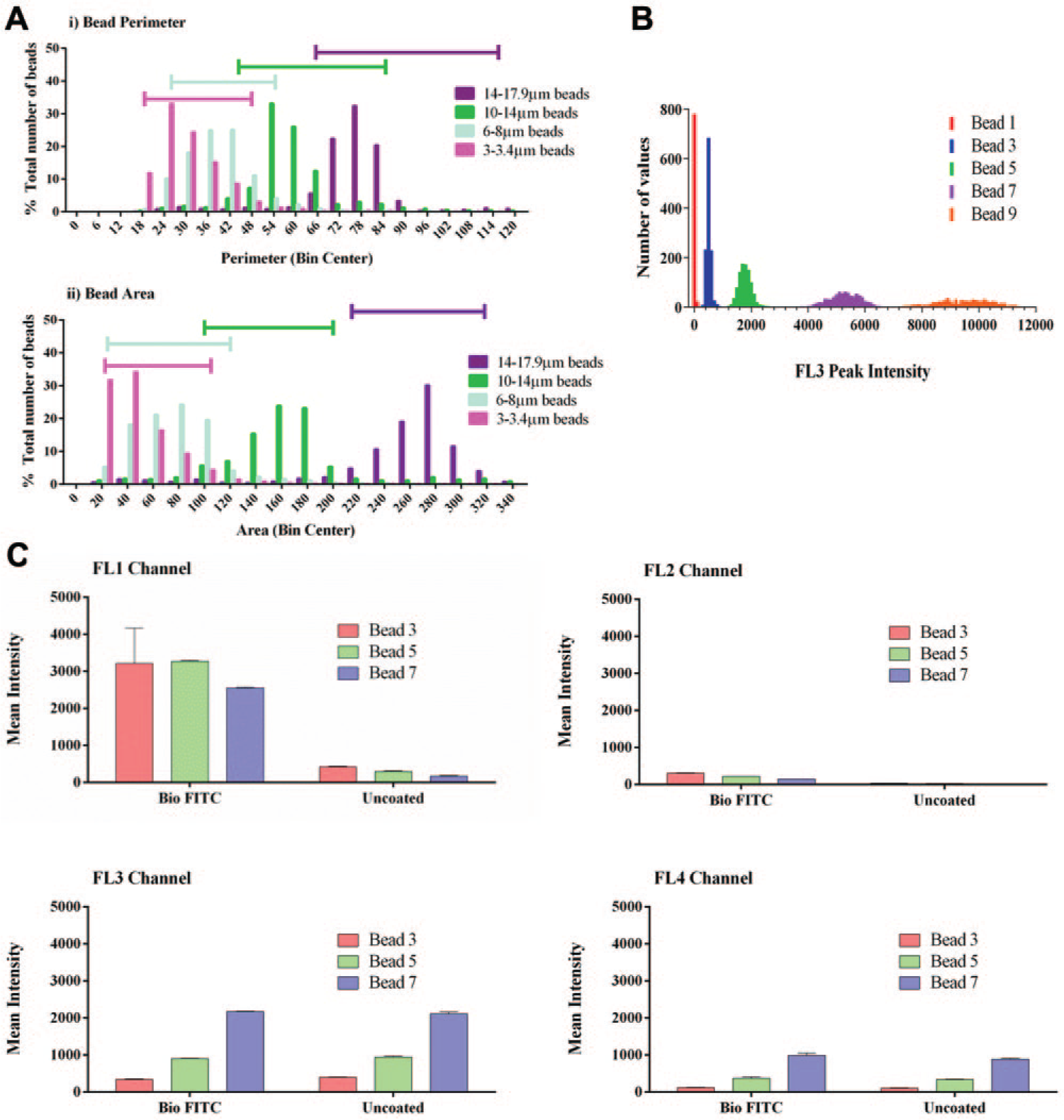
Differentiation of beads for multiplexing. (
We evaluated the use of streptavidin-coated polystyrene beads, impregnated with different intensities of a fluorophore that emits in the FL3 channel following excitation with the 640 nm laser. Four of the fluorescent bead types and the nonfluorescent beads show distinct populations based on intensity ( Fig. 2b ). Because overlapping emission wavelengths from fluorophores with asymmetrical spectra can result in fluorescence bleed-through to neighboring detection channels, we determined the amount of this bleed-through from beads with different fluorescent intensities. To mimic the combination of fluorophores in the final assay format, biotinylated fluorescein isothiocyanate (FITC) was captured on the streptavidin bead, and the emission in each of the channels compared to that of uncoated beads following 488 nm and 640 nm laser excitation ( Fig. 2c ). The fluorescence emission in the FL3 channel following 640 nm excitation was the same for the uncoated and coated beads; thus, the FITC dye did not interfere with the detection of the dye-impregnated bead. As expected, there was high emission for the biotinylated FITC–coated beads in the FL1 channel following 488 nm laser excitation. The low fluorescence in the FL1 channel (uncoated beads) and FL2 channel (all beads) showed that there was minimal bleed-through from the beads alone or from the FITC-coated beads to these channels. The increased fluorescence in the FL4 channel, particularly from the beads with higher FL3 intensity, indicated there was some bleed-through to this channel.
