
Abstract
Bromodomain protein 4 (BRD4), a member of the bromodomain and extra-terminal (BET) protein family, acts as a central element in transcriptional elongation and plays essential roles in cell proliferation. Inhibition of BRD4 binding to acetylated histone tails via its two bromodomains, BD1 and BD2, with small-molecule inhibitors has been shown to be a valid strategy to prevent cancer growth. We have evaluated and established two novel assays that quantify the interaction of transfected BRD4 BD1 with chemical inhibitors inside cultured cells. Both methods are based on the principle of ligand-induced protein stabilization by which the binding of a small-molecule inhibitor stabilizes intracellular BRD4 BD1 and protects it from proteolytic degradation. We demonstrate the universal character of this principle by using two orthogonal, highly sensitive detection technologies for the quantification of BRD4 BD1 levels in cellular lysates: enzyme fragment complementation and time-resolved fluorescence resonance energy transfer (TR-FRET). Upon optimization of both assays to a miniaturized high-throughput format, the methods were validated by testing a set of small-molecule BET inhibitors and comparing the results with those from a cell-free binding assay and a biophysical thermal shift assay. In addition, point mutations were introduced into BRD4 BD1, and the corresponding mutants were characterized in the TR-FRET stabilization assay.
Introduction
Ligand-induced protein stabilization is a well-known phenomenon already described in the early 1970s.
1
A broad range of biochemical and biophysical techniques have been developed to determine protein stability to study the structure, folding, and ligand-binding properties of proteins. The use of ligand-induced protein stabilization has been explored as a general strategy for drug discovery applications during the past decade. For instance, thermal denaturation of isolated proteins has been used to identify and characterize small-molecule inhibitors.
2
However, cell-based methods to directly monitor drug-target interaction in a physiological environment are challenging and time-consuming. Intracellular protein turnover rates vary from minutes to days, and half-lives of individual proteins are controlled by a complex degradation system that has an essential role in the regulation of numerous cellular processes. Recently, novel proteomic approaches have made use of ligand-induced protein stabilization to identify drugs and also their “off-target” effects in cell lysates by treating with proteolytic enzymes for a short period or by determining the cellular thermal stability.3–6 We here report the characterization and comparison of two cellular ligand-induced protein stabilization assays as easy and fast screening methods. The common assay principle is based on protein quantification after small-molecule compound-induced protein stabilization inside living cells (
Fragment complementation of the bacterial β-galactosidase (β-Gal) enzyme is a well-established method for a number of different assays used in drug discovery. 7 Based on the well-characterized mechanism known as α-complementation, a large, catalytically inactive β-Gal protein fragment called enzyme acceptor (EA) complements with a small enzyme donor (ED), which consists of a truncated N-terminal peptide corresponding to the deleted β-Gal region absent in EA. The two β-Gal fragments complement with high affinity (KD = ~1 nM) in the cell to reconstitute the β-Gal enzyme. This catalytically active β-Gal enzyme converts its substrate to active luminescent products with a high turnover rate, resulting in highly sensitive assays. In the new enzyme fragment complementation (EFC) cellular assay reported here, a small ED peptide (ePL = enhanced ProLink) is fused to the bromodomain 1 (BD1) of bromodomain protein 4 (BRD4). The amount of ePL-BRD4 protein can thereafter be measured by adding the inactive β-Gal EA protein to a cellular lysate.
As a second technology, we used TR-FRET to quantify the concentration of tagged BRD4 in cellular lysates. TR-FRET detection is a well-established and robust technology frequently used in drug screening. 8 It combines the advantages of FRET with a time-resolved measurement, thus enabling highly sensitive detection of proteins in cellular lysates.
BRD4 belongs to the bromodomain family with 46 human members containing in total 61 diverse bromodomains. 9 BRD4 mainly interacts with acetylated histone H3 and H4 tails and constitutes together with BRD2, BRD3, and BRDT the bromodomain and extra-terminal domain (BET) protein subfamily. This group is characterized by two N-terminal bromodomains and an extra-terminal recruitment domain. Bromodomains form a central hydrophobic cavity containing a highly conserved asparagine residue, which forms an essential hydrogen bond for the interaction with acetylated lysine residues. The implication of BET proteins and mainly of BRD4 in cancer and in inflammatory processes has led to intensive efforts toward the identification of small-molecule inhibitors.10,11 The triazolo-diazepines JQ1 and I-BET762 were among the first small molecules described to potently and selectively inhibit BET binding to acetylated histones, leading to strong antitumor activity in preclinical models. Very recently, a BET inhibitor has shown first signs of efficacy in the clinic, and several new inhibitors with different chemical scaffolds are currently being clinically characterized. Intensive efforts to identify compounds with increased target-binding properties and improved cellular activity are currently ongoing in several research groups worldwide.
For a better assessment of target engagement, we set up and compared ligand-induced protein stability assays for a quantitative determination of BRD4 protein stabilization in cells as a new technique to better understand the cellular mode of action of inhibitors. We established and compared two high-throughput compatible detection systems as easy-to-use protein quantification methods well applicable for HTS in the areas of drug discovery, protein design, and mutation analysis.
Materials and Methods
Cell Culture
The HEK293 cell line stably expressing BRD4 BD1 with an enhanced ProLabel (ePL-BRD4, InCell Hunter; DiscoveRx, Fremont, CA) and HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium including glutamine (DMEM + GlutaMax; Gibco, Life Technologies, Carlsbad, CA) and supplemented with 5% fetal bovine serum (FBS; Biochrom AG, Berlin, Germany). HEK293-6E cells (Yves Durocher, National Research Council, Ottawa, Ontario, Canada) were grown in suspension at gentle shaking in FreeStyle F17 medium (Gibco, Life Technologies) supplemented with 4 mM L-alanyl-L-glutamine (Biochrom AG) and 0.1% Pluronic (Gibco, Life Technologies). Cells were continuously grown at 37 °C in 95% humidity and a 5% CO2 atmosphere.
