
Abstract
G-protein–coupled receptors (GPCRs) are an important class of pharmaceutical drug targets. Functional high-throughput GPCR assays are needed to test an increasing number of synthesized novel drug compounds and their function in signal transduction processes. Measurement of changes in the cyclic adenosine monophosphate (cAMP) concentration is a widely used method to verify GPCR activation in the adenylyl cyclase pathway. Here, a single-label time-resolved fluorescence and high-throughput screening (HTS)–feasible method was developed to measure changes in cAMP levels in HEK293i cells overexpressing either β2-adrenergic or δ-opioid receptors. In the quenching resonance energy transfer (QRET) technique, soluble quenchers reduce the signal of unbound europium(III)-labeled cAMP in solution, whereas the antibody-bound fraction is fluorescent. The feasibility of this homogeneous competitive assay was proven by agonist-mediated stimulation of receptors coupled to either the stimulatory Gs or inhibitory Gi proteins. The reproducibility of the assays was excellent, and Z′ values exceeded 0.7. The dynamic range, signal-to-background ratio, and detection limit were compared with a commercial time-resolved fluorescence resonance energy transfer (TR-FRET) assay. In both homogeneous assays, similar assay parameters were obtained when adenylyl cyclase was stimulated directly by forskolin or via agonist-mediated activation of the Gs-coupled β2AR. The advantage of using the single-label approach relates to the cost-effectiveness of the QRET system compared with the two-label TR-FRET assay as there is no need for labeling of two binding partners leading to reduced requirements for assay optimization.
Keywords
Introduction
G-
Cyclic adenosine monophosphate (cAMP) is an important second messenger, which mediates many diverse physiological responses. The cellular cAMP levels are regulated by members of two enzyme families: adenylyl cyclases and cyclic nucleotide phosphodiesterases. cAMP is formed by the enzyme adenylyl cyclase from adenosine triphosphate and degraded by phosphodiesterases. Agonist binding to a Gs-coupled receptor leads to dissociation of the trimeric G protein and Gα-mediated activation of adenylyl cyclase and increased intracellular cAMP levels, whereas agonist stimulation of a Gi-coupled receptor inactivates the enzyme ( Fig. 1A ). The cAMP assay is a common functional assay for GPCRs. Historically, cAMP has been measured by radioisotopic methods and later on using reporter gene–based methods. 2,3 Over the years, several homogeneous assay platforms for cAMP measurements in intact cells have been developed. 4–8 HTRF (CISbio International, Bagnols-sur-Cèze, France), 5 HitHunter EFC (DiscoveRX, Fremont, CA), 6 AlphaScreen, 9 and Lance 10 (PerkinElmer Life Sciences, Inc., Boston, MA) are examples of sensitive commercially available assays for homogeneous cAMP detection.
(
We have developed a novel homogeneous high-throughput screening (HTS) assay feasible for measuring cAMP in intact cells. The assay is based on the quenching resonance energy transfer (QRET) technique 11–13 using a single-label approach and soluble quenchers affecting the fluorescence of the unbound labeled binding component. Less fluorescence quenching is observed when the labeled component binds to an antibody. The assay is based on the nonlabeled intracellular cAMP and exogenously added Eu-labeled cAMP, which compete for binding to the anti-cAMP antibody after cell lysis ( Fig. 1B ). Stably transfected human embryonic kidney (HEK) 293i cells overexpressing human β2-adrenergic receptors (β2AR, coupled to Gs) or human δ-opioid receptors (δOR; coupled to Gi) were used as model systems.