The distinct populations evident from the different intensities of fluorophore impregnated in these beads, and the minimal fluorescence bleed-through between the FL1 and FL3 channels, indicated that these beads could potentially be used to multiplex up to 5 different cytokine quantitation assays.
Comparison of Single and Multiplexed Cytokine Quantification Assays
Using the streptavidin-coated fluorescent beads, we developed single and multiplexed assays for the quantification of the cytokines IL-6, IL-8, and TNFα using the mirrorball high-sensitivity cytometer ( Fig. 3a ). A comparison of the mirrorball single and multiplexed assay formats to a commercially available ELISA method was performed, evaluating assay sensitivity and dynamic range.
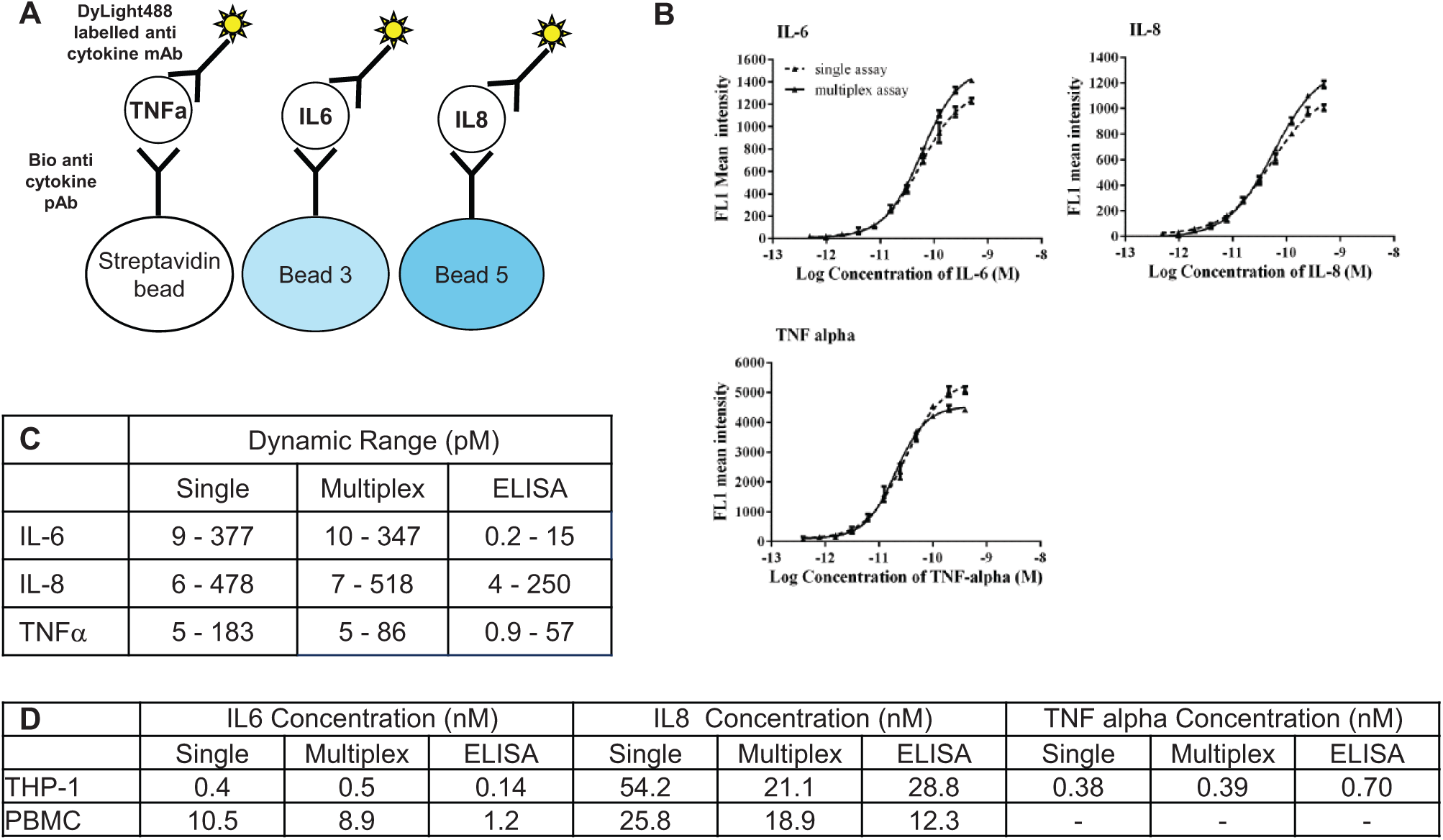
Quantification of cytokines using single and multiplexed bead-based assays. (
Standard curves generated for all cytokines tested were comparable between single and multiplexed assays on the mirrorball ( Fig. 3b ). Comparison to the ELISA quantitation method indicated that there were differences in sensitivity and dynamic range between the different assay formats ( Fig. 3c ). ELISA methods may be expected to show greater sensitivity due to the nonhomogeneous nature of the method. The IL-6 quantification ELISA method had a large increase in sensitivity over the mirrorball assay but a much reduced dynamic range. The TNFα quantification ELISA had a small increase in sensitivity over the mirrorball assay, with a similar dynamic range. The IL-8 quantification assays were comparable in terms of both assay sensitivity and dynamic range.