Compound Preparation
Compounds dissolved in DMSO were diluted with 100% DMSO in a 384-well microtiter plate (MTP) using a microplate-pipetting system (Precision; BioTek, Winooski, VT). Then, 50 nL of diluted compounds was transferred to final 384- or 1536-well MTPs by a plate replication and reformatting system (HummingBird; Cartesian Technologies, Irvine, CA).
EFC Protein Stabilization Assay
Unless otherwise described, cells were detached by accutase, resuspended in culture medium, and centrifuged at 800 g for 5 min. The supernatant was discarded and the cell pellet was resuspended in assay medium (DMEM high glucose [4.5 g/L], PAA [Pasching, Austria] supplemented with 5% FBS [Biochrom AG] and 1 mM glutamine [Sigma-Aldrich, St. Louis, MO]). After seeding 5000 cells per 12-µL assay medium into 384-well MTPs (Greiner Bio-One, Monroe, NC) containing test compounds, incubation was performed overnight for 24 h at 37 °C in 5% CO2 and 95% humidity. In the final step, cells were lysed with 10 µL InCell Hunter Detection Reagent (DiscoveRx) containing the inactive β-Gal enzyme fragment (enzyme acceptor), lysis buffer, and substrate. After a 30-min incubation, the chemoluminescence was measured with a microplate reader (Pherastar; BMG Labtech, Offenburg, Germany) in luminescence mode. Dose-dependent stabilization by JQ1 or other compounds was evaluated using the four-parameter nonlinear regression algorithm (“log(Agonist) vs. response – Variable slope”) of the GraphPad Prism software (GraphPad Software, La Jolla, CA).
TR-FRET Protein Stabilization Assay
Unless otherwise described, transiently transfected HEK293 cells (1000 ng/ml DNA coding for tagged BRD4 BD1 or mock DNA containing pBluescript SKII; Stratagene, La Jolla, CA), transiently transfected HEK293-6E cells (10 ng/mL DNA or mock DNA), or a stable HEK293 cell pool (transfected with 1000 ng/mL DNA and selected with 400 µg/mL hygromycin B, PAA) were used for the TR-FRET assay. Then, 5000 cells per 10 µL assay medium (DMEM high glucose [4.5 g/L], PAA supplemented with 5% FBS; Biochrom AG) were seeded into a 384-well MTP containing test compounds. The 384-well MTP was incubated for 24 h at 37 °C in 5% CO2 and 95% humidity. Then, 10 µL lysis buffer (HTRF conjugation/lysis buffer; Cisbio HTRF, Marcoule, France) containing Anti Tag1 D2 (final molarities: 6.67 nM mAB Anti His D2/Flag D2/Myc D2; Cisbio HTRF) and Anti Tag2 Tb (final molarities: 0.73 nM mAB Anti His Tb, 0.43 mAB Anti Flag Tb, 0.50 nM mAB Anti Myc Tb; Cisbio HTRF) was added per well to the cells. After a 2-h incubation, the 384-well MTP was measured with a TR-FRET–compatible plate reader (e.g., Pherastar, BMG Labtech; excitation: 337 nm, emission I: 620 nm, emission II: 665 nm). Dose-dependent stabilization by JQ1 or other compounds was evaluated using the four-parameter nonlinear regression algorithm (“log(Agonist) vs. response – Variable slope”) of the GraphPad Prism software (GraphPad Software).
Biochemical Binding Assay
A biochemical TR-FRET assay described previously 12 was used to assess inhibitor affinity. The method quantifies the inhibition by test compounds of the binding between N-terminal His6-tagged human BRD4 bromodomain 1 and a synthetic tetra-acetylated peptide derived from human histone 4. Briefly, the experiments were performed in 384-well black small-volume MTP (Greiner Bio-One) in a buffer composed of 50 mM HEPES (pH 7.5) (Applichem, Darmstadt, Germany), 50 mM NaCl (Sigma), 400 mM KF (Sigma), 0.5 mM CHAPS (Sigma), and 0.05% bovine serum albumin (BSA; Sigma) in a final volume of 5 to 10 µL (n = 4). BRD4 BD1 protein (50 nM) was first added to 50 nL of test compound in the MTP, subsequently mixed with 200 nM of the biotin-labeled H4(Ac)4 K5K8K12K16 tetra-acetylated peptide, and incubated for 30 min at room temperature. The protein-peptide complexes at equilibrium were then detected with 5 nM of Eu3+ cryptate-conjugated streptavidin (CisBio) and 10 nM of anti-6His-XL665 (Cisbio). After 3 h of further incubation at 4 °C, the plates were measured in a PheraStar reader (BMG Labtech) using the homogeneous time-resolved fluorescence (HTRF) module (excitation: 337 nm with 10 flashes; emission: 620 and 665 nm). IC50 values were calculated by fitting the data to the equation “log(inhibitor) vs. response – Variable slope” using the GraphPad Prism software.
Thermal Shift Assay
Thermal shift assay (TSA) was used to measure the thermal stability of the BRD4 wild-type and variant proteins and also to assess the stabilizing effects of compounds. In a typical TSA, 0.4 µg of BRD4 BD1 protein was mixed with 5× Sypro Orange (Molecular Probes, Eugene, OR) and adjusted to a final volume of 5 µL with 100 mM HEPES buffer (pH 7.5) (Applichem) and 150 mM NaCl (Sigma). For binding experiments, 1% DMSO (Sigma) or 100 µM compounds was added to the mixture. To protect samples from evaporation, they were overlaid with 1 µl Silicone Oil DC200 (Sigma). Melting curves were recorded using a ThermoFluor system (Johnson & Johnson Pharmaceutical Research and Development, Spring House, PA) by heating the samples from 25 °C up to 95 °C while measuring the fluorescence intensity of the dye with the excitation and emission filters set to 465 nm and 590 nm, respectively. Melting points were calculated with the midpoint method, using the evaluation software provided by the manufacturer of the instrument.