Materials and Methods
Materials
Dulbecco’s Modified Eagle’s Medium (DMEM) with high glucose, fetal bovine serum (FBS), penicillin/streptomycin solution, L-glutamine, and trypsin/EDTA solution was purchased from Euroclone (Milano, Italy). Blasticidin S and hygromycin were from Invitrogen (Carlsbad, CA). Tetracycline, forskolin, 3-isobutyl-1-methylxanthine (IBMX), γ-globulin, and the β2AR agonists metaproterenol, terbutaline, and epinephrine were obtained from Sigma-Aldrich (St. Louis, MO). Phosphate-buffered saline (1×PBS) for cell culture was obtained from Lonza (Basel, Switzerland). The δOR agonist SNC-80 [(+)-4-[(αR)-α-((2S,5R)-4-Allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N,-diethylbenzamidin] was purchased from Tocris (Ellisville, MO). Triton X-100 was from Fluka (Buchs, Switzerland). The quencher was obtained from QRET Technologies (Turku, Finland). Anti cAMP antiserum, europium(III)-labeled cAMP (cAMP-Eu), AD0262 LANthanide Chelation Excitation (LANCE) cAMP 384 kit, and Optiplate 384F were obtained from PerkinElmer, Wallac (Turku, Finland).
Cell culture
Preparation of stably transfected HEK293i cell lines overexpressing human β2ARs or human δORs by transfecting Flp-In-293 cells (Invitrogen) have been described elsewhere. 14,15 We used δOR construct codes as receptor carrying phenylalanine at position 27. 15 Cells were cultured in DMEM, containing 10% FBS, 100 U mL−1 penicillin, 0.1 mg mL−1 streptomycin, 2 mM L-glutamine, 1 µg mL−1 blasticidin, and 100 µg mL−1 hygromycin in a humidified atmosphere of 5% CO2 at 37 °C. Cells were cultured to 80% to 90% confluency in 100-mm culture plates and then passaged approximately every 3 days with the use of trypsin/EDTA solution. The δOR cells were induced to express the receptor at 50% to 60% confluency by adding 0.5 µg mL−1 tetracycline to the growth medium 24 h before harvesting. The β2AR cells were not inducing due to the loss of the Tet repressor expression. These cells express the receptor in a constitutive manner (~7 pmol/mg). 14
cAMP assays
Cells overexpressing β2ARs were cultured to near confluency on 10-cm culture plates, harvested in the growth medium with a scraper, and washed once with the growth medium and counted under a microscope after centrifugation (100 × g, 5 min). For the experiment, the cell growth medium was changed to the stimulation buffer containing 0.16 mM IBMX and 1 mg mL−1 γ-globulin in PBS. To a 384-microplate, 6 µL of the stimulation buffer including metaproterenol at concentrations of 0.1 or 100 nM, 6 µL of the cell suspension with a varying number of cells (1000, 3000, 6000, 9000, or 12 000 cells per well), and the anti cAMP antibody (3 nM) were added. The reaction mixture was incubated for 30 min at room temperature in the dark with gentle shaking. Thereafter, 12 µL of detection buffer including 1.5 nM cAMP-Eu, 12 mg mL−1 Triton X-100, and the quencher in 50 mM Hepes pH 7.4 was added, and the final reaction solution was incubated for another 20 min at room temperature in the dark with gentle shaking to achieve the reaction equilibrium. The time-gated fluorescence signal was measured at 615 nm using 340-nm excitation wavelength, 400-µs delay, and 400-µs integration times with a Victor 2 1420 multilabel counter (PerkinElmer, Wallac).
For the agonist stimulation assay, cells were cultured and assayed using the same protocol as above with 9000 cells per reaction. Varying concentrations (0.003–10 000 nM) of metaproterenol, terbutaline, or epinephrine were added in the stimulation buffer to activate β2AR. For the measurement of the cAMP calibration curve, the same assay conditions were applied without added agonists or cells using exogenously added cAMP at concentrations from 0.1 to 10 000 nM.