The comparison between ELISA and mirrorball methods was extended to include quantification of cytokine levels in supernatants from stimulated cells. THP-1 cells, a human monocytic cell line, and a PBMC preparation were stimulated with lipopolysaccharide (LPS), and the concentration of IL-6, IL-8, and TNFα in the cell supernatant was measured using ELISA, the single mirrorball assay, or the multiplexed mirrorball assay ( Fig. 3d ). The THP-1 cells released quantifiable levels of IL-6, IL-8, and TNFα, and PBMCs released quantifiable levels of IL-6 and IL-8. The concentration of these cytokines determined using all of the alternative methods was comparable.
The 3-plex mirrorball assay that has been developed has several benefits over existing cytokine quantification methods. Three cytokines are quantified within 1 well; therefore, more information is gained from a reduced sample volume. In addition, the increase in dynamic range for certain cytokines may allow a truly homogeneous cytokine quantification assay to be performed. Often, multiple dilutions of sample are required to ensure that cytokine concentrations are within the linear range of the assay. Due to the larger dynamic range of the mirrorball assay, sample dilution is not necessary, and the quantification assay components can be added directly to the stimulated cell, thereby reducing the need for liquid-handling and dilution steps and increasing throughput.
A Multiplexed Antibody–Cell Binding Assay
Bead-based antibody binding assays and cytokine quantification assays are both useful assay formats for the discovery of antibody therapeutics; however, there are occasions when an antibody–cell binding assay is preferred. These assays measure binding of an antibody to a receptor target in its native conformation, within the extracellular membrane environment, and they can be particularly useful for the identification of antibodies to receptors in which the ligand is unknown or when it is not possible to use a functional assay in the primary screen.