Results
Optimization and Miniaturization of a Protein Stabilization Assay with Enzyme Fragment Complementation Readout
To find out whether the interaction of small molecules with proteins can be assessed by intracellular protein stabilization, we tested and optimized a novel assay system based on β-galactosidase enzyme fragment complementation (
First, we determined the optimal seeding densities by varying the cell number per well in a 384-well MTP between 1250 and 10,000. As shown in Figure 1A , the BRD4 inhibitor JQ1 significantly increased the relative luminescence signal for all cell numbers in a dose-dependent manner. As expected, the baseline luminescence signal increased with higher cell numbers. The calculated EC50 values for JQ1 were in the submicromolar range and showed a tendency to higher values at low cell numbers. The lowest cell number showing a robust sigmoidal response was 5000 per well, which was therefore used in further experiments.
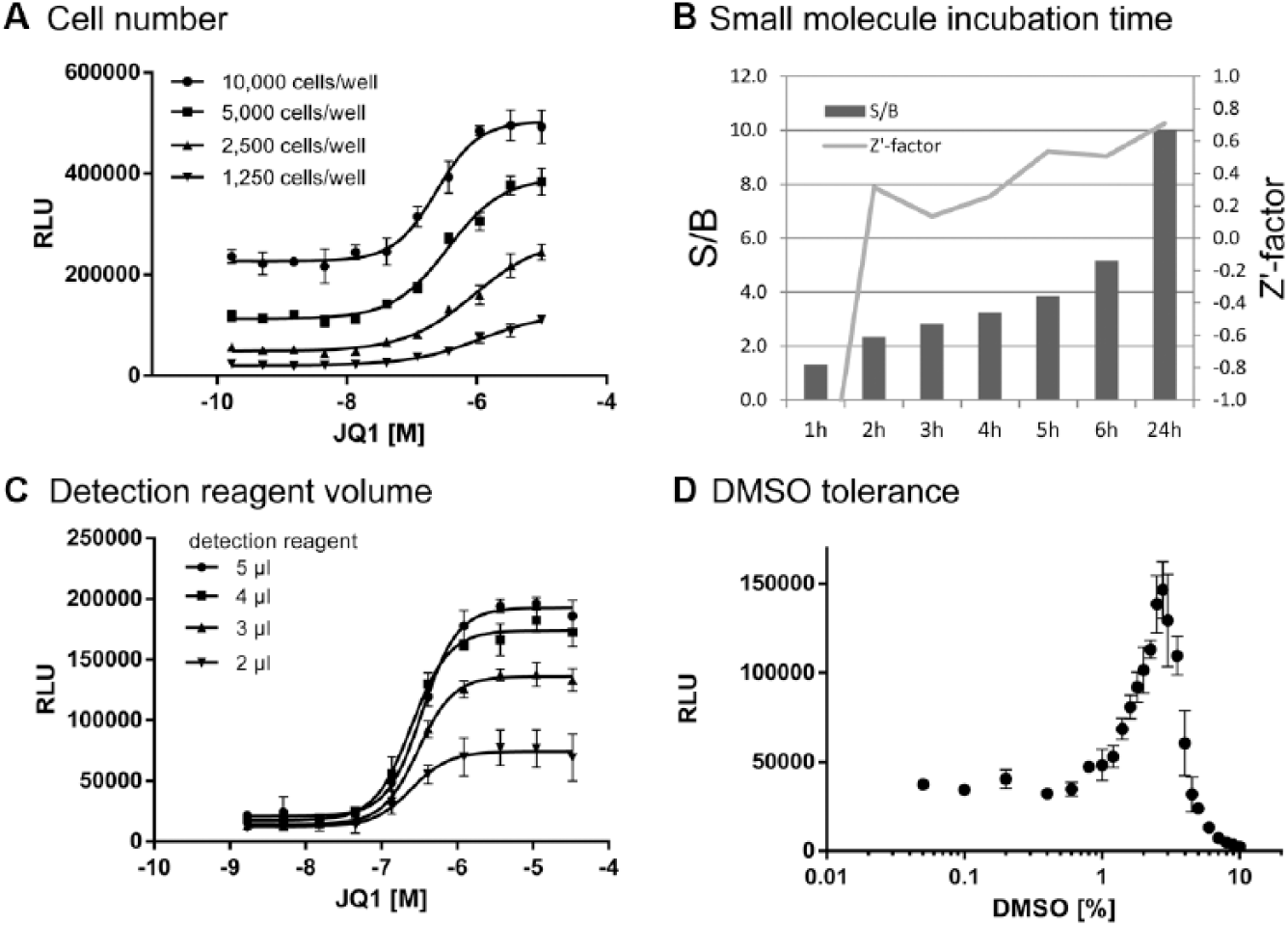
Enzyme fragment complementation (EFC) assay development. (
To establish the optimal small-molecule incubation time, we treated 5000 cells with 1% DMSO (control) or with 10 µM JQ1 before adding detection reagent at different incubation times (1–6 h and 24 h, Fig. 1B ). Within the first 6 h, there was a constant increase in the signal-to-background (S/B) ratio. The Z′ factor 13 reached acceptable levels above 0.4 after an incubation time of 5 h or more. Overnight incubation further increased the signals and the overall assay quality, which is why an incubation time of 24 h was chosen for further experiments.
Miniaturization is an important factor in the development of HTS-compatible assays. It enables assays in high-density formats for the screening of large compound collections, and ideally, it reduces costs by saving detection reagents, cells, and consumables at equal assay quality. To test if the assay could be miniaturized, we seeded 5000 cells in 3 µL assay medium into wells of a 1536-well MTP containing 50 nL compound and varied the detection reagent volume (2, 3, 4, and 5 µL). As Figure 1C shows, the conversion from 384-well to 1536-well MTPs was easily accomplished. The S/B ratio depended on the volume of the detection reagent, with a minimal amount of 3 µL being sufficient to reach an excellent assay quality in the 1536-well format.