The developed QRET assay was compared with the time-resolved fluorescence resonance energy transfer (TR-FRET) LANCE cAMP 384 assay from PerkinElmer for their assay parameters. The same assay protocol was adapted to the QRET assay as above. The LANCE assays were carried out according to the manufacturer’s protocol 10 in parallel with the QRET assay using the same ligand dilution for both assays. Metaproterenol-mediated stimulation of β2AR was monitored at concentrations from 0.011 to 10 000 nM. Adenylyl cyclase was chemically activated with forskolin at concentrations from 1 to 100 000 nM in addition to receptor stimulation. The statistical Z′ value is commonly used as an indicator of assay robustness and value should exceed 0.5 to be adaptable to HTS. The Z′ values were calculated for both experiments by using the equation 1-[(3σmax + 3σmin) / (µmax − µmin)], where µ is the signal mean and σ the standard deviation. 16
Upon activation of Gi-coupled receptors, the adenylyl cyclase activity was reduced. This was tested using the HEK293i cell line overexpressing δORs. Cells were cultured and harvested in the same manner as the cells overexpressing β2ARs. The stimulation buffer contained both 10 µM forskolin and the δOR agonist SNC-80 at concentrations ranging from 0.03 to 30 nM. The assay conditions were the same as described above for β2AR using 9000 cells per reaction.
Results
The principle of the cAMP-QRET assay is outlined in Figure 1B . Agonist-mediated activation of GPCRs coupled to either the stimulatory Gs protein (β2AR) or the inhibitory Gi protein (δOR) leads to modulation of adenylyl cyclase activity and cAMP production. In the QRET assay, the endogenous cAMP competes with the exogenously added Eu-labeled cAMP for binding to the cAMP-specific antibody, and a high fluorescence signal is observed when the Eu-labeled cAMP binds to the antibody. This occurs when adenylyl cyclase is activated via β2AR and intracellular cAMP levels are increased. In comparison, an increased fluorescence signal is detected following inactivation of adenylyl cyclase via δOR, which reduces the intracellular cAMP production. The fluorescence of the unbound Eu-labeled cAMP is efficiently quenched in solution by a soluble quencher. High fluorescence signal corresponds to low cAMP concentration and the low signal to high cAMP concentration because of the competitive nature of the assay.