It is advantageous to incorporate evaluation of species cross-reactivity and specificity into the antibody screening process as early as possible to facilitate drug development. Often, a cell line transfected with target will be used for screening, and therefore selectivity over the nontransfected cell line must be evaluated. In addition, antibodies to a particular target can demonstrate such a high level of specificity that attaining species cross-reactivity is challenging. Having the ability to evaluate antibody–cell binding to multiple cell lines in the same assay would increase the assay efficiency, potentially reduce assay-to-assay variability, and save time and costs. As with multiplexed bead-based assays, the key challenge for the successful development of a multiplexed antibody–cell binding assay is the ability to distinguish events in each cell line.
We have investigated the possibility of achieving cell differentiation using fluorescence and exploiting the multilaser capabilities of the mirrorball. Cell identification can be achieved using the lipophilic cell membrane stains DiO and Dil (Invitrogen). These dyes absorb at wavelengths of 484 nm and 549 nm and emit at wavelengths of 501 nm and 565 nm, respectively, and therefore can be detected in the FL1 and FL2 channels. Stained cells can then be combined with an antibody detection reagent containing a fluorophore that is measurable in the FL3 channel ( Fig. 4a ) to determine on which cell the antibody is binding.
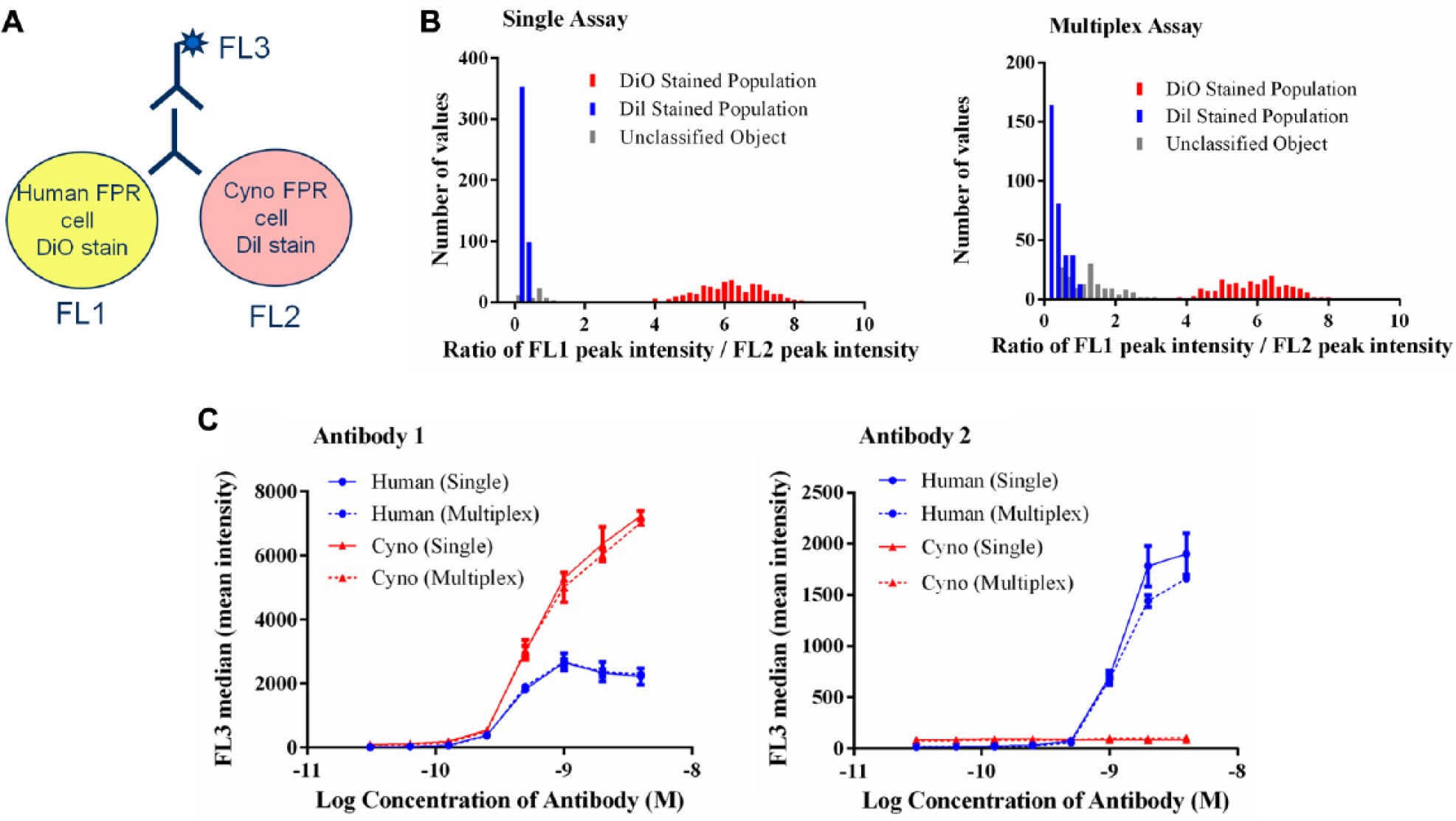
A multiplexed antibody–receptor binding assay. (
Due to the slightly overlapping emission spectra for DiO and Dil, there is a risk of fluorescence bleed-through between the FL1 and FL2 channels. To ensure the identification of separate cell populations, the ratio of the peak intensity of FL1–FL2 is analyzed using the mirrorball. Higher fluorescence in the FL1 channel (DiO-stained cells) will yield a high ratio value, and conversely higher fluorescence in the FL2 channel (Dil-stained cells) will yield a low ratio value. We have demonstrated that gating based on the FL1–FL2 ratio allows differentiation between the 2 cell populations ( Fig. 4b ). In a single assay format, there are 2 clear populations and when multiplexed there is some degree of overlap, but this does not preclude differentiation of populations.
We have used the DiO and Dil cell stains to develop multiplexed antibody–cell binding assays using the mirrorball. Two antibodies, a human–cynomolgus cross-reactive antibody (Antibody 1) and a human-only binding antibody (Antibody 2), were evaluated for binding to cells expressing human or cynomolgus FPR, and a comparison of single and multiplexed assay formats was performed. Populations were defined in the Cellista software for the