
Abstract
Autophagy is an evolutionarily conserved homeostasis process through which aggregated proteins or damaged organelles are enveloped in a double-membrane structure called an autophagosome and then digested in a lysosome-dependent manner. Growing evidence suggests that malfunction of autophagy contributes to the pathogenesis of a variety of diseases, including cancer, viral infection, and neurodegeneration. However, autophagy is a complicated process, and understanding of the relevance of autophagy to disease is limited by lack of specific and potent autophagy modulators. ATG4B, a Cys-protease that cleaves ATG8 family proteins, such as LC3B, is a key protein in autophagosome formation and maturation process. A novel time-resolved fluorescence resonance energy transfer (TR-FRET) assay measuring protease activity of ATG4B was developed, validated, and adapted into a high-throughput screening (HTS) format. HTS was then conducted with a Roche focus library of 57,000 compounds. After hit confirmation and a counterscreen to filter out fluorescence interference compounds, 267 hits were confirmed, constituting a hit rate of 0.49%. Furthermore, among 65 hits with an IC50 < 50 µM, one compound mimics the LC3 peptide substrate (-T
Introduction
Autophagy is a revolutionarily conserved catabolic process that plays a housekeeping role by digesting aggregated proteins or damaged organelles in a lysosome-dependent manner. Originally identified by transmission electron microscopy and named by its function of “self-eating,” 1 autophagy was later extensively studied. It was found that autophagy is a process involving multisteps of membrane formation, trafficking, lysosome fusion, and ultimately degradation of its cargo. This complicated and well-orchestrated process relies on many players, including multiprotein complexes that possess various enzymatic activities and functionalities. Many of these proteins encoded by autophagy-related genes (Atg) have been identified and characterized. 2
It is now widely recognized that autophagy starts at the phagophore assembly site (PAS) where the initial sequestering structure called the phagophore/isolation membrane expands and the ATG proteins are recruited. Subsequently, membrane elongation leads to the formation of a double-layer structure called the autophagosome, which sequesters portions of cytosol. During this process, two interrelated ubiquitin-like (Ubl) conjugation systems, ATG12-ATG5-ATG16 and ATG8-PE (phosphatidylethanolamine), play an essential role in regulating the membrane elongation and expansion of the forming autophagosome.3,4 The ATG12-ATG5-ATG16 complex serves as an E3-like enzyme to conjugate ATG8 to PE. 5 ATG8 must undergo a proteolytic process to expose a C-terminal Gly for PE conjugation. ATG4, a cysteine protease of the C54 family, is required for processing ATG8 to initiate the autophagosome formation. ATG4 is also required for deconjugating PE from the C-terminal Gly of ATG8-PE during a later stage of the autophagic process, which is also critical for a normal autophagy process.
Unlike yeast, where there are only one atg4 and one atg8 gene, there are four ATG4 (ATG4A, ATG4B, ATG4C, and ATG4D) and six ATG8 (LC3A, LC3B, LC3C, GABARAP, GATE-16/GABARAPL2, and ATG8L/GABARAPL1) homologues in mammals. The six ATG8 homologues belong to two subfamilies: the MAP1A/B/LC3 subfamily (LC3A, LC3B, and LC3C) and the GABARAP subfamily (GABARAP, GATE-16/GABARAPL2, and ATG8L/GABARAPL1). These homologues all contain a conserved Gly residue proximal to the cleavage site and contribute to an autophagy process in different stages. 6 Recent studies showed that ATG4B had the broadest substrate spectrum, followed by ATG4A. Surprisingly, ATG4C and ATG4D had minimal activities until they were activated by the cleavage of their N-termini via a caspase. On the other hand, GATE-16 was the best substrate for all ATG4 homologues. 7
The role of autophagy extends beyond the general homeostasis. Based on extensive research during the past decade, autophagy is now widely implicated in pathophysiological processes (e.g., cancer, metabolic and neurodegenerative disorders, and infectious diseases) and in physiological responses to exercise and aging.8,9 However, controversy remains for the role of autophagy in diseases. To have a better understanding how autophagy affects cellular homeostasis and pathogenesis, an assay development for high-throughput screening (HTS) may provide a powerful platform for identifying novel modulators of autophagy pathways.
A novel time-resolved fluorescence resonance energy transfer (TR-FRET) assay measuring protease activity of ATG4B was developed, validated, and adapted into an HTS format. HTS was conducted to a Roche (Basel, Switzerland) focus library. Promising hits were successfully identified, including the ones that mimic peptides in natural substrates. Furthermore, some hits are active in a cell-based luciferase release assay. Overall, the novel TR-FRET ATG4B protease assay provides a robust platform to identify specific ATG4B inhibitors, which would be used as tool compounds to elucidate the mechanism of the autophagy pathway and to be further optimized for therapeutic treatment.
Materials and Methods
Cell Culture
An HEK-293T (human embryonic kidney) (ATCC, CRL-11268) cell line was grown in Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F12 50/50 Mix (cat. 10-092-CVR; Corning, Corning, NY) supplemented with 10% fetal bovine serum (cat. 10099-141; Gibco, Grand Island, NY) and penicillin-streptomycin (cat. 15073-063; Gibco). Cells were incubated at 37 °C in a humidified atmosphere with 5% CO2.