The robustness of cell-based assays can be influenced by the DMSO sensitivity of the cells. We incubated cells with varying DMSO concentrations for 24 h. A DMSO concentration under 1% had no impact on luminescence signals. Unexpectedly, we saw a bell-shaped concentration-response curve with DMSO. Increasing luminescence signals were observed from 1% DMSO onward with a peak at 3%, whereas above 3% DMSO, the signals dramatically decreased ( Fig. 1D ).
Enzyme Fragment Complementation Mechanism of Degradation
Intracellular protein levels are influenced by protein synthesis and degradation. To further characterize the assay, we investigated the effects of inhibitors of these pathways. Using cycloheximide (CHX), an inhibitor of protein synthesis, we studied the time-dependent changes of the concentration of ePL-BRD4 in the HEK293 cells. The accumulation of ePL-BRD4 seen from 120 min after treatment start with 20 µM JQ1 was prevented in the additional presence of CHX ( Fig. 2A ). DMSO (1%) alone had no significant impact on the ePL-BRD4 protein concentration. Likewise, addition of CHX to the cells did not change the luminescence signal significantly. This supports the notion that there is a steady state of synthesis and degradation of the ePL-BRD4 protein inside the cells and shows that treatment with an inhibitor of protein synthesis such as CHX can counteract the JQ1-induced accumulation of ePL-BRD4 in cells.
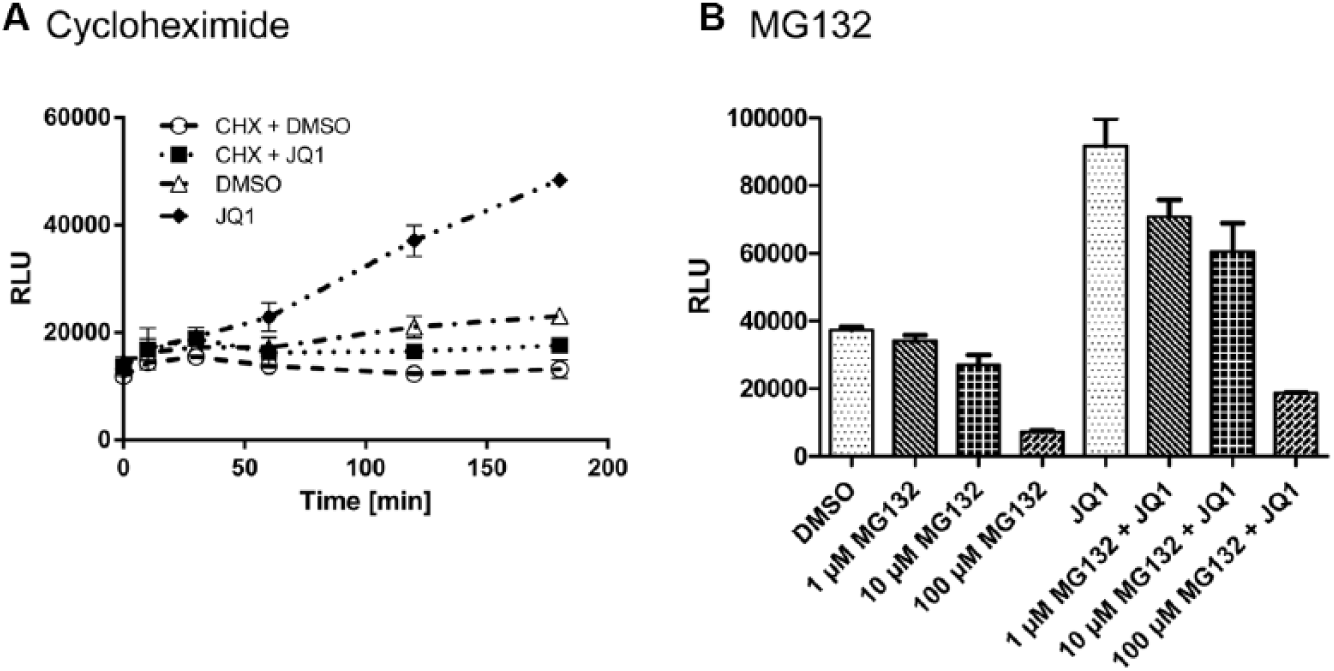
Enzyme fragment complementation (EFC) mechanism of degradation. DMSO (1.1%) and JQ1 (2E-5 M, 1.1% DMSO) were used as controls. (
MG132 is a highly potent and reversible proteasome inhibitor. It reduces the degradation of ubiquitin-conjugated proteins by inhibiting the 26S proteasome complex. 14 To block proteasomal degradation, we incubated the cells with MG132 for 3 h using different concentrations (100 µM, 10 µM, and 1 µM) and used DMSO (1%) and JQ1 (20 µM) as single agents or in combination. As shown in Figure 2B , JQ1 alone again showed a stabilizing effect on the ePL-BRD4 protein levels. Unexpectedly, MG132 treatment did not increase the concentration of ePL-BRD4 measured in the cellular lysate. At higher concentrations, MG132 rather led to a strong decrease of the luminescence signal, probably due to toxic effects. A similar result was obtained when the cells were treated with a combination of MG132 and JQ1.
Taken together, the inhibitory effect of CHX on the JQ1-induced ePL-BRD4 protein stabilization could clearly be shown, but the mechanism of degradation seems to be independent of the proteasomal pathway.