DiO-stained cells with a high FL1–FL2 ratio and for the Dil-stained cells with a low FL1–FL2 ratio. Fluorescence from the AlexaFluor647 secondary detection, in the FL3 channel, was measured for each population to differentiate the binding signals. Antibody 1 produced an FL3 signal in both FL1 and FL2 populations, confirming this antibody bound both human and cynomolgus FPR ( Fig. 4c ). Antibody 2 gave an FL3 signal in the FL1 population only, confirming this antibody bound human FPR only. A comparison of the single and multiplexed assays demonstrated that there was no discernable difference between the 2 assay formats.
With this multiplexed antibody–cell binding assay, we have shown the mirrorball can be used to evaluate binding to multiple cell lines in 1 well. This leads to the possibility of evaluating antibody cross-reactivity and selectivity with increased throughput, a reduced timeframe using fewer cells, less sample, and reduced amounts of reagents. An increased throughput assay for cross-reactivity and selectivity will increase the efficiency of the screening process, ensuring only antibodies with the desired properties are taken forward and minimizing the requirement for secondary characterization assays.
In summary, we have demonstrated how the mirrorball, and in particular the multilaser capability of the mirrorball, can be used for antibody drug discovery. We have shown how assays can be optimized for improved sensitivity using independent definition of objects, using either scatter for beads or fluorescent dyes for cells. This could enable the development of assays in which the binding signal may be expected to be low, such as for primary cell lines. In addition, the 2 excitation lasers and 4 detection channels have enabled the development of both bead-based and cell-based multiplexed assays amenable to high-throughput screening. Here, we demonstrated the multiplexing of 2 antibody–cell binding assays (using different dyes) and 3 cytokine quantitation bead assays (using the same dye at different intensities) in biologics discovery. Such multiplexed assays may be applied to screen for and characterize potential therapeutics for biological function, species cross-reactivity, and selectivity. Time is of the essence within drug discovery, in which more sensitive, multiplexed assays, as made possible by the mirrorball, may lead to more efficient, more streamlined screening processes and earlier identification of antibody therapeutics.
Footnotes
Acknowledgements
We would like to acknowledge the help given from TTP Labtech, in particular from Paul Wylie, with the development of assays on the mirrorball and analysis of multiplexed data.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Financial support was through authors employment at MedImmune.