Protein Expression and Purification
The open reading frame encoding the fusion of LC3B-GST or GATE-16–GST was inserted into the Pet30a vector (Merck, Kenilworth, NJ) individually, resulting in two constructs each containing an N-terminal His6-Avi tag and C-terminal GST tag. His-Avi-LC3B-GST or His-Avi-GATE-16–GST was expressed in Escherichia coli BL21 DE3 cells. The expressed His-Avi-LC3B-GST was purified by affinity chromatography using a Ni-chelating affinity column and concentrated to 1 mg/mL in 20 mM phosphate buffer (PB), 150 mM NaCl, 10% glycerol, and 2 mM EDTA (pH 7.4). His–Avi–GATE-16–GST was purified by the affinity chromatography using the His Trap_HP column (GE Healthcare, Piscataway, NJ) and Q_HP column (GE Healthcare) and then concentrated to 5 mg/mL in 20 mM Tris, 150 mM NaCl, and 0.5 mM tris(2-carboxyethyl)phosphine (TCEP) (pH 7.4).
The open reading frame encoding the full length of His-ATG4B was inserted into the Pet30a vector, which fused a His6 tag to the N-terminus. His-ATG4B was expressed in Escherichia coli strain BL21 DE3 cells. After cell lysis, the expressed protein was first purified by affinity chromatography using the His Trap_HP column (GE Healthcare), and the His tag was cleaved by TEV protease (Novoprotein, Summit, NJ). Further purification was performed using the Q_HP column (GE Healthcare) followed by Superdex 75 columns (GE Healthcare). The purified protein was concentrated to 11.2 mg/mL in 0.15 M NaCl with 20 mM Tris-HCl (pH 7.4) and 2 mM dithiothreitol (DTT).
Measurement of ATG4B Kinetics Using the FRET-Based Assay
Purified ATG4B was mixed with His–GATE-16–GST or His-Avi-LC3B-GST in assay buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM DTT, and 0.2 mg/mL albumin; cat. A5503-1; Sigma, St. Louis, MO), and the total volume was 15 µL (384-well format; ProxiPlate-384, cat. 6008280; Perkin Elmer, Waltham, MA). After incubation at room temperature (RT) for a given time, 5 µL mixture of Eu-anti-His (cat. AD0111; PerkinElmer) and ULight-anti-GST (cat. TRF0104-R; PerkinElmer) in detection buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 0.2 mg/mL albumin) was added and incubated at RT for another given time. After incubation, the fluorescence signal was measured with EnVision 4 (PerkinElmer; ex: 340 nm; em: 665/615 nm).
The cleavage activity of the ATG4B was thus determined based on the proportion of the fluorescence resonance energy transfer (FRET) signal change, which is calculated as follows: normalized FRET signal = (615Eu − 615buf) * (665sample − 665buf −crosstalk factor * (615sample − 615buf))/(615sample − 615buf), where 615 is a raw reading at 615 nm, 665 is a raw reading at 665 nm, Eu represents the signal of Eu-anti-His only (15 µL reaction buffer and 5 µL detection solution), buf represents signal of the buffer (15 µL reaction buffer and 5 µL detection buffer), and crosstalk factor = (665Eu − 665buf)/(615Eu − 615buf). Percentage inhibition is calculated as follows: Inhibition% = 100 − [(normalized FRET signal of test compound − normalized FRET signal of DMSO)/(normalized FRET signal of positive compound − normalized FRET signal of DMSO)] × 100.
HTS of the ATG4B Inhibitors Based on the TR-FRET Assay
In total, 5 µL compound (final 1% DMSO) in assay buffer was added to 5 µL (final 0.1 nM) ATG4B in a 384-well plate and incubated at RT for 30 min, then, 5 µL/well of His–GATE-16–GST (final 20 nM) was added after incubation and incubated at RT for another 30 min. A 5-µL/well detection solution (final 2 nM Eu-anti-His and 20 nM ULight-anti-GST) was then added, and the resulting mixture was incubated at RT for 40 min. After incubation, signal was read with an EnVision Multilabel Plate Reader (PerkinElmer; ex: 340 nm, em: 665/615 nm). Data were analyzed using software Genedata Screener (Genedata AG, Basel, Switzerland).
To filter out interference caused by the autofluorescence or quenching of compounds, a parallel counterscreen experiment was carried out. Purified ATG4B was mixed with His–GATE-16–GST in assay buffer, resulting in a total volume of 10 µL. After incubation at RT for 30 min, a 5-µL/well detection solution (final 2 nM En-anti-His and 20 nM ULight-anti-GST) was added, and the resulting mixture was incubated at RT for 40 min. Finally, 5 µL compound was added, and TR-FRET signal was measured with an EnVision Multilabel Plate Reader (PerkinElmer; ex: 340 nm, em: 665/615 nm). Since compounds were added after enzymatic reaction of substrate cleavage, the interference of compounds to the readout signal was assessed.
Western Blotting
The cell lysate was prepared in RIPA buffer and quantified by the bicinchoninic acid (BCA) method (Pierce, Rockford, IL). In total, 30 µg of protein per sample was loaded onto a 4% to 12% NuPAGE Novex SDS gel (Invitrogen, Carlsbad, CA). The protein was transferred by an iBlot dry blotting device (Invitrogen) onto nitrocellulose membranes. After blocking nonspecific binding with Tris-buffered saline (TBS)/Tween-20 (0.1%) (TBS/T) containing 5% nonfat milk for 1 h at room temperature, the membrane was incubated in the antibody of LC3B (L7543; Sigma), or ATG4B (cat. 5299; Cell Signaling, Danvers, MA) (1:1000 in TBS/T containing 3% bovine serum albumin [BSA]) and gently shaken at 4 °C overnight. The membrane was washed with TBS/T three times to remove the unbound antibody and then incubated with the secondary antibody (horseradish peroxidase [HRP]–conjugated goat anti-mouse IgG or goat anti-rabbit IgG, 1:5000; KangChen Biotech, Shanghai, China) for 1 h at RT. Protein bands were visualized with an enhanced chemiluminescence (ECL) kit (Pierce).
