
Abstract
Identification of potential lead antibodies in the drug discovery process requires the use of assays that not only measure binding of the antibody to the target molecule but assess a wide range of other characteristics. These include affinity ranking, measurement of their ability to inhibit relevant protein-protein interactions, assessment of their selectivity for the target protein, and determination of their species cross-reactivity profiles to support in vivo studies. Time-resolved fluorescence resonance energy transfer is a technology that offers the flexibility for development of such assays, through the availability of donor and acceptor fluorophore-conjugated reagents for detection of multiple tags or fusion proteins. The time-resolved component of the technology reduces potential assay interference, allowing screening of a range of different crude sample types derived from the bacterial or mammalian cell expression systems often used for antibody discovery projects. Here we describe the successful application of this technology across multiple projects targeting soluble proteins and demonstrate how it has provided key information for the isolation of potential therapeutic antibodies with the desired activity profile.
Introduction
Monoclonal antibodies have become an important class of therapeutics for a wide range of diseases indications. 1 They can be generated using immunization in vivo 2 or in vitro technologies such as phage display.3,4 Each of these methods presents its own challenges for the development of assays to identify antibodies with the desired activity profile.
In vivo approaches use the immune system of a host animal, for example, a wild-type or a transgenic mouse that expresses human immunoglobulin genes, to generate antibodies to the target antigen of interest. 5 Isolated B cells from the immunized mouse are stably fused with immortalized myeloma cells to yield single antibody producing hybridomas. The antibodies are secreted into the culture medium, which can then be tested for the presence of an antibody that is specific for the target of interest. A robust, high-throughput assay that is able to identify antibodies with the desired binding and activity profile and is tolerant to other components of the cell culture media is therefore required for the screening.
Phage display uses bacteriophages that express antibody fragments (e.g., single-chain fragment variable [scFv]), in fusion with minor coat proteins on their surface, providing a link between genotype and phenotype. Immunoglobulin variable heavy chain (VH) and light chain (VL) genes isolated from human donors are used to generate phage libraries of up to 1011 in size. 6 Multiple rounds of selection by binding of the phage-displayed antibody fragments to an antigen of interest, followed by elution of the antigen-antibody complex, allow for the enrichment of antigen-specific antibodies from these libraries. The scFv are expressed in Escherichia coli through a system that mimics eukaryotic folding and assembly by using the periplasmic space of E. coli instead of the lumen of the endoplasmic reticulum. Periplasmic extracts are prepared from the E. coli using osmotic shock buffers, and these crude preparations are screened for activity. These samples will contain antibody fragments, bacterial contaminants, and endotoxin produced by the E. coli. Thus, choice of assays that can tolerate these sample types can be limited. Further challenges are provided by the concentration of the scFv in the crude samples (<10 µg/mL) 7 and their relatively low affinity. Although it is possible to isolate antibodies with subnanomolar affinity directly from phage display libraries, during the early stages of antibody “lead isolation,” many of the scFv are of low affinity (>100 nM).8,9 Therefore, the assay used for the screening of these crude samples must be of high sensitivity.
A high-throughput assay format that is robust, sensitive, cost-effective, and adaptable is desirable for both antibody generation methods. 10 Enzyme-linked immunosorbent assays (ELISAs) have been used routinely for antibody screening to isolate “hits” that could then be purified for further profiling. However, these are relatively low throughput due to the multiple wash steps, and in most cases, they only assess the binding of the antibody-antibody fragment to the target antigen. Prompt fluorescence-based homogeneous assays formats generate strong assay signals with good sensitivity, but the signals can be affected by optical interference from assay media (serum and biological fluids), resulting in variable background and light scattering. 11 Time-resolved fluorescence resonance energy transfer (TR-FRET) is a technology that can overcome some of these challenges.
Förster fluorescence resonance energy transfer occurs when an excited donor fluorophore comes into close proximity to an acceptor fluorophore molecule that has overlapping excitation spectra to the donor emission spectra and a compatible dipole orientation. The result is a nonradiative energy transfer from donor to acceptor, which then emits a fluorescent signal. TR-FRET uses the long-lived fluorescence properties of the lanthanide metals for measurement of time-resolved fluorescence.12,13 Lanthanide complexes such as those using europium, terbium, samarium, or dysprosium have long fluorescent lifetimes in the microsecond to millisecond range compared with the nanosecond lifetime of other fluorescent molecules. Thus, a time delay from excitation of the donor lanthanide to measurement of the acceptor emission minimizes interference from nonspecific fluorescence of buffers, media components, and proteins. They also have large Stokes shifts, further reducing background signals. 14 Binding partners are directly labeled with the lanthanide donor and acceptor fluorophores or detected using fluorescently labeled secondary reagents, making the system accessible for the measurement of many different binding interactions. 15 Europium or terbium cryptate or chelates are the most commonly used for TR-FRET–based assays. In 1996, Kolb et al. 16 described the use of these assays for high-throughput screening (HTS) where the donor was a lanthanide cryptate and the fluorescent acceptor molecule was XL665 in a system called homogeneous time-resolved fluorescence (HTRF).
HTRF has become widely implemented for small-molecule HTS as it provides a platform for robust, reproducible mix-and-read assays, with reduced reagent usage through use of higher density plate formats. 17 The commercially available tool box reagents and availability of protein labeling kits allow the flexibility of assay adaptation for the interrogation of interactions involving many different soluble proteins. Here we describe how different configurations of the assays can be used for HTS of IgG and scFv in crude (unpurified) sample preparations and for profiling purified IgG and scFv to provide information on biological activity, species cross-reactivity, specificity, relative affinity, and epitope. Example data are shown from antibody projects targeting PrPc (cellular prion protein), PlGF (placental growth factor), FGFR (fibroblast growth factor receptor), CXCL13 (chemokine C-X-C motif ligand 13), and IL-17 (interleukin-17) to demonstrate how these assays provide key information to enable isolation of a potential therapeutic antibody.