Development of a Protein Stabilization Assay with TR-FRET Readout
We set out to study intracellular stabilization of BRD4 by small molecules in more detail using another highly sensitive detection method based on TR-FRET. For this approach, we expressed the BRD4 BD1 protein region flanked by N- and C-terminal affinity tags to enable detection via specific antibodies. To verify the feasibility of the approach, various BRD4 BD1 expression constructs with different combinations of N- and C-terminal affinity tags were employed. We chose three different short tag sequences (FLAG, His, Myc) that should not have large secondary structures and be suitable for detection with standard reagents. Expression constructs containing the BRD4 BD1 sequence (amino acids 44–166, codon-optimized for mammalian expression) flanked by these three tags in six different combinations were generated and introduced into a pTT5 expression vector.
Initial experiments using transiently transfected, adherent HEK293 cells indicated the need for DNA concentrations of 1 µg/mL (
On the basis of the initial experiments, we then established a stable cell pool expressing the His-BRD4-Flag construct by selecting transfected HEK293 cells with hygromycin. We confirmed the dose-dependent signal increase by JQ1 for the stable cell pool in the TR-FRET assay and also by semi-quantitative Western blot ( Fig. 3 ). These results were nicely corroborated in the Western blot where a concentration-dependent signal increase upon JQ1 treatment was observed.
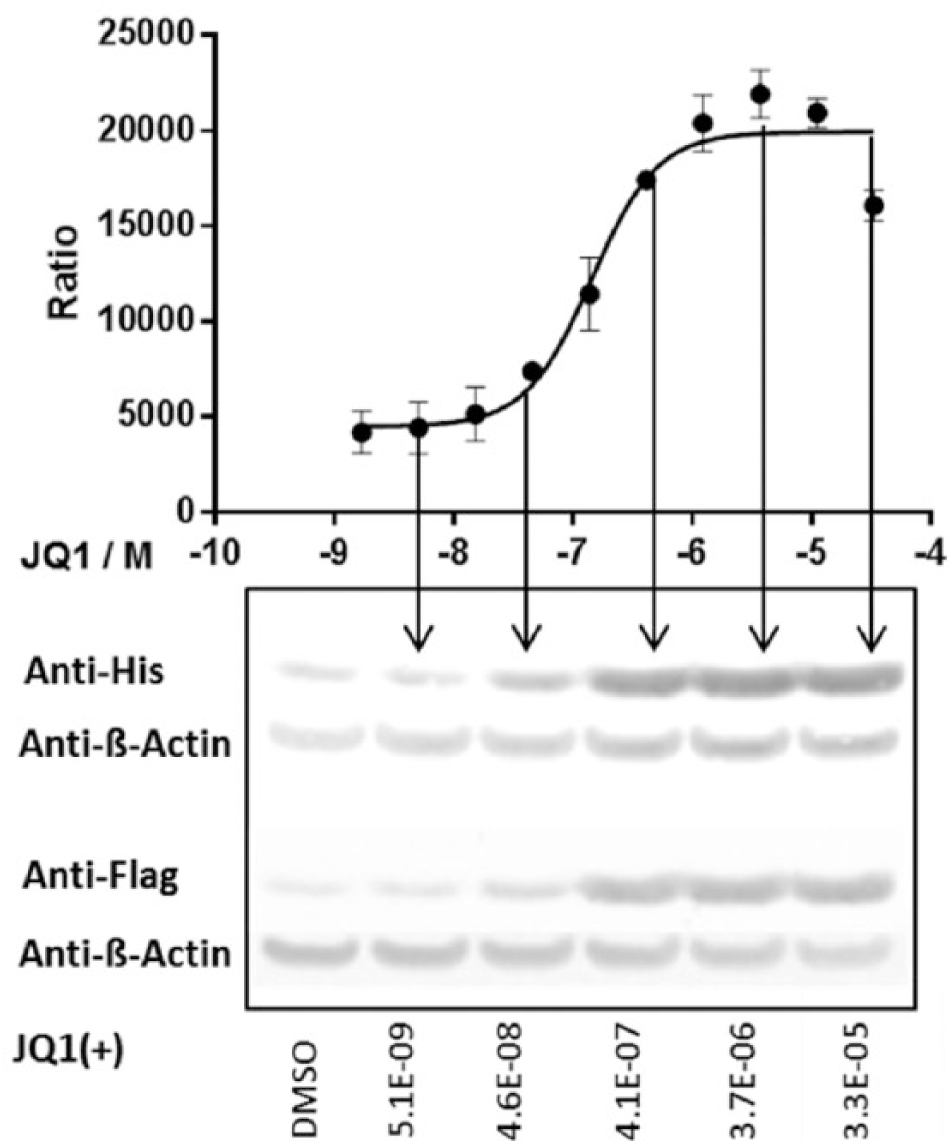
Time-resolved fluorescence resonance energy transfer (TR-FRET) assay using HEK293 cells expressing the His-BRD4-BD1-Flag construct. The cells were treated with JQ1, and the amount of tagged BRD4 was measured with the TR-FRET assay and by Western blot analysis using anti-His and anti-FLAG antibodies. As loading control, β-actin was analyzed. Data points represent mean ± SD calculated from four replicate data points.