Analysis of the ATG4B-Mediated Cleavage of Substrates by SDS-PAGE
Assay buffer used was 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM DTT, and 1 mM EDTA. For substrate comparison, purified ATG4B (final 0.03 mg/mL, diluted with assay buffer) was incubated with His–Avi–GATE-16–GST or His-Avi-LC3B-GST (final 0.3 mg/mL) separately for 5 min, and the products were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed with Coomassie Brilliant Blue (CBB) staining. For ATG4B inhibitor testing, ATG4B (final 0.67 µM) was preincubated with compounds (various concentrations for titration) for 4 h, and then His–Avi–GATE-16–GST was added to the ATG4B mixture for another 30 min and full-length or cleaved GATE-16–GST was detected by SDS-PAGE and CBB staining.
Cell-Based ATG4B Activity Detection Using Luciferase Release Assay
A dNGLUC sequence optimized for Home sapiens was subcloned into the pCDH9 vector. The open reading frames encoding the full length of actin-LC3B or actin were inserted into the pCDH9 vector, which fused a dNGLUC to the actin-LC3B or actin C-terminus. The open reading frames encoding the full length of ATG4B or HTRA4 were inserted into the pCDNA3.1 vector. All the dNGLUC, actin-dNGLUC, actin-LC3B-dNGLUC, ATG4B, and HTRA4 plasmids were expressed in 293T cells. pCDH9 and pCDNA3.1 vectors were used as control.
HEK-293T was seeded in a 6-well plate with a density of 70% confluence. Then, 1 µg enzyme expression plasmid and 2 µg substrate expression plasmid were cotransfected using 9 µL X-tremeGENE HP DNA Transfection Reagent (cat. 06366236001; Roche Diagnostics, Risch-Rotkreuz, Switzerland) separately. The three combinations were pCNDA3.1 vector and dNGluc, HTRA4 and actin-dNGluc, and ATG4B and actin-LC3-dNGluc. After 24 h, transfected cells were trypsinized, suspended, and transferred to 96-well plates (cat. 3599; Corning), and compounds were added into the plates at the same time. After 24 h of treatment, 50 µL cell supernatant was transferred to a 96-well white plate (cat. 3917; Corning), and luciferase signal was detected using the BioLux Gaussia Luciferase Assay Kit (cat. E3300L; New England BioLabs, Ipswich, MA) and measured with the EnVision Multilabel Plate Reader (PerkinElmer).
LC/MS/MS
Preparation of the trypsin-digested sample: 20 µL sample was mixed with 200 µL of 100 mM DTT and 8M urea in 50 mM ammonium bicarbonate (ABC), and the mixture was incubated at 37 °C for 10 min and then centrifuged in an Amicon Ultra-0.5 centrifugal filter (10K cutoff; Millipore, Billerica, MA) at 14,000 g for 15 min. This was repeated one more time before trypsin was added and incubated at 37 °C overnight. The resultant peptide mixture was centrifuged, dried in a speed vac, and reconstituted with ~30 µL of 5% DMSO/5% formic acid/5% acetonitrile (ACN).
Liquid chromatography/mass spectrometry (LC/MS) for a molecular weight (MW) measurement: a 3-µL sample was injected onto a reverse-phase C4 column (BEH300, 150 × 2.1 mm, 1.7 µm; Waters, Milford, MA) equilibrated at 60 °C at a flow rate of 160 µL/min. Reverse-phase chromatography was performed using ultra-performance liquid chromatography (Dionex UltiMate 3000; Thermo Fisher, Waltham, MA). The gradient was generated by using 0.1% FA for mobile phase A and acetonitrile containing 0.1% FA for mobile phase B. The eluted species were then analyzed online by an Orbitrap mass spectrometer (Q-Exactive; Thermo Fisher) operating in the positive ion mode from m/z 500 to 2000 and calibrated according to the manufacturer’s procedure. The LC/MS data files were deconvoluted using ProMass (Thermo Scientific, Waltham, MA) to get the MW information.
LC/tandem MS (LC/MS/MS) to determine the modified sites: an aliquot of the digestion representing an amount of 3 µg C74 protein was injected onto a reverse-phase C18 column (BEH300, 150 × 2.1 mm, 1.7 µm; Waters) equilibrated at 35 °C at a flow rate of 160 µL/min. LC/MS/MS was performed in an information-dependent mode using the same system as above. For each cycle, one full MS scan from m/z 200 to 2000 was followed by up to 10 MS/MS for the most intense ions. The LC/MS/MS data files were manually analyzed according to the reference sequence of C74 and the protease specificity.
Results
Development of the ATG4B TR-FRET Assay Using GATE-16 Substrate
To set up a robust and efficient assay to identify ATG4B inhibitors, a TR-FRET assay was designed and implemented. In principle, the substrates of ATG4B, ATG8 protein (including LC3B and GATE-16) were fused at the N- and C-terminal with two epitopes, His-tag and GST, respectively. Their corresponding antibodies, anti-His Ab and anti-GST Ab, were labeled with fluorophores, Europium and ULight, respectively, serving as a FRET donor and receptor pair ( Fig. 1A ). FRET occurs when the substrate is intact so that binding of two antibodies to their respective antigen would bring two associated fluorophores close. However, FRET signal decreases when the substrate is cleaved by ATG4B, and thus the two fluorophores are not in proximity. Compounds that can inhibit ATG4B activity would show an enhanced FRET signal in this system ( Fig. 1A ).