Material and Methods
Selections and Preparation of Bacterial Periplasmic Extracts
Phage display selections were performed according to standard methods6,18 using naive libraries and recombinantly expressed proteins. Multiple rounds of selection were carried out prior to screening of the scFv in crude bacterial periplasmic extracts prepared in osmotic shock buffer (50 mM 4- morpholinepropanesulfonic acid [MOPS], 0.5 mM EDTA, and 500 mM sucrose, pH 7.4) according to previously described methods. 19
Preparation of Purified scFv and IgG
Purified scFv with C terminal His and Myc tags were produced by cloning into the phagemid vector pCantab6 and expression in E. coli TG1 cells. Expression was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) induction and cells harvested by centrifugation. The protein was removed from the periplasm using osmotic shock buffers and the scFv purified by affinity chromatography using Histrap FF (GE Healthcare, Little Chalfont, UK) and then buffer exchanged using NAP10 columns (GE Healthcare), according to the manufacturer’s protocol. Purified scFv were quantified using a bicinchoninic acid (BCA) assay (Perbio Science, Cramlington, UK). Reformatting of scFv fragments to IgG formats was performed as described by Persic et al. 20 Heavy and light chain IgG-expressing vectors were transfected into Chinese hamster ovary (CHO) or human embryonic kidney (HEK) mammalian cells. Supernatants were pooled and filtered, and then IgG was purified using a Ceramic Protein A column (Pall Life Sciences, Portsmouth, UK). The IgG was eluted using 0.1 M sodium citrate (pH 3.0). The eluant was neutralized by the addition of Tris-HCl (pH 9.0), and buffer was exchanged into phosphate-buffered saline (PBS) using NAP10 columns and the concentration of IgG determined spectrophotometrically.
Europium Cryptate Labeling
Antibodies (100–200 µg) in 0.1 M sodium hydrogen carbonate/PBS were incubated with trisbipyridine-Eu3+ cryptate-N-hydroxysuccinimide (NHS) (Cisbio, Codolet, France) at a cryptate to protein ratio of 13–14:1 (pmol) for 100 min at room temperature in the dark. The solution was applied to a PBS-equilibrated D-Salt Dextran Desalting column (Perbio Science, Cramlington, UK) and the appropriate fraction analyzed by mass spectrometry to confirm labeling. Recombinant soluble human FGFR1β (IIIc)–Fc and FGFR2β (IIIc)–Fc (R&D Systems Europe, Abingdon, UK) were labeled using a trisbipyridine-Eu3+ cryptate N-hydroxysuccinimide labeling kit (Cisbio) according to the manufacturer’s instructions.
Biotinylation of Antibodies
Antibodies (150 µg) in 0.1 M sodium hydrogen carbonate/PBS were incubated with the NHS-LC-Biotin (Perbio Science, Cramlington, UK) at a ratio of 1:3 to 1:4 protein/biotin (pmol). Protein was purified from the reaction mixture using PBS-equilibrated Zeba Desalt Spin columns (Perbio Science) according to the manufacturer’s instructions and biotin incorporation measured by mass spectrometry.
HTRF Assays
All assays were performed in 384-well black shallow-well plates in assay buffer containing PBS, 0.1% (v/v) bovine serum albumin (BSA) (Sigma, St. Louis, MO), and 0.4 M Potassium Fluoride (KF) (VWR, Lutterworth, UK) unless otherwise stated. Samples were prepared in 96-well or 384-well plates and transferred to the assay plates using a MiniTrak (PerkinElmer, Waltham, MA). All other reagents were added using a Multidrop (Thermo Electron Corporation, Waltham, MA). Plates were read on an EnVision (PerkinElmer, Waltham, MA) plate reader by measurement of time-resolved fluorescence emission (50-µs delay) at 665 nm and 590 nm, measured following excitation at 320 nm. Ratio values of (665 nm emission/590 nm emission) × 10,000 were used to calculate Delta F% according to the following equation:
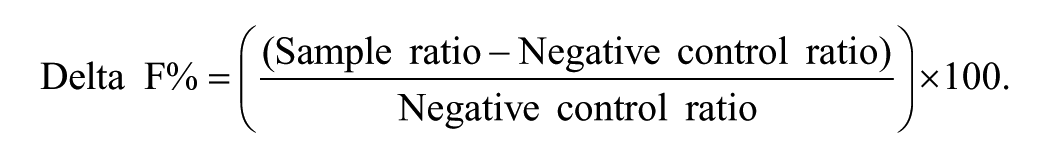
The negative control ratio was derived from nonspecific binding (NSB) control wells.
IC50 curves were analyzed using GraphPad Prism software (GraphPad, La Jolla, CA) with a 4-parameter logistic curve-fitting equation.
To optimize the HTRF assays, we assessed different configurations with respect to donor/acceptor-labeled binding partners, use of fluorophore-labeled secondary detection, concentration of reagents, and tolerance to crude bacterial periplasmic extracts and hybridoma supernatants. The final HTS format was selected based on a minimal target antigen concentration, sensitivity to reference binders/inhibitors (where available), and, for competition assays, a minimum assay window of 200 Delta F% with Z′ factor values of >0.6.
PrPc Direct Binding Assays
ScFv in crude periplasmic preparations or purified IgGs (2.5 µL) were incubated with 5 nM human, cynomolgus, rat, or mouse PrPc (Flag-His tagged linear fragment prepared in-house), 1 nM anti–Flag-cryptate (Cisbio), 15 nM anti–Myc-XL665 (CisBio), or anti–human-Fc-XL665 (CisBio) in a total assay volume of 10 µL for 4 h at room temperature.
VEGFR-PlGF1/2 Receptor-Ligand Inhibition Assay
Detection reagents were prepared by preincubating 0.53 nM Flag-tagged human PlGF1 or PlGF2 (produced in-house) with 1.3 nM anti–Flag-cryptate and 0.8 nM VEGFR-Fc (R&D Systems Europe) with 4 nM anti–human-Fc-XL665 for 1 h at room temperature. Hybridoma supernatants (2.5 µL), 7.5 µL PlGF/anti–Flag-cryptate, and 10 µL VEGFR-Fc/anti–human-Fc-XL665 were added to white 384-well low-volume assay plates. Fluorescence emission was measured after either a 4-h incubation at room temperature (PlGF1 assay) or overnight at 4 °C with reequilibration to room temperature prior to reading (PlGF2 assay). Total binding was defined using assay buffer, and NSB was defined using 30 nM PlGF1 (Peprotech, Rocky Hill, NJ).