For further assay development, we also tested HEK293-6E suspension cells and varied the cell number and DNA concentration (
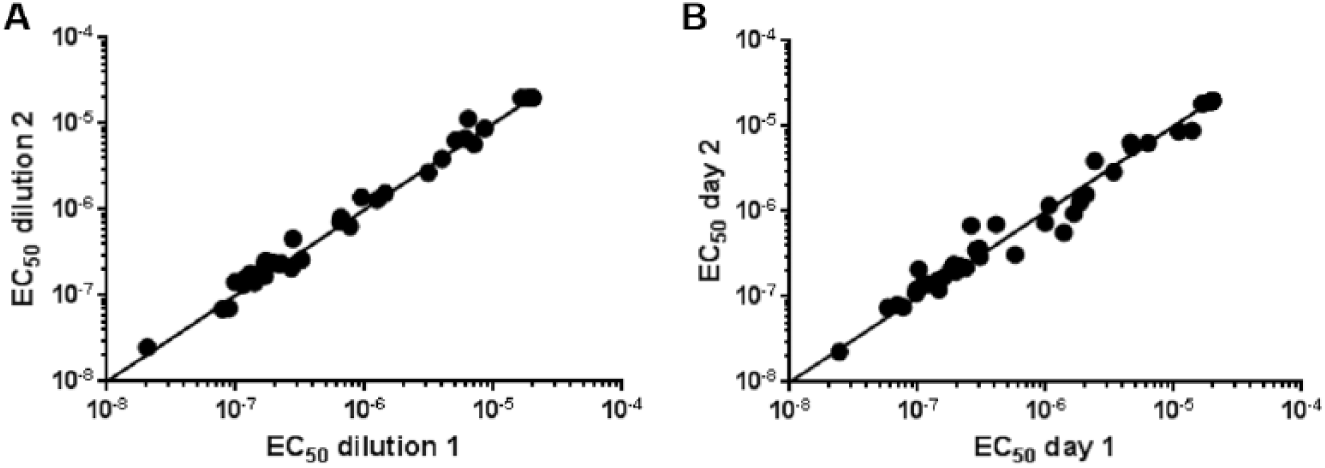
Intra- and interassay variance of the time-resolved fluorescence resonance energy transfer (TR-FRET) stabilization assay. EC50 values were determined for a set of test compounds on (
Comparison of Cellular, Biochemical, and Biophysical BRD4 Assays
Having optimized several parameters in the EFC assay and established the TR-FRET assay, we compared a set of 48 small molecules in dose response in both cellular stabilization assays. The compound set contained small molecules with BRD4 binding activities ranging from double-digit nM to µM and included six reference compounds
10
(JQ1, I-BET762, I-BET151, CPI203, PFI-1, and the PFI-1 analogue Cpd 18; structures shown in
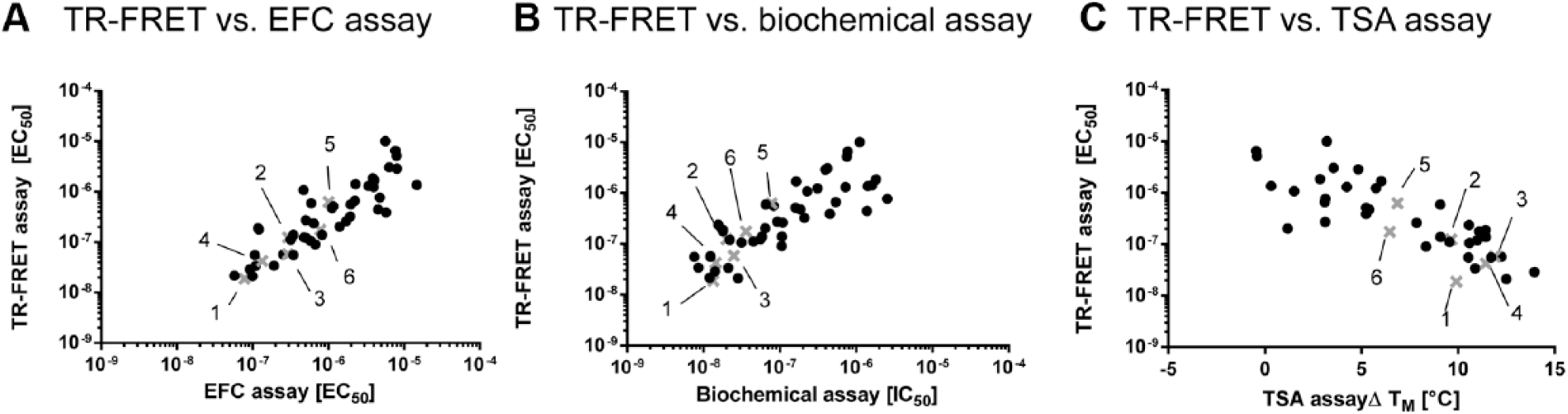
Assay correlation upon treatment with BRD4 binding compounds. EC50 values of the intracellular protein stabilization assay with time-resolved fluorescence resonance energy transfer (TR-FRET) detection are plotted against (
To further validate the cell-based assays, we performed two additional independent assays with isolated BRD4 BD1. First, a biochemical binding assay measuring protein peptide interactions between BRD4 BD1 and a tetra-acetylated H4 peptide using a competitive TR-FRET approach was employed. The same set of compounds and the same dilution series as before were used. The calculated IC50 values of the competitive biochemical assay showed a medium positive correlation (r = 0.41) with the cell-based EC50 values of the TR-FRET stabilization assay ( Fig. 5B ). The absolute values were in some cases lower for the biochemical assay, for example, in the case of PFI-1 and PFI-1 analogue (Cpd 18). This might be due to incomplete cellular permeability of some compounds. Second, we tested the compounds in a TSA, a biophysical method interrogating the melting temperature point shift of an isolated protein in the absence and presence of a test compound. The melting point of the BRD4 BD1 domain used in our assay in the absence of compound was 49.9 ± 0.35 °C. Upon binding of a compound, the melting point of the protein shifts toward higher temperatures. Figure 5C shows the correlation of the EC50 values obtained in the TR-FRET stabilization assay and the melting point shifts (ΔTM) of the compound set tested at a single-compound concentration of 100 µM. We found a high negative correlation between both methods (r = −0.58). The most potent compounds induced a strong ΔTM of up to 12 °C, whereas inactive compounds did not change the TM of BRD4 BD1.
Overall, we could show a high correlation between the two cell-based BRD4 stabilization assays as well as medium to good correlation between the cell-based and the biochemical and biophysical methods.