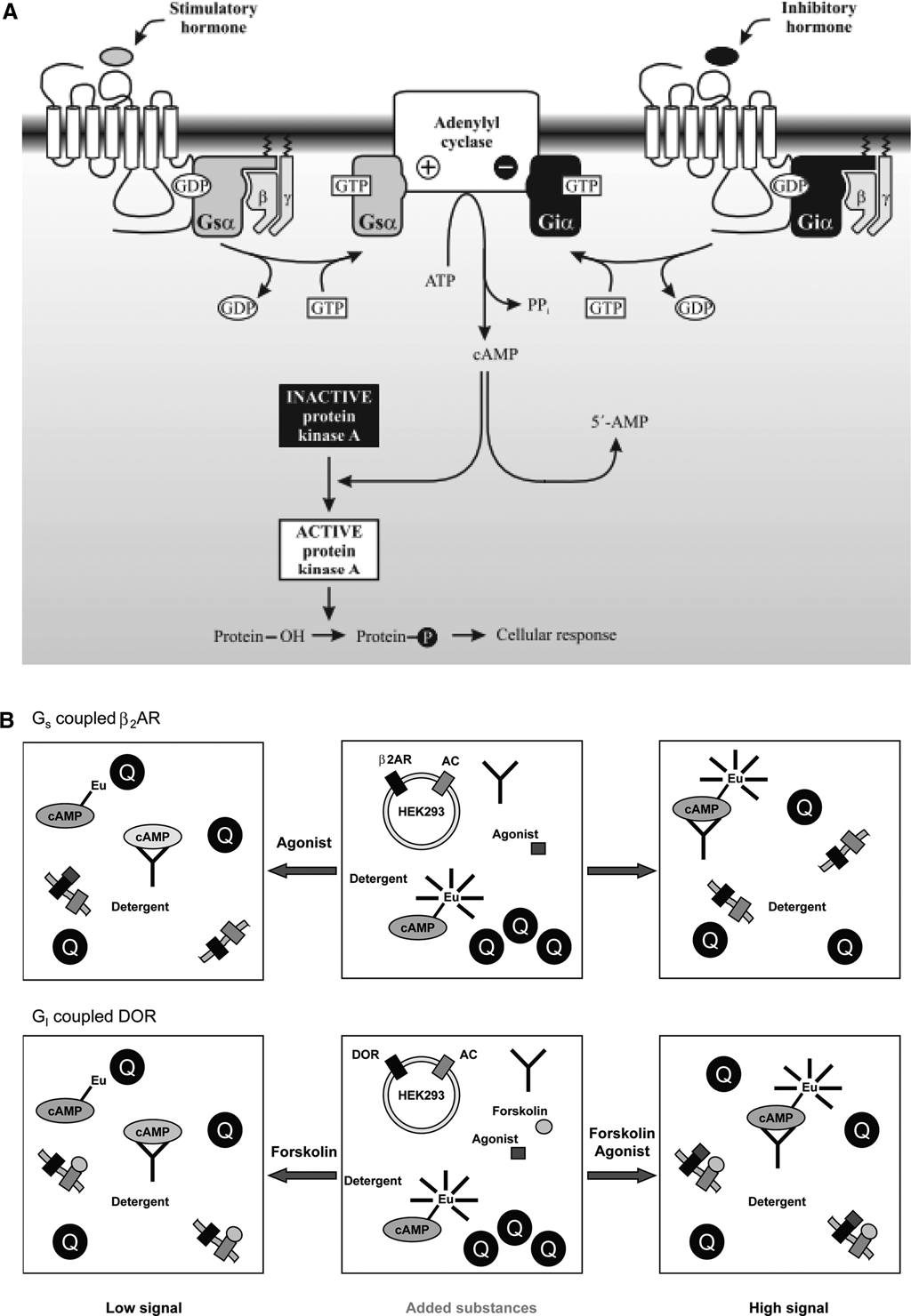
Prior to GPCR activation by receptor-specific agonists, we measured the calibration curve for cAMP at equilibrium under optimized conditions. This was carried out using increasing concentrations of added cAMP (0.1-10 000 nM) in a biochemical system without cells (
Fig. 2
). The EC50, signal-to-background (S/B) ratio, and the average assay signal variability (CV) were 44 nM, 12.6 and 5.1%, respectively. Varying concentrations of DMSO (0-10 % of the assay total volume) was tested in the developed assay. When DMSO concentration increased from 0 to 10 %, the time-resolved fluorescence signal decreased by 24% and increased by 27% at 0 and 10 µM concentrations, respectively (see
Calibration curve for cyclic adenosine monophosphate (cAMP) in a biochemical assay without cells. cAMP was used at concentrations from 0.1 to 10 000 nM. The EC50 value was 44 nM.
Cell number optimization for the QRET assay
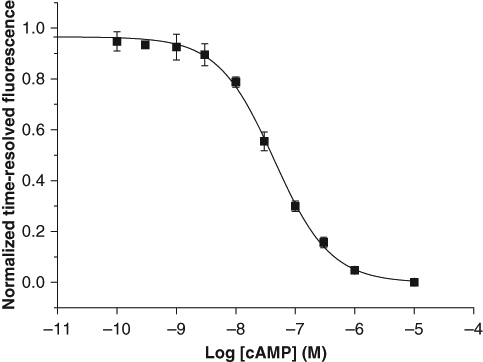
The cAMP assay was developed for a stably transfected HEK293i cell line overexpressing human β2ARs. The number of cells required for the cAMP-QRET assay was assessed by stimulating the receptor with an agonist metaproterenol at concentrations of 0.1 and 100 nM ( Fig. 3 ). The saturation of the S/B ratio was identical at cell counts higher than 3000 cells per assay for both tested ligand concentrations. This led us to employ 9000 cells in a reaction in all subsequent experiments.
Optimization of the cell number. HEK293i cells overexpressing β2-adrenergic receptors were activated with metaproterenol at concentrations of 100 pM (●) or 100 nM (■). Identical saturation levels for signal-to-background ratios were measured at low and high metaproterenol concentrations when the number of cells varied from 0 to 12 000 cells per assay.