FGFR1/2-FGF2 Receptor-Ligand Inhibition Assay
Purified scFv (10 µL) were added to assay plates containing 2.5 µL 40 nM fibroblast growth factor 2 (FGF2)–10HisFlag ligand (in-house), 2.5 µL 26 nM anti–Flag-XL665, and 5 µL europium cryptate–labeled FGFR1-Fc or FGFR2-Fc and incubated for 3 h at room temperature. The final concentration of the labeled FGFR was determined on a batch-to-batch basis depending on the europium cryptate incorporation. Total binding was defined using assay buffer, and NSB was defined using excess untagged FGF2 (Prospec, Rehovot, Israel).
CXCL13 Epitope Competition Assay
Detection reagents containing 1 nM biotinylated CXCL13 (Dictagene, Geneva, Switzerland) and 2 nM streptavidin-XL665 (CisBio) for DOS-G01 and DP2-B09 epitope competition assays or 2 nM biotinylated CXCL13 and 4 nM streptavidin-XL665 for the C6G-D07 epitope competition assay were preincubated for 30 min at room temperature. IgG (10 µL), biotinylated CXCL13/streptavidin-XL665 mix (5 µL), and europium cryptate–labeled antibodies (5 µL) were added to the assay plate and incubated for 2 h at room temperature. Final assay concentrations of cryptate-labeled antibodies were 3.2 nM for DOS-G01, 1.2 nM DP2-B09, and 2 nM for C6G-D07. Total binding was defined using assay buffer and NSB using unlabeled antibody at a final assay concentration of 100× that of the cryptate-labeled antibody.
PlGF Epitope Competition Assay
For the parent antibody epitope competition assay, detection reagents were prepared by preincubating 1.2 nM biotinylated parent anti-PlGF1 antibody with 8 nM streptavidin-XL665 and 0.15 nM Flag-tagged human-PlGF1 with 1 nM anti–Flag-cryptate for 1 h at room temperature prior to addition to the assay plate. For the partially optimized antibody assay, detection reagents were prepared by preincubating 0.6 nM biotinylated partially optimized anti-PlGF1 antibody with 8 nM streptavidin-XL665 and 0.05 nM Flag-tagged human-PlGF1 with 1 nM anti–Flag-cryptate detection. Crude periplasmic extracts or IgG (5 µL), preincubated biotinylated antibody/streptavidin XL665 (5 µL), and Flag-tagged human PlGF/anti–Flag-cryptate (10 µL) were added to 384-well assay plates and incubated overnight at 4 °C. The plates were then warmed to room temperature prior to measurement of fluorescence emission.
IL-17 Epitope Competition Assay
IL-17B (#200-28), IL-17D (#200-27), and IL-17F (#200-25) were obtained from Peprotech. IL-17C (#1234-IL), IL-17E (#1258-IL), and mouse IL-17A (#421-ML) were obtained from R&D Systems. Human Flag-tagged IL-17A was prepared in-house. Untagged human IL-17A, cynomolgus IL-17A, and canine IL-17A were a kind gift from AstraZeneca (Macclesfield, UK). All reagents were prepared in buffer containing 0.1% (v/v) BSA, 0.4 M KF, and 50 mM MOPS (pH 7.4). Detection reagents were prepared by preincubating 20 nM anti–human-Fc-XL665 with 0.6 nM anti–IL-17A IgG and 1.6 nM anti–Flag-cryptate with 4 nM Flag-tagged IL-17A for 1 h at room temperature. Samples (10 µL), 5 µL anti–human-Fc-XL665/anti–IL-17A IgG mix, and 5 µL IL-17A/anti–Flag-cryptate mix were added to the assay plate and incubated for 2 h at room temperature prior to measurement of time-resolved fluorescent emission. Total binding was defined using assay buffer, and NSB was defined using untagged IL-17A.
Results
HTRF Direct Binding Assays
The simplest form of HTRF high-throughput antibody screening assay that can be developed is one that directly measures a binding interaction of an antibody to a target protein of interest. Running parallel assays that measure binding to species orthologues or closely related proteins can enable identification of antibodies with the desired species cross-reactivity/selectivity profiles early on in a screening cascade.
We used direct binding assays to isolate antibodies from naive phage display libraries to a noninfectious linear peptide derived from PrPc for which the binding partner is unknown. A broad species cross-reactivity was considered a key property for a lead antibody targeting PrPc to support preclinical studies. Therefore, assays were developed to measure the binding to linear fragments of human PrPc and its species orthologues 21 from cynomolgus monkey, mouse, and rat, which share 97%, 93%, and 94% identity to humans, respectively, within the linear fragment sequence. Outputs from phage display selections were initially screened as crude periplasmic extracts to identify scFv that bound to human and cynomolgus biotinylated PrPc ( Fig. 1A ). A signal of >100 Delta F% was considered a positive binder in each assay. Of a total of 551 unpurified scFv tested at a single dilution, 345 (62.6%) bound both human and cynomolgus PrPc with less than a 2-fold difference in signal in each assay. In total, 167 bound more strongly to human (>2-fold higher binding signal compared to cynomolgus PrPc) and 3 bound more strongly to cynomolgus PrPc than human ( Fig. 1B ). Combining these data with antibody sequence information enabled selection of samples for characterization as purified scFv or IgG for binding to human, cynomolgus, rat, and mouse PrPc. Figure 1C shows example data from four antibodies that displayed different binding profiles in these assays. In this case, the assay used was a modified version of the scFv binding assay with anti–human-Fc-XL665 used for detection of the IgG. Ab-1 had comparable binding to human, cynomolgus, rat, and mouse. The other three antibodies showed differential binding. Ab-2 showed strong binding to human, cynomolgus, and mouse but reduced binding to rat; Ab-3 gave comparable signal for binding to human, rat, and mouse but weaker binding signal for cynomolgus PrPc; and Ab-4 showed reduced binding to rat and mouse compared with human and cynomolgus PrPc. Thus, using the direct binding HTRF assays, we were able to identify an antibody (Ab-1) with comparable binding to PrPc from four different species that was suitable for progression.