Impact of BRD4 BD1 Mutations on Intracellular Stability and Compound Interaction
The TR-FRET assay described in this study also allowed us to study the effects of BRD4 BD1 point mutations on the compound-induced stabilization. Especially the assay system comprising transiently transfected HEK293-6E cells is exceptionally well suited as a fast and efficient way to generate data for mutated variants of target proteins. Previous studies have indicated certain point mutations in BRD4 and related bromodomain proteins to be important for binding of the acetylated histone peptide. We selected W81, P82, Y97, N140, and M149 as mutational sites for this study, in view of their role in the recognition by acetylated histone peptides and JQ1. 12 In total, 48 small molecules were tested against wild-type and five different mutant BRD4 BD1 constructs.
As shown in
Figure 6A
, all mutant BRD4 variants could be expressed and were stabilized by JQ1 to different extents. The EC50 values for JQ1 were strongly increased for the mutants Y97F, N140A, and M149A, whereas those for W81A and P82A were less affected, in comparison to the stabilization measured in presence of wild-type BRD4. Comparable profiles were observed for the five BRD4 BD1 mutants in presence of all six reference compounds (
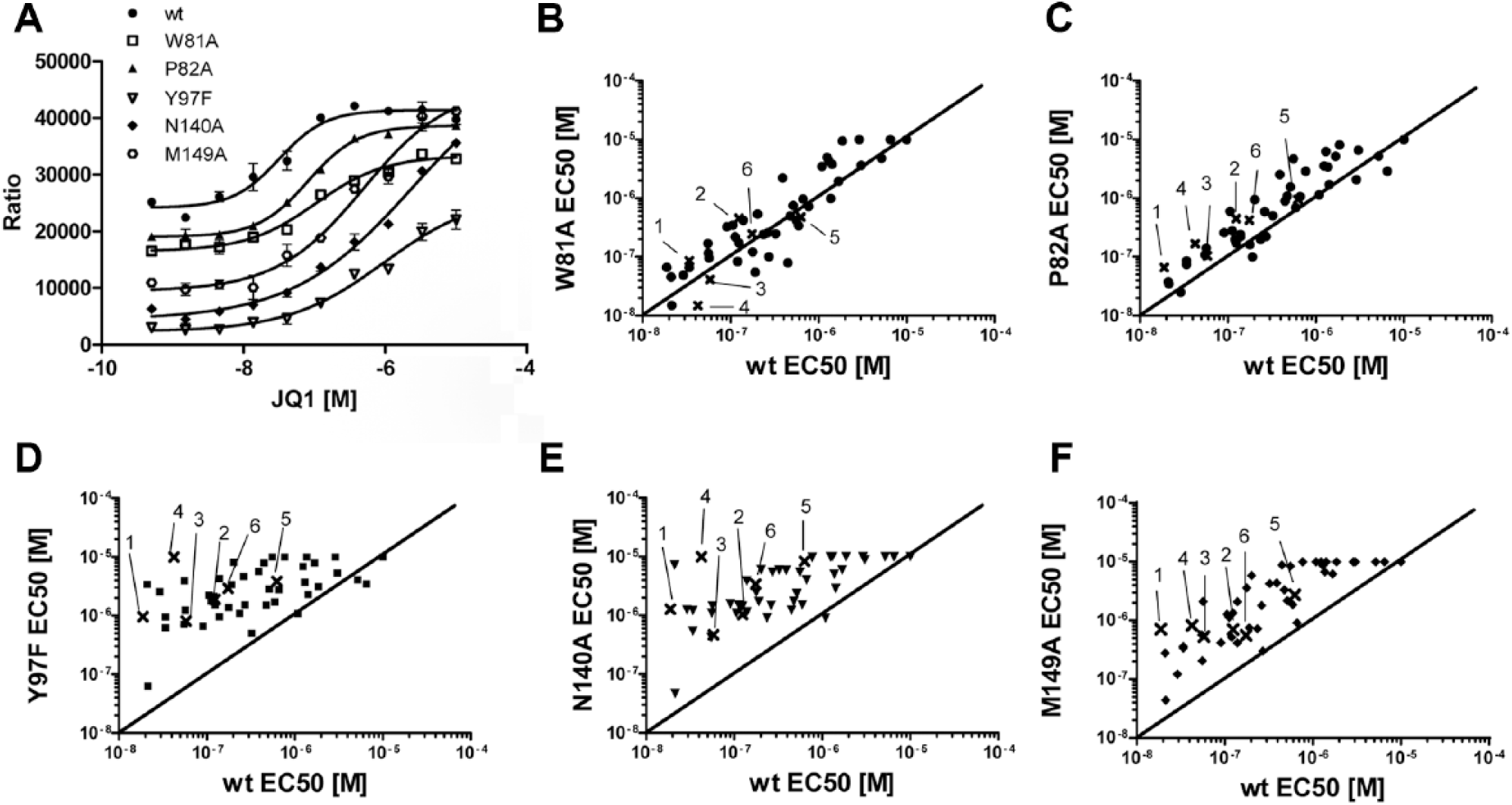
Mutation analysis of BRD4 BD1. (
Discussion
Ligand-induced protein stabilization in cell-free systems has been extensively explored during the past decade for drug discovery applications.2,4,5,15,16 Measuring ligand interaction with purified targets has several drawbacks linked to the fact that proteins are usually found in complexes and undergo posttranslational modifications. Also, information linked to some of the pharmacokinetic properties of the ligand such as cell penetration is lost. More recently, first attempts to determine target engagement in the cellular environment using an adaptation of the TSA have been reported. 3 This technology, however, has some limitations, so improvements in the quantification and collection of data are needed. Our aim was therefore to evaluate the principle of cell-based protein stabilization by small molecules in an intact cellular environment and develop an assay amenable to screening. For this, we used the target protein BRD4 and focused on its first bromodomain, BD1, which is known to play the main role in histone binding and in tumor cell progression. 11
Two independent methods, InCell Hunter enzyme fragment complementation and homogeneous time-resolved fluorescence, were used for quantitative analysis. The EFC assay was easily established using JQ1 as a tool compound. High-quality concentration-response curves were obtained, and we were successful in optimizing the EFC assay for performance in a 1536-well MTP. Also, the workflow was simplified by direct cell seeding into assay plates prefilled with 50 nL of test compound, facilitating the use of assay-ready plates often applied in HTS approaches. 17 The EFC assay was sensitive toward elevated concentrations of DMSO, resulting in an increased luminescence signal between 1% and 3% DMSO, which was initially surprising. On the other hand, we also observed an influence of DMSO at similar concentrations in the biochemical peptide-binding assay (not shown), suggesting that DMSO actually binds into the BRD4 pocket and thus leads to a stabilization of ePL-BRD4 inside cells. A similar finding has recently been made in a crystallographic study, where DMSO was found to bind in the acetylated lysine-recognition pocket of BRD4. 18
We were interested in the mechanism of compound-induced protein stabilization and used CHX and MG132 as tools to interfere with protein synthesis or protein degradation, respectively. We found that the inhibitor of protein synthesis CHX completely blocked the accumulation of BRD4 by JQ1. The picture was somewhat different for the proteasome inhibitor MG132. Unexpectedly, MG132 rather showed the opposite effect by increasing the degradation, resulting in a reduced ePL-BRD4 concentration. We conclude that ePL-BRD4 is not degraded by the proteasomal pathway in our cellular system. A possible explanation for this unexpected finding might be that ePL-BRD4 degradation is mediated by a different proteolytic pathway (e.g., the lysosomal proteolytic pathway). It has been reported that both the proteasomal and the lysosomal pathways “communicate” with each other. 19 The observed increase in degradation of ePL-BRD4 using MG132 might be explained by an upregulation of the lysosomal pathway by proteasome inhibition. Due to the short incubation times used in this experiment, unspecific effects on the cell viability cannot be excluded but are rather unlikely, since similar CHX (100 µg/mL) and MG132 (10 µM) concentrations are usually used.14,20