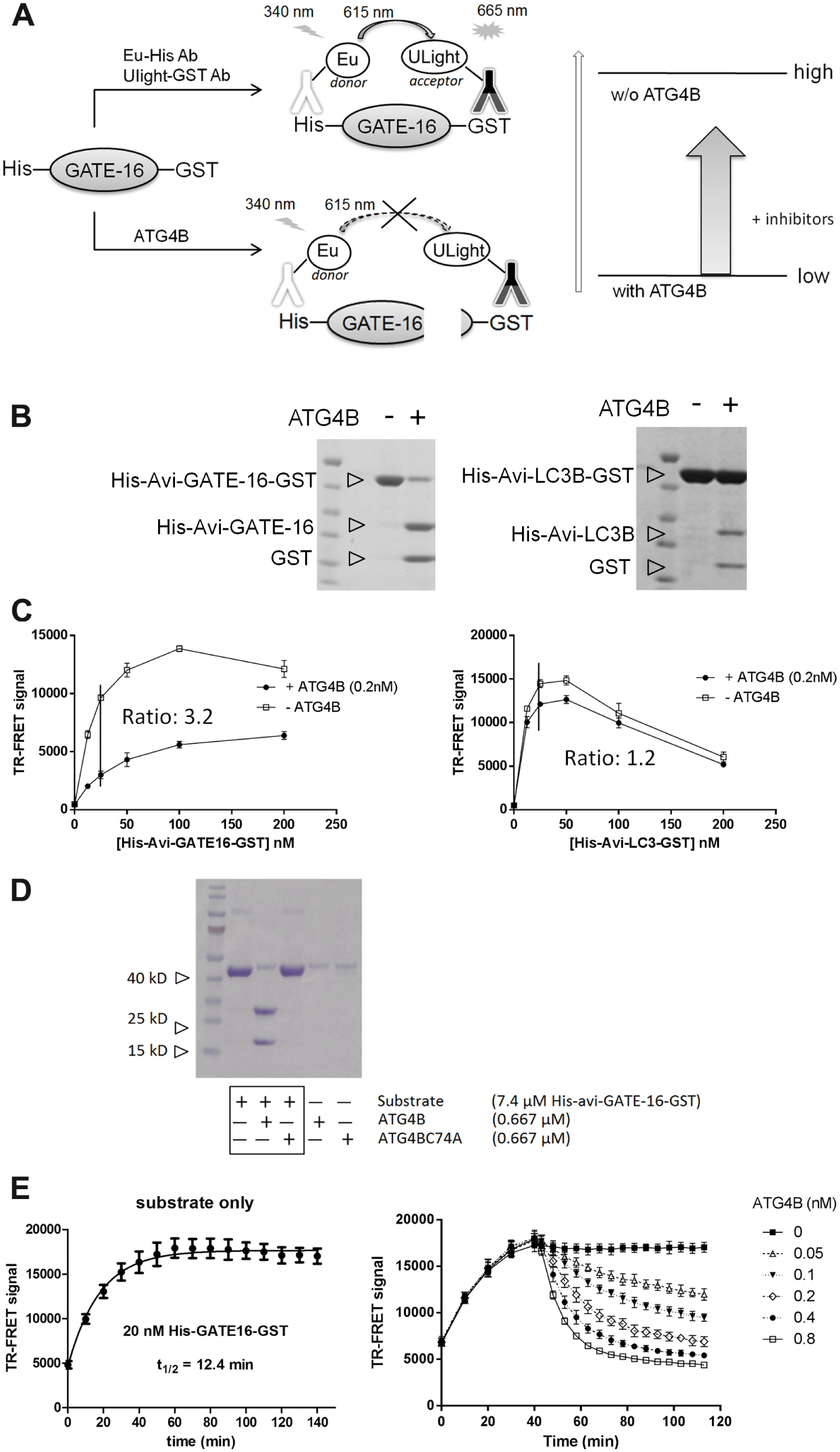
Assay development of a time-resolved fluorescence resonance energy transfer (TR-FRET) high-throughput screening (HTS) assay to identify ATG4B inhibitors. (
Mammalian cells express various forms of ATG8. Here we tested two different forms, LC3 and GATE-16, to choose an appropriate substrate for the in vitro ATG4B activity assay. Purified proteins were cleaved by ATG4B in a time-dependent manner, generating the His-Avi-LC3B or His-GATE-16 and the GST fragments ( Fig. 1B ). Under the experimental conditions, nearly 90% of GATE-16 and only 10% of LC3B could be processed within 5 min. GATE-16 was cleaved by ATG4B more efficiently, which was consistent with a previous report about substrate specificity of ATG4B: GATE-16 > LC3B ≥ Atg8L ≥ GABARAP. 7 Additional experiments showed that GATE-16 provided a better assay window in the TR-FRET assay. The 25-nM GATE-16 cleaved by 0.2 nM ATG4B showed an assay window of 3.2-fold. However, substrate LC3B exhibited an assay window of 1.2-fold ( Fig. 1C ). Based on these observations, substrate GATE-16 was selected for the HTS assay. To further validate this assay, ATG4BC74A (an inactive ATG4B mutant 10 ) was shown to be inactive in cleavage activity ( Fig. 1D ).