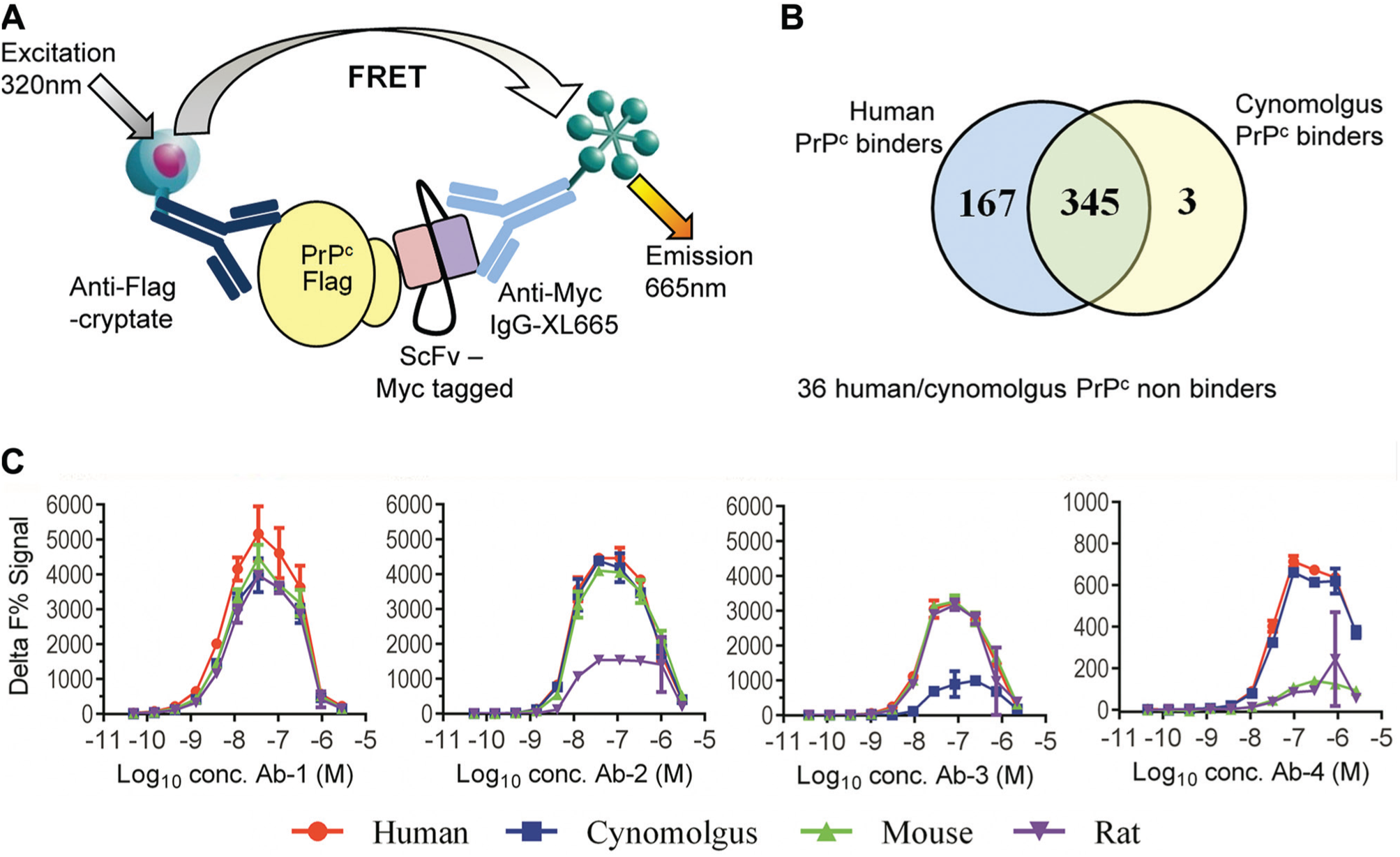
Identification of species cross-reactive antibodies to cellular prion protein (PrPc) using homogeneous time-resolved fluorescence (HTRF) direct binding assays. (
Receptor-Ligand Inhibition Assays
While the direct binding assays are relatively easy to develop and require minimal optimization, they provide a limited amount of information on the ability of an antibody to modulate a relevant functional response. Where a lead antibody is required to block a receptor-ligand interaction, then an assay measuring antibody inhibition of this interaction can be established. This approach was taken for development of an antibody to human PlGF. Supernatants from 14,080 hybridomas, generated by immunization of mice with Flag-tagged human PlGF1, were tested for their inhibition of the binding of either PlGF1 or PlGF2 (a splice variant with an additional 21 amino acids at the C-terminus 22 ) to an Fc-tagged vascular endothelial growth factor receptor (VEGFR) extracellular domain in an HTRF assay ( Fig. 2A ). In total, 363 samples (2.6% hit rate) were identified that showed >50% inhibition of binding of both human PlGF1 and PlGF2 in the assay ( Fig. 2B ). As Flag-tagged versions of PlGF1 were used for both immunization and screening, an additional direct binding screen was performed using biotinylated PlGF1 to confirm that the antibodies were specific for PlGF and did not recognize the Flag tag. This further reduced the number of hits to 229 (1.6% final hit rate). The next stage of the screening cascade required maintenance of the hybridomas in culture to enable production of sufficient quantities of purified IgG for further characterization. Through the use of the receptor-ligand competition assays, it was possible to triage the initial tens of thousands of hybridomas to several hundred, a manageable number for progression.