To study the assay principle of ligand-induced stabilization in cells in more detail, we established an alternative detection method using TR-FRET. A set of BRD4 expression constructs with dual N- and C-terminal affinity tags was designed for antibody-based TR-FRET detection. The JQ1-induced BRD4 stabilization obtained in the TR-FRET and the EFC assays could be nicely confirmed by Western blot detection, validating the general concept by three independent methods. Having established the EFC and TR-FRET assays, we were able to compare and validate the new formats using a compound test set composed of 48 BRD4 inhibitors. We found a high correlation between the EC50 values of the EFC and TR-FRET assays. Furthermore, the EC50 values in the cellular protein stabilization assays closely matched the IC50 values derived from a biochemical competition assay. Finally, we observed a high correlation with the melting temperature point shifts measured in a biophysical TSA. Altogether, these data substantiate the concept that in our cellular assay system, by measuring the increase of the protein concentration in the cellular lysate, we indeed can quantify a direct impact of small-molecule inhibitors known to bind to BRD4. This concept is further supported by reports showing that in cell-free systems, small-molecule ligands shift the protein unfolding equilibrium, resulting in a change of the susceptibility to proteases.4,5 Our finding that not the proteasome but rather the lysosome is involved in the degradation of the ePL-BRD4 also fits well with this concept.
The expression of BRD4 mutants in the TR-FRET stabilization assay again demonstrates the validity of the approach. In addition, the results exclude the possibility that the compound-induced signal increase is mediated by an interaction with endogenous BRD4 in our cells and not with the recombinant tagged version. If the stabilizing effects of the compounds were due to endogenous BRD4, one would expect similar EC50 values for all expressed mutant BRD4 proteins, which was not the case in our study. Indeed, the EC50 values of JQ1 for the mutant BRD4 variants in our cellular assay matched well with the affinities obtained in biochemical and biophysical assays performed with these mutants. 12 Moreover, we showed that the loss of stabilization induced by JQ1 for each mutant was similar to the loss of binding affinity of BRD4 to acetylated H4 peptide and that the highest impact was found with mutants Y97F and N140A. These residues are known to recognize acetylated histones by forming direct (N140) and water-mediated (Y97) hydrogen bonds with acetylated peptides and to interact with inhibitors mimicking acetyl-lysines such as JQ1 and I-BET-762. 12 The cellular protein stabilization assay turned out to be a very fast and efficient screening tool for the differentiation of novel BRD4 inhibitors. It may help to study resistance mutants arising from patients treated with BRD4 inhibitors to develop novel treatments for these patients in the future.
In summary, we describe a new powerful cellular HTS tool capable of identifying BRD4 inhibitors that interact with the hydrophobic binding pocket of bromodomain BD1. It remains to be seen whether this methodology can be transferred to other target proteins and whether it is applicable to larger proteins with a more complex structure. In fact, the described assays are not necessarily restricted to the identification of stabilizing compounds to protein domains but may also be instrumental in the identification of novel drugs for treatment of diseases originating from deregulated protein levels (e.g., age-related diseases such as Alzheimer disease, in which proteases play an important role in the regulation of amyloid-β protein levels). 21 Another interesting option is to use such assays for the identification and characterization of protein destabilizers (e.g., estrogen receptor antagonists). 22 Finally, our assays may be applied to identify or characterize pharmacological chaperones, which have been described to correct enzyme deficiencies. 23 Our results revealed that protein stability, which is a fundamental cellular mechanism, can be directly influenced by small-molecule ligands. More high-throughput assays that quantify other target proteins in cells will therefore be very useful to identify and characterize drug candidates in many different research fields.
Footnotes
Acknowledgements
We thank Sabrina Reimann, Marina Isernhagen, and Dagmar Zeggert-Springer for their excellent technical assistance.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.