To determine the optimal assay conditions, a titration of enzyme (ATG4B), substrate (His-GATE-16–GST), and two detection reagents (Eu-anti-His and ULight-anti-GST) was carried out (data not shown). The time course experiment was also conducted to determine the optimal reaction time ( Fig. 1E ). The binding kinetics of the TR-FRET detection antibody reagents to their corresponding antigens was assessed in the presence of only substrate, showing that half-time of binding was around 12.4 min. ATG4B cleavage of substrate was dependent on the concentration of ATG4B ( Fig. 1E ). Assay condition was finalized based on all these optimization processes. With optimized assay conditions, good assay stability and consistence can be achieved with a Z′ factor of 0.72, which was obtained in a 384-well plate format during test experiment (data not shown). Thus, HTS was ready and then employed to screen a library composed of ~57,000 compounds, including LOPAC, Roche Xplore 50 library, and other compounds selected from a diverse subset of covalent reversible Cys-protease inhibitors or a biostructure-guided selection ( Fig. 2A ).
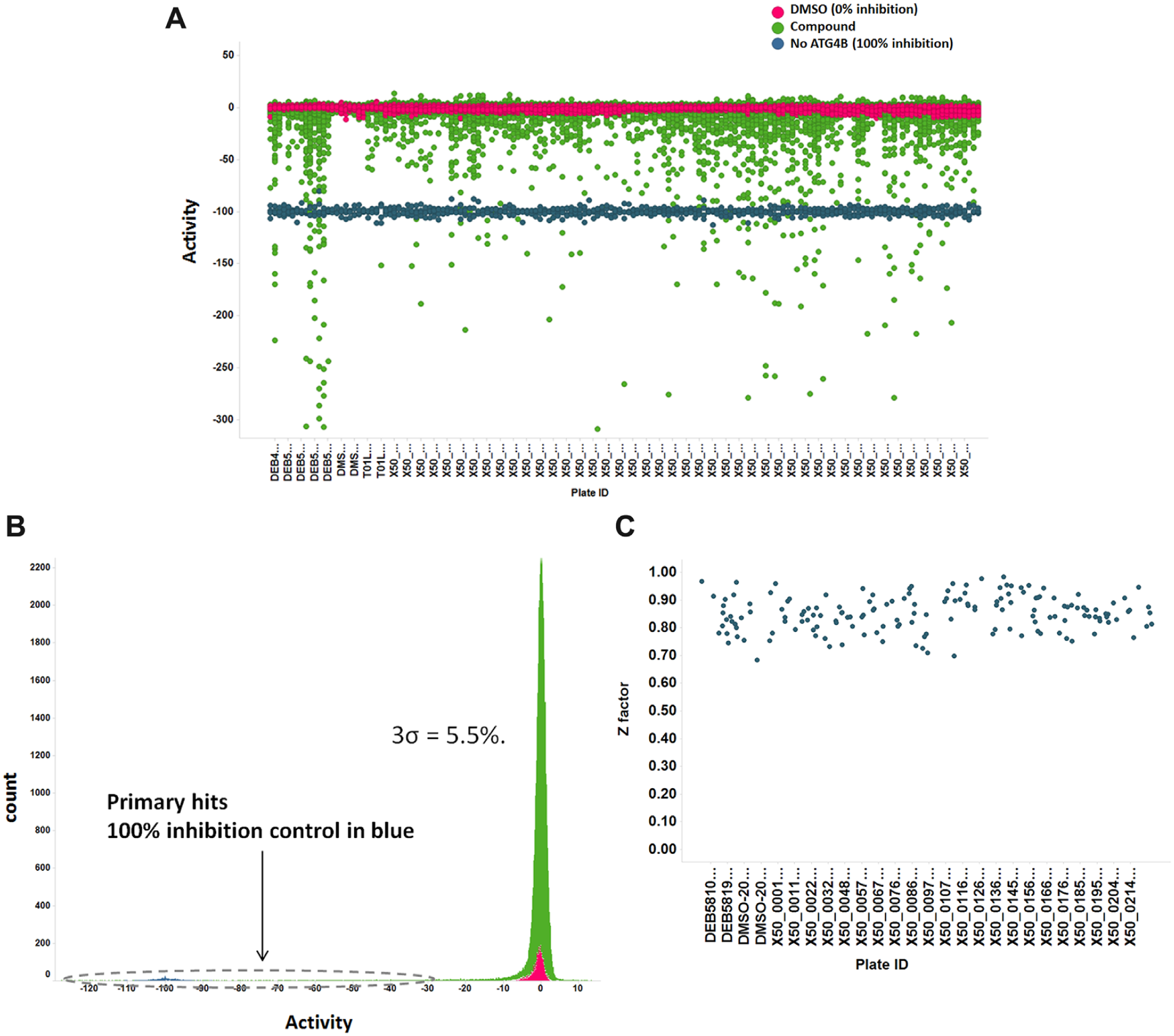
Primary screening result: 57,000 compounds were screened using the time-resolved fluorescence resonance energy transfer (TR-FRET) assay. High-throughput screening (HTS) was very robust and had high-quality performance, with 3σ = 5.5% and Z′ = 0.90. (
To filter out interference caused by the autofluorescence or quenching of compounds, a parallel counterscreen experiment was set up similarly. Basically, all assay conditions remained same, expect that compounds were added at the end so that compounds would not affect cleavage reaction, and thus their interference to the signal readout could be assessed.