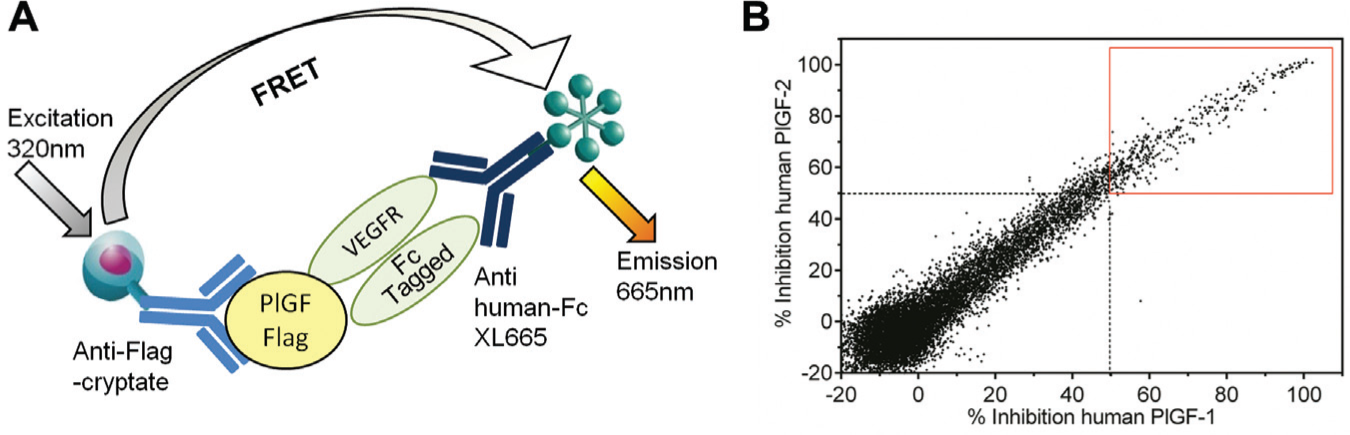
Screening of hybridoma supernatants using a vascular endothelial growth factor receptor (VEGFR)/placental growth factor (PlGF) receptor-ligand inhibition assay. (
FGFR was the target of a second antibody project for which receptor-ligand assays were established. This receptor consists of an extracellular ligand binding domain, a single transmembrane domain, and an intracellular tyrosine kinase domain. 23 The extracellular domain (ECD) can be expressed in recombinant soluble protein form. ECD Fc-fusion proteins were used for the development of two HTRF assays measuring the binding of FGF2 to either FGFR1β or FGFR2β (IIIc splice variants), which share 70% amino acid identity ( Fig. 3A ). The FGFR β splice variant used in these assays lacks the first immunoglobulin domain in the ECD, which is involved in heparin binding and binds FGF2 with higher affinity than the α splice variant. The assays were used to profile purified sequence unique scFv to identify antibodies that could inhibit the binding of FGF2 to both receptor subtypes through interaction with a common epitope. Representative data from seven scFv ( Fig. 3B , C ) show the different activity profiles against the two receptor subtypes that were obtained. Fgfr0028 completely inhibited the interaction of FGF2 with the FGFR1 assay but did not inhibit its binding to FGFR2. Fgfr0038 was more active in the FGFR1 assay than in the FGFR2 assay, while fgfr0034 was more active in the FGFR2 assay than in the FGFR1 assay. Fgfr0029 was unable to inhibit binding in either assay format, while the remainder of the panel demonstrated different binding profiles. ScFv that inhibited the interaction of FGF2 with both receptor subtypes were selected for conversion to full IgG format for further profiling.
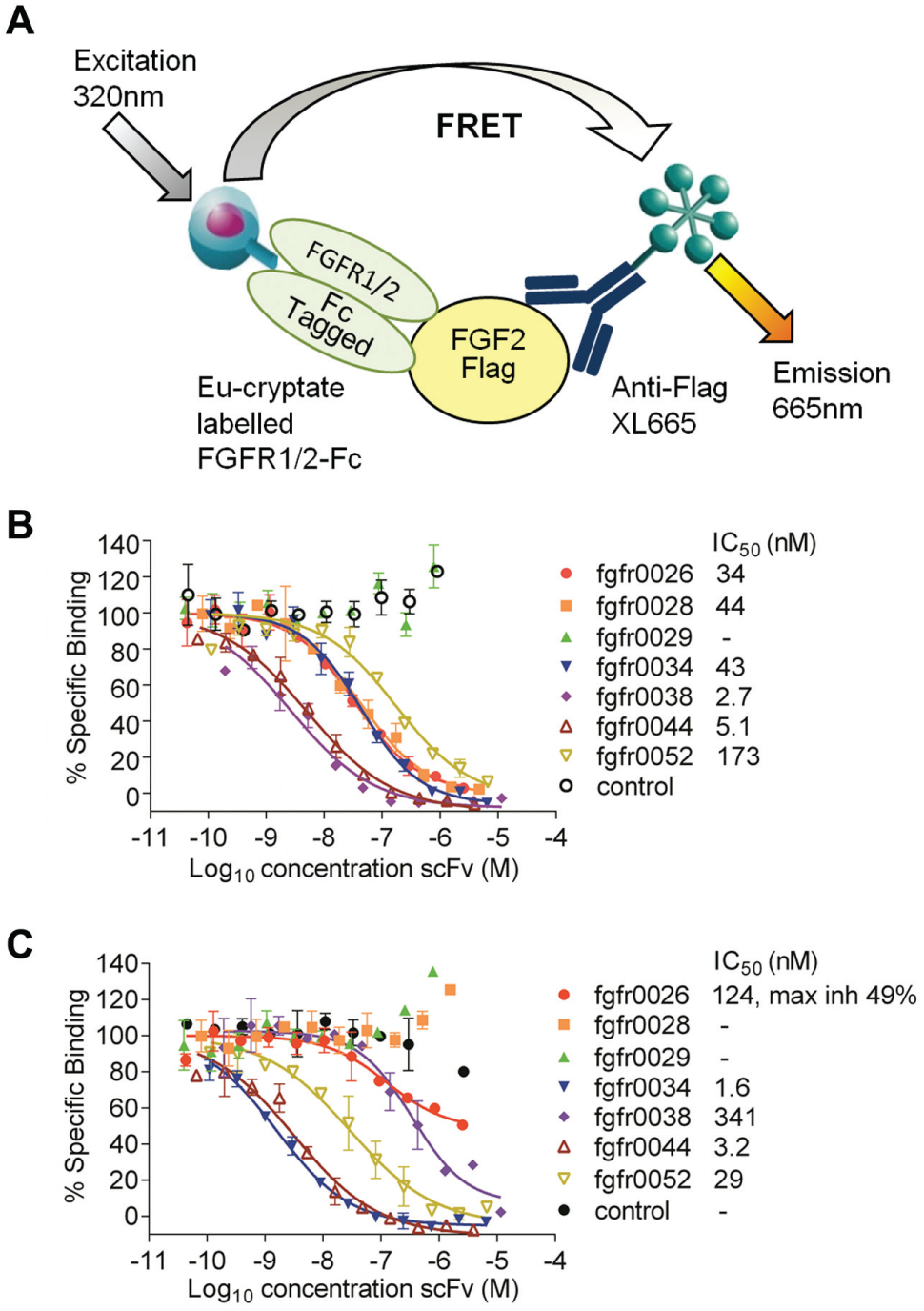
Profiling of purified single-chain fragment variable (scFv) in the fibroblast growth factor receptor 1/2 (FGFR1/2)/fibroblast growth factor 2 (FGF2) receptor-ligand inhibition assay. (
Epitope Competition Assays
Epitope competition assays measure the ability of antibodies to compete with each other for binding to a target antigen. In antibody lead isolation campaigns, they can be used to broadly categorize antibodies according to the epitope they bind, thus ensuring diversity in a panel of antibodies. Figure 4A shows a schematic for an HTRF epitope assay used for binning of antibodies against the human B-cell chemotractant chemokine CXCL13. 24 In the assay, a FRET complex is formed from the binding of an antibody that has been directly labeled with europium cryptate to biotinylated CXCL13, which is detected using streptavidin-XL665. Unlabeled antibodies that bind an overlapping epitope to that of the fluorescently labeled antibody will compete for binding to the biotinylated CXCL13, resulting in a reduced fluorescence emission. A panel of 16 potential lead antibodies was tested for its ability to compete against three fluorescently labeled antibodies ( Fig. 4B ). The antibodies could be divided into two categories: those that competed with both DP2-B09 and DOS-G01 but did not compete with C6G-D07 and those that competed with C6G-D07 but not with DP2-B09 or DOS-G01. This identified antibodies that could bind distinct epitopes, and the most active within each category were progressed for further characterization in functional assays.