The cAMP-QRET assay and Gs-coupled β2AR
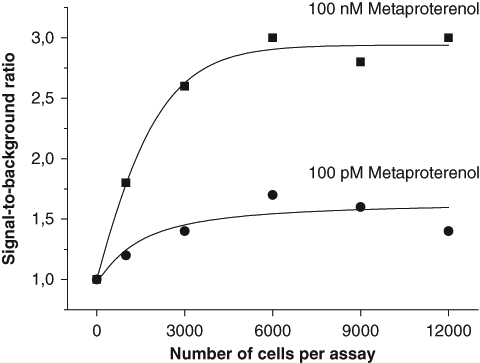
The functionality of the developed cAMP method was assayed using β2AR-expressing HEK293i cells that were stimulated with β2AR agonists with differing efficacies and potencies: epinephrine, metaproterenol, and terbutaline. As expected, both the EC50 and Emax values differed for the three ligands tested. The EC50 values were 0.056, 3.6 and 2.9 nM and the detection limits (90% of signal) 4.2, 450 and 400 pM for epinephrine, metaproterenol, and terbutaline, respectively ( Fig. 4 ). Epinephrine acted as a full agonist having the highest Emax value. The partial agonists metaproterenol and terbutaline had Emax values that were 75 and 74% of that of epinephrine, respectively. The signal variability (CV) of the assays varied from 1 to 15 %.
Stimulation of cyclic adenosine monophosphate (cAMP) accumulation by β2-adrenergic receptor (βAR) agonists in HEK293i cells expressing the Gs-coupled β2AR. Partial agonists metaproterenol (■) and terbutaline (●) and the full agonist epinephrine (▲) were used to test the functionality of the assay. The agonists were investigated at 0.03 to 10 000 nM concentrations. The EC50 values were 0.056, 3.6, and 2.9 nM, and the detection limits were 7.5, 610, and 536 pM for epinephrine, metaproterenol, and terbutaline, respectively.
Comparison of the QRET and TR-FRET methods
To compare the developed QRET assay with the commercial LANCE assay, the effect of the adenylyl cyclase activator forskolin on intracellular concentrations of cAMP was measured. The adenylyl cyclase in β2AR expressing HEK293i cells was stimulated by varying concentrations of forskolin (1-30 000 nM) and changes in the time-resolved fluorescence signal were monitored. Following the commercial LANCE kit protocol and the optimized assay conditions for the QRET method, the assays were performed in parallel using the same forskolin solutions ( Fig. 5 ). The EC50 values of 89 and 32 nM, the detection limits of 6.6 and 6.3 nM and the S/B ratios of 5.1 and 3.3 were calculated for the QRET and the LANCE assays, respectively ( Fig. 5 ). This indicates that both methods measure cell-based cAMP levels with high efficiency. The Z′ values were calculated to study the robustness of the assay. The Z′ values for the QRET and LANCE assays were 0.72 and 0.85, respectively. The signal variability for the QRET assay varied from 1 to 12 % and for the LANCE assay from 1 to 7 %.
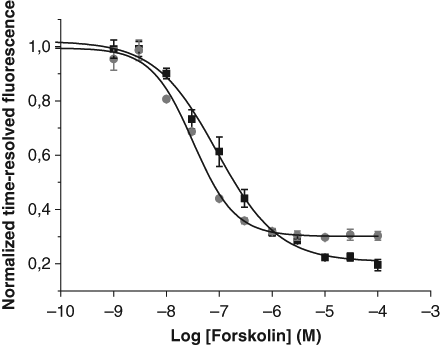
Comparision of quenching resonance energy transfer (QRET) (■) and LANthanide chelation excitation (LANCE) time-resolved fluorescence resonance energy transfer (TR-FRET) (●) assays following forskolin-mediated stimulation of adenylyl cyclase. Adenylyl cyclase in HEK293i cells overexpressing β2 adrenergic receptors was activated with forskolin at concentrations ranging from 0.01 to 100 000 nM. The EC50 values for the cyclic adenosine monophosphate (cAMP)–QRET and the LANCE cAMP assays were 89 nM and 32 nM, respectively. The detection limits were 8.4 and 6 nM, and the S/B ratios were 5.1 and 3.3 for the QRET and the LANCE assays, respectively.