The compound library was screened with both ATG4B activity and interference TR-FRET assays at 50 µM at room temperature, with final DMSO concentration of 0.1%. The primary HTS was accomplished with very high quality: 3σ = 5.5% ( Fig. 2B ) and average Z′ = ~0.90 ( Fig. 2C ). From the screening of ~57,000 chemical compounds, we obtained 696 hits (cutoff of 27%, inhibition ≥15σ). These hits were cherry picked for dose-response testing to get IC50 values. Of these hits, 267 were confirmed, constituting a hit rate of 0.49%, while 65 hits showed an IC50 < 50 µM. In addition, the HTS system was highly consistent with stable Z values in multiple plates ( Fig. 2C ).
Natural substrates of ATG4B contain peptide sequence SQE
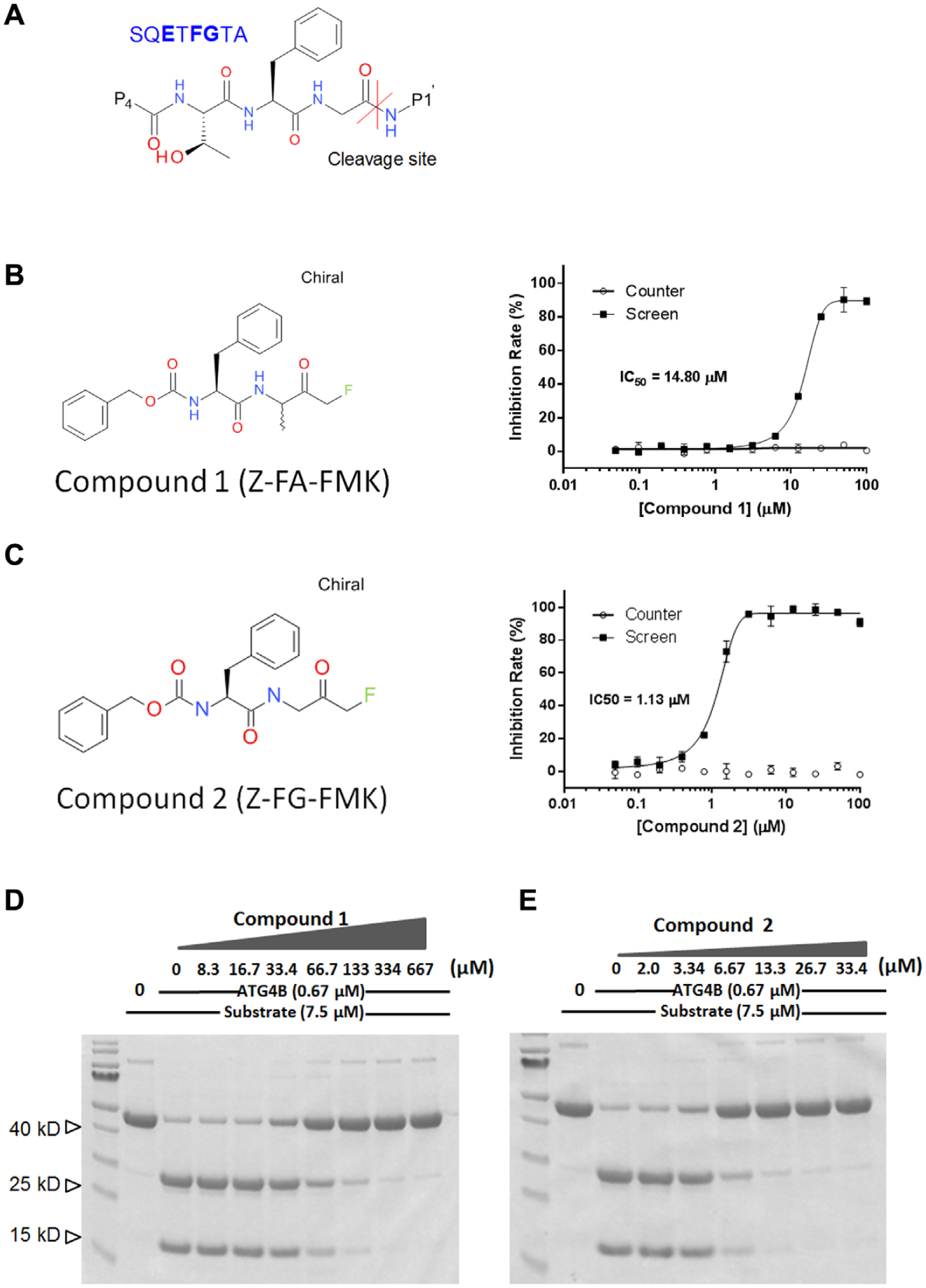
High-throughput screening (HTS) hits that mimic substrate sequence (
LC/MS/MS Confirmed That Compound 2 Covalently Modifies ATG4B Catalytic Cysteine
Both compound
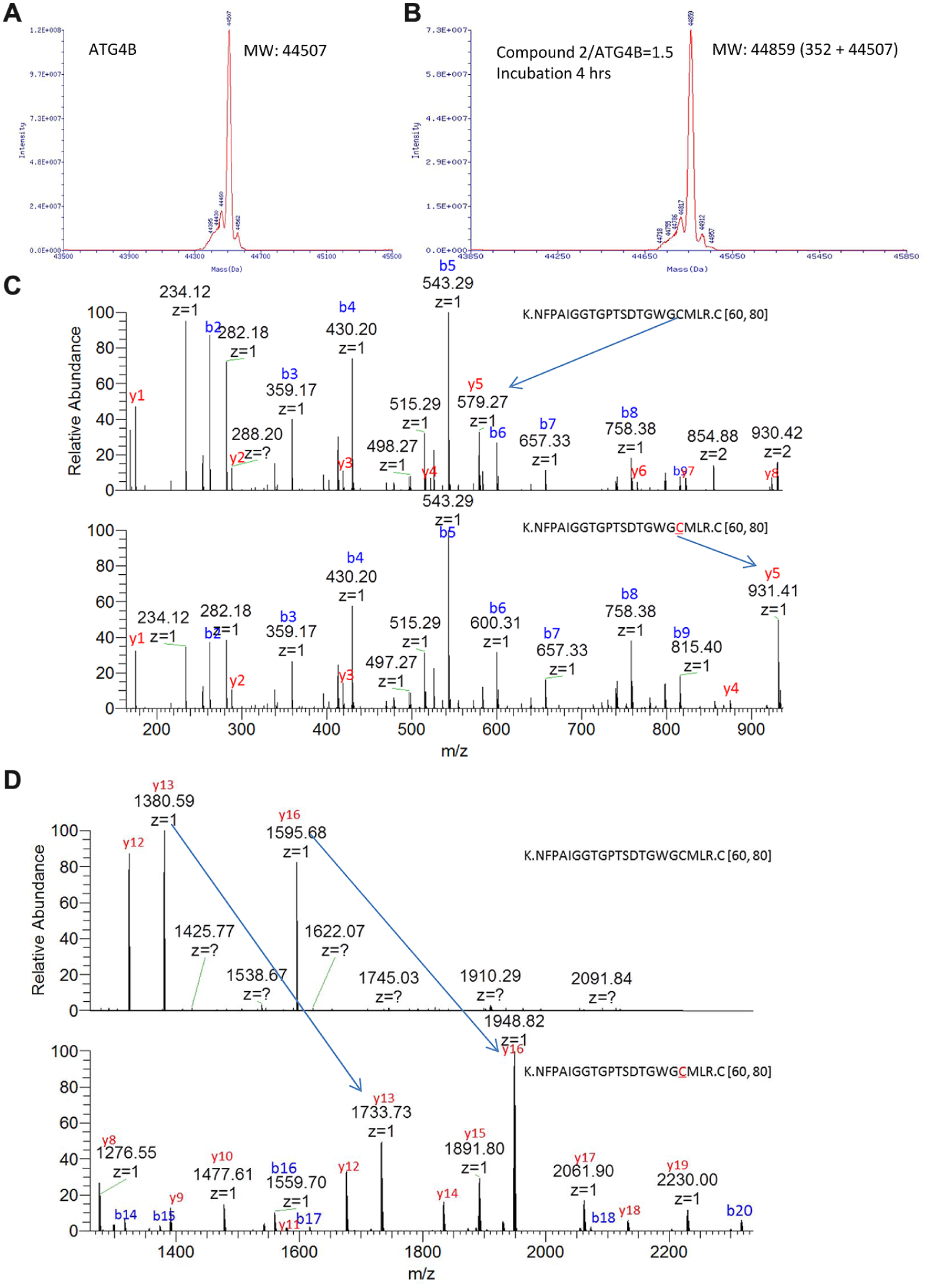
Liquid chromatography/tandem mass spectrometry (LC/MS/MS) data showed that compound
HTS Hits Showed Cellular Activity Monitored by Cell-Based Luciferase Release Assay
After careful validation of the luciferase release assay (data not shown), we used this system to assess the cellular activity of the HTS hits. The principle of this assay was to monitor the level of released dNGLuc in the medium, which reflected the cellular cleavage activity of ATG4B. Gaussia luciferase without an N-terminal fragment, dNGLuc, had an intrinsic tendency to be secreted into cell culture medium, while it could be retained in cells when conjugated to actin. Construct of actin-LC3B-dNGLuc was generated and used as an ATG4B cleavage activity reporter (
Fig. 5A