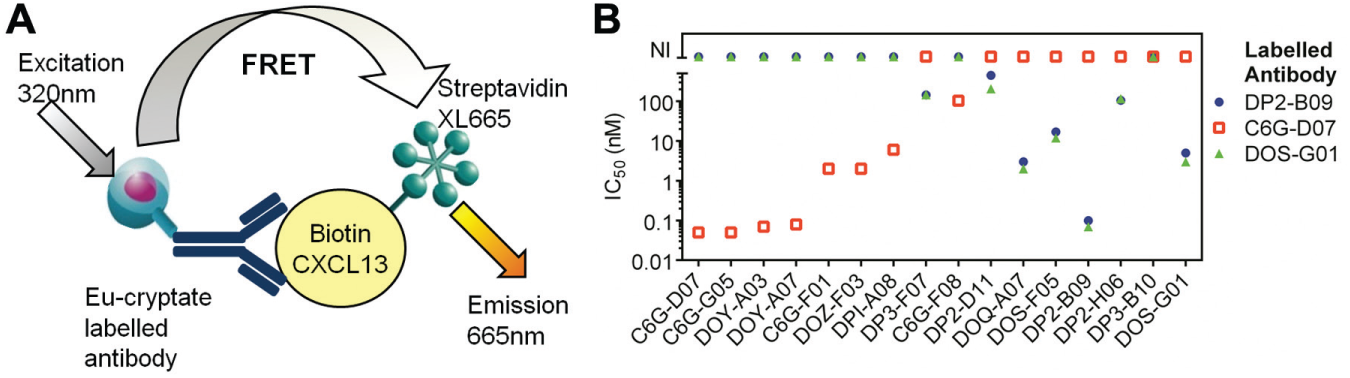
Epitope binning of antibodies to chemokine C-X-C motif ligand 13 (CXCL13) using an epitope competition assay. (
During lead optimization, the sequence of an antibody (parent antibody) is mutated to introduce amino acid changes that may result in improved affinity for the target antigen. An HTRF epitope competition assay format can be established to measure binding of the fluorescently labeled parent antibody, or partially optimized antibody derived from this parent, to the target antigen and used to screen for antibodies with improved affinity. These antibodies will show greater inhibition in the assay compared with that of the labeled parent antibody. This approach was applied to the optimization of an antibody generated to human PlGF. Phage display libraries were derived from the parent antibody by introduction of mutations, either through mutagenesis of blocks of amino acids in the complementarity determining regions (CDRs) or by use of error-prone PCR to randomly introduce mutations across the variable heavy and light chains. Selections using decreasing concentrations of human PlGF1 enriched for high-affinity binders in these libraries. The outputs of these selections were then screened as crude periplasmic extracts in an HTRF epitope competition assay ( Fig. 5A for assay schematic). A representative data set for percent control antibody binding observed for error-prone and VH CDR3 and VL CDR3 block mutagenesis selection outputs is shown in Figure 5B . These data helped to guide the strategy for further affinity optimization. ScFv with mutations in regions of the antibody likely to be involved in antigen binding gave the highest degree of inhibition in the assay compared with the parent scFv. These regions were then prioritized for further mutation analysis or for recombination with other advantageous mutations. ScFv with improved inhibition were chosen as “hits” and sequenced, and the unique scFv were purified for further profiling in an assay that used a partially optimized biotinylated antibody ( Fig. 5C ). This resulted in the identification of antibodies against human PlGF1 and PlGF2 with an affinity improvement of over 10,000-fold compared with that of the parent antibody.