The functionality of the QRET and TR-FRET cAMP assays were further investigated and compared in a cell-based assay, in which β2ARs were activated by the partial agonist metaproterenol ( Fig. 6 ). The EC50 value for the QRET assay was 3.2 nM and the detection limit was 0.35 nM; for the LANCE assay, the EC50 value was 1.5 nM and the detection limit was 0.42 nM ( Fig 6 .). The signal imprecision for the QRET assay was from 0 to 12 % giving a Z′ value of 0.76, and the S/B ratio was 4.5. The corresponding values for the LANCE assay were 1-3 % for CV, 0.95 for the Z′ value and 3.3 for the S/B ratio.
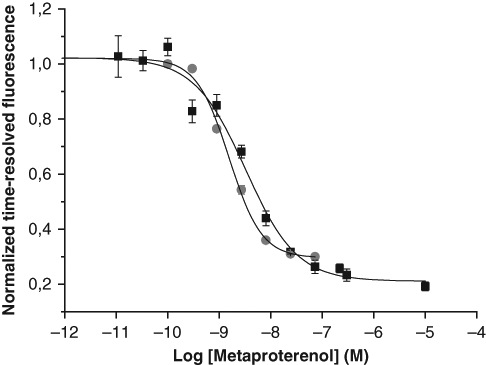
Comparision of quenching resonance energy transfer (QRET) (■) and LANthanide chelation excitation (LANCE) time-resolved fluorescence resonance energy transfer (TR-FRET) (●) assays following agonist-mediated stimulation of β2-adrenergic receptors (β2ARs) in HEK293i cells. Metaproterenol-mediated activation of β2ARs was studied at concentrations ranging from 0.01 to 10 000 nM. The EC50 values were 3.2 and 1.5 nM, the detection limits 0.14 and 0.45 nM, and the signal-to-background ratios 4.5 and 3.3 for the QRET and TR-FRET assays, respectively.
The cAMP-QRET assay and Gi-coupled δOR
Having proven the capability of the cAMP-QRET assay for a Gs-coupled GPCR that stimulates adenylyl cyclase, assay parameters were investigated for an inhibitory Gi-coupled GPCR. To this end, the response of stably transfected HEK293i cells overexpressing human δORs to a selective opioid agonist SNC-80 was measured. Cells were incubated with increasing concentrations of agonist in the presence of 10 µM forskolin to enhance the response window. As expected, an increasing fluorescence signal was observed upon SNC-80 activation of the receptor ( Fig. 7 ), proving the negative effect on cAMP production. The EC50 value of 0.68 nM was measured for SNC-80 and the signal variability varied from 1 to 15%.
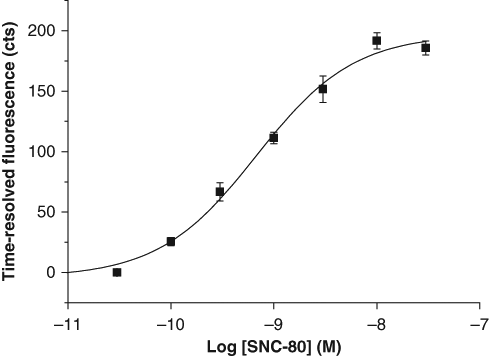
Inhibition of cyclic adenosine monophosphate (cAMP) accumulation by SNC-80 in HEK293i cells overexpressing the Gi-coupled δ-opioid receptors (δOR). The cAMP production by activation of adenylyl cyclase with forskolin (10 µM) was inhibited with increasing concentrations (0.03–30 nM) of the δOR-specific agonist SNC-80. The EC50 value of 0.68 nM and the detection limit of 0.1 nM were calculated for the inhibition curve.