). A control of dNGLuc was included to filter out the effect of compounds on the secretion process. Since some endogenous serine proteases such as HTRA4 can cleave the C-terminal part of actin, controls of actin-dNGLuc and HTRA4 were included to demonstrate the cleavage specificity of effects observed in the experiment (
Fig. 5A
). As expected, dNGLuc can be detected in the cell culture medium. Actin fusion helps to anchor it in a cellular environment. HTRA4 helps to release luciferase by cleaving the actin part, while ATG4B increases the luciferase release of the construct containing LC3B (
Fig. 5B
). When tested at 10 µM, compound
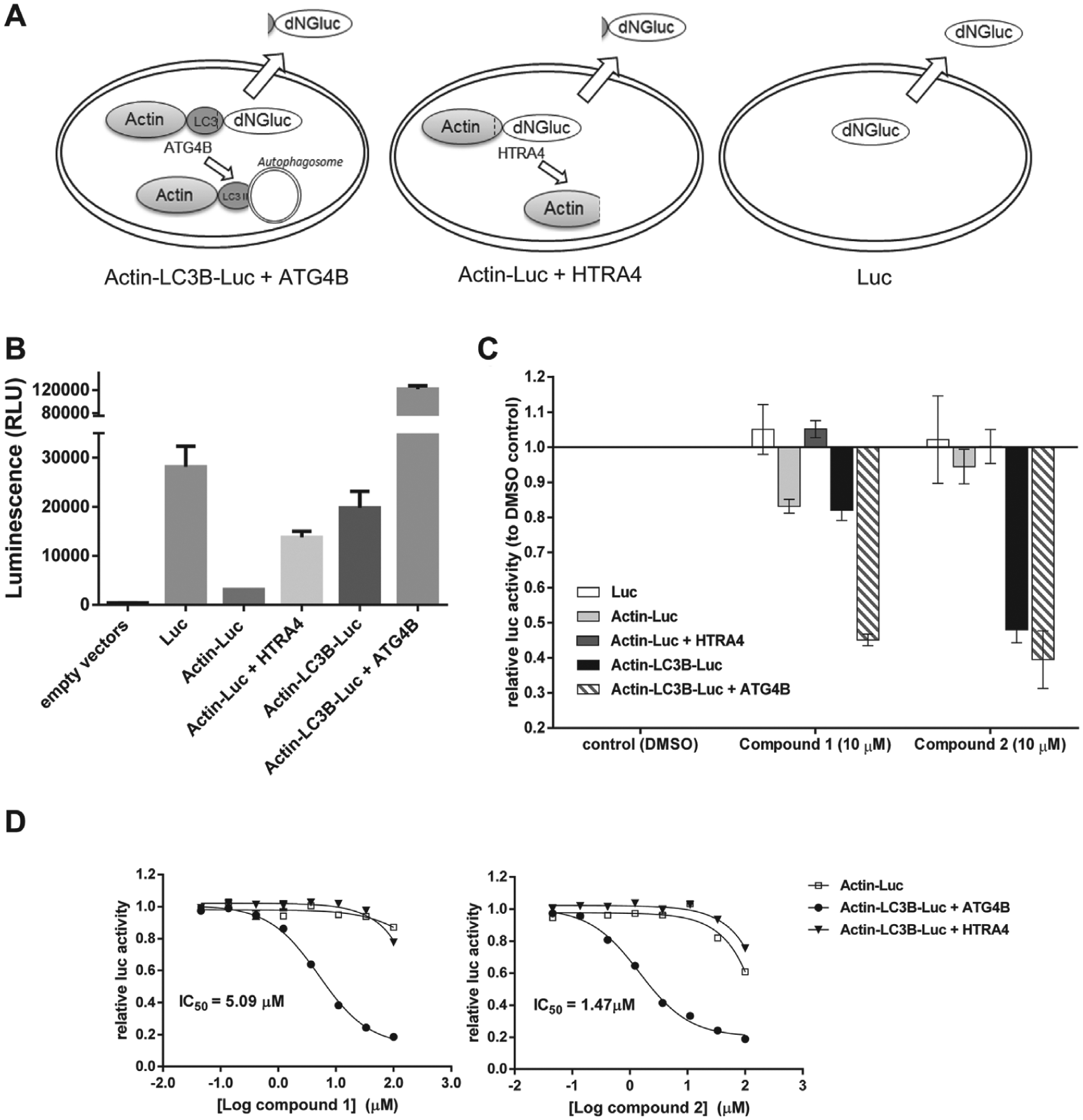
Cell-based luciferase release assay was used to assess the cellular effect of high-throughput screening (HTS) hit compounds. (
In summary, our data showed that we have established a novel and efficient HTS platform to screen ATG4B inhibitors. These screen platforms contained two different screen methods: (1) the TR-FRET screening using His–GATE-16–GST with a parallel counterscreen to filter out the autofluorescent interference and (2) the cell-based ATG4B luciferase release assay. By using these two assays, we managed to identify several hits from 57,000 compounds. With the chemical modification, we obtained compound
Discussion
Autophagy has been studied extensively during the past decade. It has been found that autophagy plays diverse roles in multiple diseases, including neurodegenerative diseases, infectious diseases, metabolic diseases, and cancer. However, the relevance and mechanism of autophagy in pathogenesis remain controversial. To better understand the mechanism of autophagy and its role under normal and pathogenic conditions, access to specific and potent modulators is highly desirable.