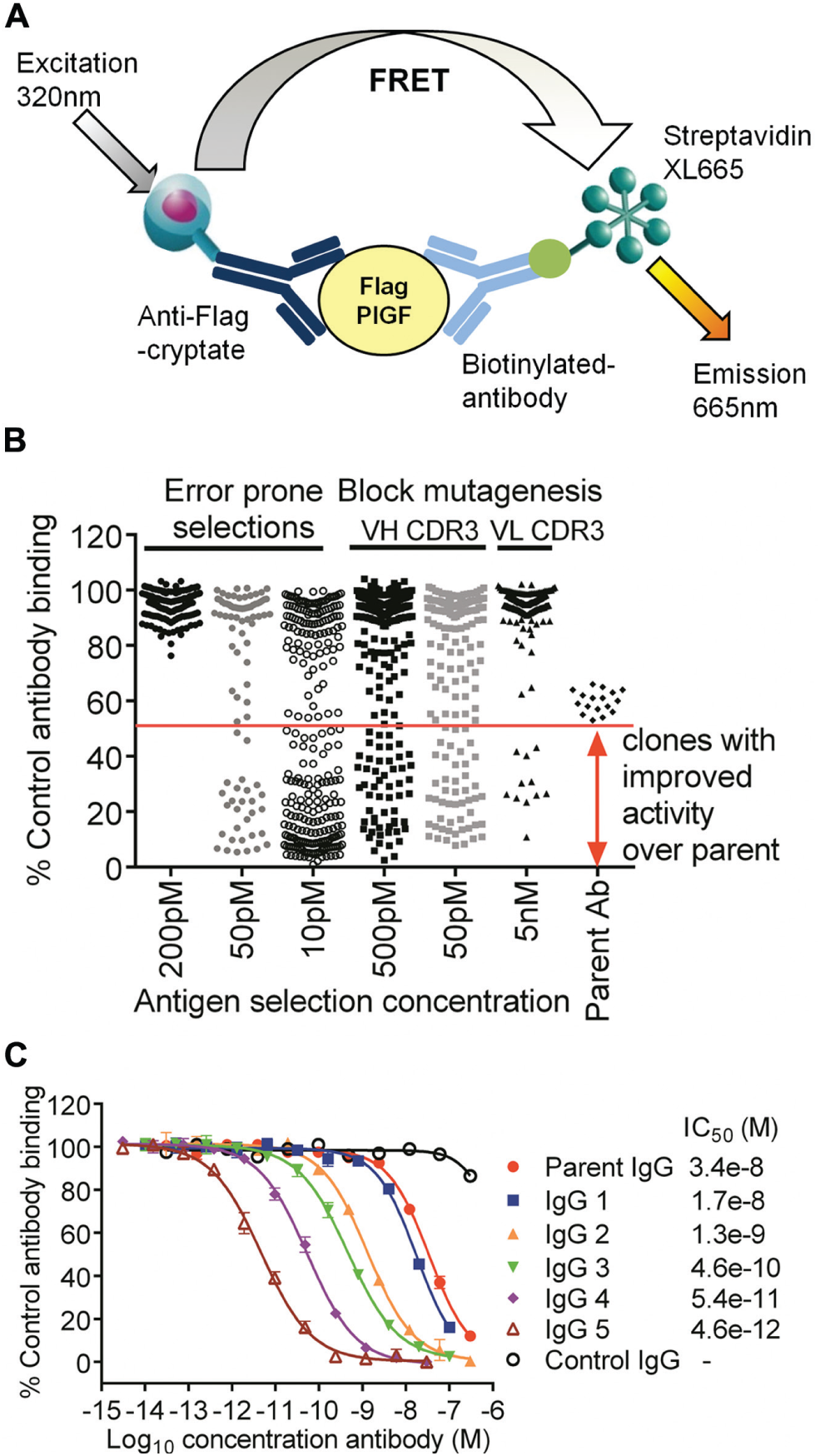
Epitope competition assays to identify high-affinity antibodies to placental growth factor (PlGF). (
Activity against species orthologues and closely related proteins can also be assessed using an epitope competition assay with the final lead antibody. An example of this type of assay has been briefly described previously for IL-17A antibody isolation. 25 In the assay, a FRET complex is formed between Flag-tagged IL-17A, anti–Flag-cryptate, the lead IL-17A IgG, and anti–human-Fc-XL665. Here we show the full concentration curves ( Fig. 6 ) obtained for profiling of this antibody against structurally related family members (IL-17B-F) 26 that share an amino acid sequence identity of 17% to 55% with IL-17A and murine, canine, and cynomolgus orthologues (62%–95% amino acid identity). The assay was validated using untagged human IL-17A, which competed with an IC50 of 1 nM equivalent to the concentration of the Flag-tagged IL-17A in the assay. IL-17B, C, D, E, and F were unable to compete with the Flag-tagged IL-17A, even at concentrations 500-fold higher than that of the IL-17A, confirming the selectivity of the antibody for IL-17A. Human and cynomolgus IL-17A IC50 values were within 2-fold. Canine IL-17A competed with the tagged human IL-17A for antibody binding with an IC50 of 15 nM, indicating that the affinity of antibody for canine IL-17A was lower than that for human IL-17A. The antibody was not mouse cross-reactive, as demonstrated by the lack of competition observed for murine IL-17A. Thus, using a single-epitope competition assay format, it was possible to directly compare the binding of the lead antibody to IL-17 family members and species orthologues.
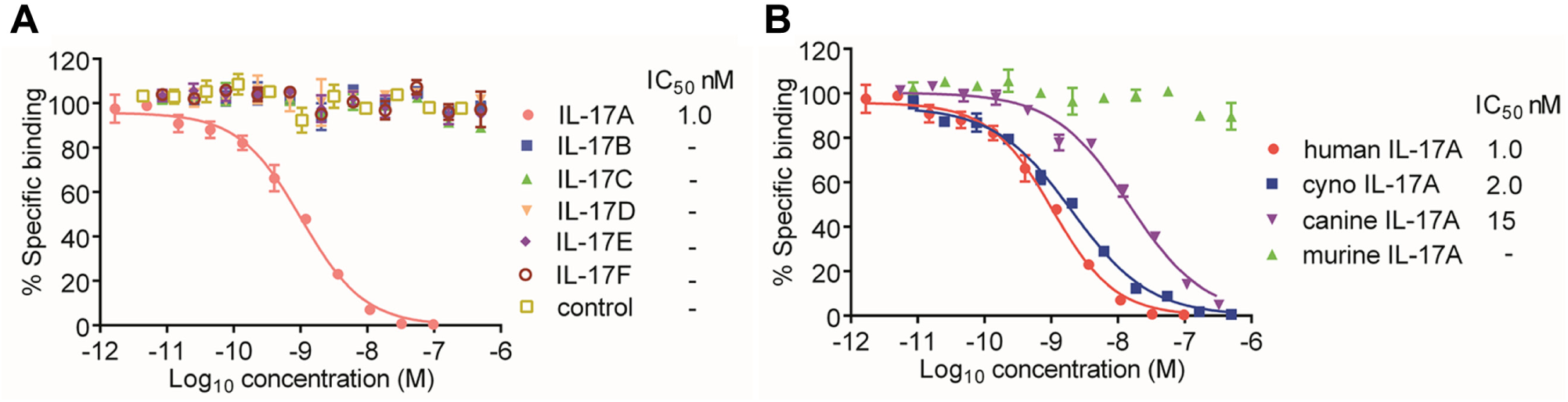
Selectivity and species cross-reactivity testing using an interleukin-17 (IL-17) lead antibody epitope competition assay. Inhibition profiles for (
Discussion
Robust, reproducible sensitive assays are required throughout the antibody discovery process for isolation of a “lead” antibody that has the in vitro properties of a potential drug candidate. Here we have provided examples of how HTRF assay formats can be adapted for use at different stages of this process, from the initial HTS of crude antibody preparations to the downstream characterization of the antibody activity profile.