Discussion
Previously, the potential of the QRET technique has been demonstrated for receptor-ligand and GTP binding studies on cells and cell membranes. 11,13 Here we have developed a cell-based homogeneous assay for the second messenger, cAMP, using a time-resolved fluorescence single-label approach. The cAMP assay was developed using a stably transfected HEK293i cell line overexpressing human β2ARs. The number of receptors 240 000 per cell was determined previously in a saturation ligand-binding assay for the cell line. 14 In this study, the optimized cell count in an assay was investigated, and 3000 cells per assay were sufficient to obtain saturation of S/B ratio although 9000 cells in a reaction were eventually employed. DMSO tolerance is one of the important parameters needed in the evaluation any method for the HTS use. Our data suggests that DMSO is well tolerated by the cAMP immunoreaction. The cell-based QRET method has also been shown to tolerate DMSO well in a receptor-ligand binding assay. 11 HTS applications also require stable signal to enable off-line detection. This has been studied in a cell-based QRET method earlier, and a stable signal up to 72 hours has been reported. 11
The feasibility of the developed method was proven by comparing the assay performance with the commercial homogeneous TR-FRET assay (LANCE cAMP kit from PerkinElmer). The choice of the reference method is justified because the two techniques share the same detection features: excitation at 340 nm and time-resolved fluorescence detection above 600 nm with a temporal resolution leading to reduced background fluorescence. This also enables the use of a single instrument equipped with a red sensitive detector for both assays, although different emission filter setups must be applied. TR-FRET-based homogeneous assays have been proven to be one of the most sensitive and applied methods for the measurement of intracellular cAMP levels, enabling a direct evaluation of the developed method for HTS use. 17 The performance of the developed assay was similar to the commercial assay whether adenylyl cyclase was stimulated directly with forskolin or β2AR was stimulated with agonists. The obtained EC50 values for the agonists are in accordance with those reported in previous studies. 18-20 Minor differences in the concentration response curves and calculated assay parameters were considered insignificant. This is not surprising as the same cAMP-specific antibody was applied in both assays.
The functionality of the cAMP-QRET assay was further proved in the inhibitory Gi-coupled GPCR model. The response of the stably transfected HEK293i cells overexpressing human δOR to a selective opioid agonist SNC-80 was successfully measured. The determined EC50 value of 0.65 nM was comparable to previously reported values in other heterologous expression systems. 21-23
The main advantage of the QRET assay is the use of a single label, making this method more cost-effective for assay development and end-users. In this particular TR-FRET assay, labeling of the soluble antibody is relatively straightforward but requires optimization, adding extra efforts to the assay development. However, in homogeneous methods such as cell-based receptor-ligand and GTP-binding assays, the distance-dependent approach is not readily applicable because membrane-embedded receptors may not be accessible to labeling or highly specific antibodies are difficult to produce to target molecules such as GTP. Labeling procedures always add another factor of variation between batches, including possible reduction of binding sites (e.g., of the cAMP antibody). These assay-related problems can be solved using a single-label approach. We have previously demonstrated that receptor-ligand and GTP binding assays can be constructed on the QRET concept. 11,13 Fluorescence polarization is another known single-label approach for drug-screening purposes. However, the drawback of this method is the use of conventional fluorescence having 3-4 orders of magnitude less sensitive detection limit than time-resolved fluorescence 24 and a low S/B ratio, 4,12,25,26 reducing the applicability of the method for many target molecules.
In conclusion, the changes in intracellular cAMP levels were successfully measured following agonist activation of both Gs- and Gi-coupled GPCRs using the single-label QRET technique. The developed assay requires no separation or washing steps, making it simple and fast to use and highly suitable to HTS. The single-label approach of the QRET technique provides a powerful tool for screening purposes in a cost-effective manner as there is no need for labeling of a receptor or antibody, reducing optimization steps and cost of the assay. The feasibility of the method was proven in a comparative study with the commercial LANCE cAMP method. Similar parameters for both assays were measured for the β2AR agonist- and forskolin-mediated stimulation of adenylyl cyclase. This is the first demonstration of the QRET method in a cell-based signal transduction assay with assay parameters equal to well-established commercial methods, showing its capability for efficient monitoring of intracellular events.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.