ATG4B is a critical player of the autophagy pathway, and its enzymatic activity is essential to initiate the autophagosome formation process. Thus, ATG4B inhibitors might provide novel therapeutic potential in disease treatment. Previously, a LC3B-PLA2 reporter assay was developed for the identification and characterization of Atg4B-specific inhibitors. 11 Unfortunately, no tractable hits were identified, which might be due to the limited size and diversity of the library. In this study, we developed a novel assay that directly detected the enzymatic digestion product of ATG4B and adapted this assay into a robust HTS format. Besides this biochemical assay, a cell-based luciferase assay was also combined to further demonstrate the enzymatic activity of ATG4B in a cellular environment.
Interestingly, one of the most promising HTS hits (compound
The ATG4B TR-FRET assay was a robust HTS platform and could be applied to a large compound library to identify more novel chemical scaffolds. This biochemical assay is straightforward and cost-effective but not able to reflect the membrane permeability and cellular effect under physiological conditions. Combination of a counterassay and a secondary cellular assay would help to minimize the promiscuous effects of compounds in cells. Using the combined platform of the TR-FRET assay and the cellular luciferase release assay, we have successfully identified a chemical series that inhibits ATG4B activity both in the biochemical and in the cellular context. The platform we described in this study could potentially provide a powerful tool to identify inhibitors amenable to further optimizations for therapeutic purposes.
Footnotes
Acknowledgements
The authors thank Rui Niu, Meifang Zhang, and Zhaohu Lin for their help with screening.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.