HTRF direct binding assays are easy to set up due to the minimal number of reagents that need to be optimized in the assay. They can be used to confirm binding of an antibody to a target protein but provide limited information on the ability of an antibody to block a relevant interaction or modulate a functional response. Use of direct binding assays for screening of antibody fragments derived from phage display approaches can result in large numbers of “hits” as the libraries have been already enriched for scFv that bind to the target protein during the selection process. Parallel screening approaches where the binding to multiple targets is assessed (either different species or closely related proteins) can help to rapidly reduce the number of antibodies that are selected for further characterization. The exquisite specificity of antibodies means that even a single amino acid difference in the epitope can confer selectivity. 27 Therefore, even when there is high percent sequence identity between species orthologues, cross-reactivity may not necessarily be achieved as described for the primate-specific (nonrodent cross-reactive) 17E6 antibody against αV-integrin. 28 Despite amino acid sequence identity of 93% to 97% between human, cynomolgus, rat, and mouse PrPc, we observed clear differences in antibody binding, which provided criteria for selection of leads for progression. The ability to run the direct binding assay in low volumes (e.g., 10 µL total volume for the PrPc assay) minimized the sample requirement and assay costs, making the running of the multiple screens feasible.
Direct binding assays have a limited dynamic range due to the fixed concentration of acceptor fluorophore used for detection of scFv or antibody bound to the target antigen. Therefore, sample dilution factors need to be carefully considered when screening preparations of unknown scFv or antibody concentration. If the samples are too dilute, poorly expressed scFv or antibodies will not be identified as binders. Screening of undiluted samples can increase the number of false negatives. This is a result of the “hook effect,” where the concentration of acceptor fluorophore, which binds both antigen bound and free scFv or antibody, becomes limiting. Under these conditions, not all of the scFv or antibody bound to the antigen is associated with an acceptor fluorophore, resulting in erroneously low binding signals. Screening at multiple sample dilutions can limit these effects but affects time and costs.
Competition assay formats reduce the potential for false negatives and can provide more information about the potential of an antibody to block a relevant functional response. Receptor-ligand inhibition assays such as those we have described for VEGFR-PlGF and FGFR-FGF interactions can be used to provide an indication of this functional inhibition and for selection of high-affinity antibodies that block the receptor-ligand interaction. We have shown their utility for triaging a large number of samples in the PlGF HTS. Screening of hybridoma supernatants for their ability to block the PlGF1/2-VEGFR interaction reduced the number of hybridomas for progression from over 14,000 to 229. However, when running these assays, it should be considered that they are often performed under conditions of ligand depletion to achieve a suitable assay signal window.29,30 This can lead to the requirement for higher concentrations of competitor to observe inhibition, resulting in right-shifted dose-response curves. During antibody optimization, very high-affinity antibodies with slow dissociation rates can be isolated, resulting in competitor depletion in the assays (i.e., tight binding). In this situation, the assays are unable to discriminate between these high-affinity antibodies that appear to have comparable IC50 values, often with steep concentration curves (steep Hill slopes). When using the assays to determine selectivity, conversion to Ki is preferable to account for differing affinities of the ligand for the receptors and differences in assay concentrations. Through the use of assays that assessed binding to both FGFR1β (IIIc) and FGFR2β (IIIc), we were able to identify antibodies that inhibited the interaction of FGF2 with both these receptors. We also isolated antibodies that showed differences in their ability to block the interaction with the two receptors. We used the assays to facilitate the identification of antibodies with the desired inhibition profiles rather than to determine absolute values for selectivity.
Epitope competition assays provide a format for the broad classification of antibodies according to the epitope they bind. When functional assays cannot be performed early in the screening cascade, the generation of a panel of epitope diverse antibodies can help to maximize the likelihood of isolating one with the desired functional activity. It can provide information that may be relevant to patent claims, particularly when antibodies have been previously isolated to the target. 31 Furthermore, it may be the preferred option to optimize two antibodies that bind discrete epitopes to reduce the risk of not achieving a high-affinity target that has been predicted to be required for in vivo efficacy. The generation of antibodies that recognize distinct epitopes can also have application as a detection antibody pair for measurement of target levels in enzyme-linked immunosorbent assays (ELISAs) to support pharmacokinetic modeling. The types of HTRF epitope competition assays we consider here can be used to broadly “bin” antibodies by epitope. The cross-competition of antibodies, such as that described for CXCL13 antibodies, can be due to them binding overlapping epitopes or due to steric hindrance effects. Alternative techniques are required for a more refined definition of the epitope bound by an antibody. 32
Epitope competition assays are also particularly useful during the antibody optimization process. They can be used to identify antibodies that bind an overlapping epitope to that of the parent with improved affinity. They do, however, rely on the mutations in the antibody sequence not causing a significant shift in the epitope as this will affect their ability to compete with the labeled antibody in the FRET complex. We have shown data for optimization of an antibody to PlGF in which an HTRF epitope competition assay was used for HTS of crude periplasmic preparations. This assay guided the mutagenesis strategy, resulting in the isolation of a high-affinity antibody. Throughout the optimization process, it is possible to modify the assay to incorporate partially optimized labeled antibodies. Replacement of the parent labeled antibody with one of improved affinity can facilitate discrimination of high-affinity clones by reduction of the target concentration in the assay, resulting in a sensitive assay with a wide dynamic range. Once a final lead antibody has been isolated, a single epitope competition assay using the fluorescently labeled lead (or indirect detection of the lead antibody) can be used to provide selectivity and species cross-reactivity data as shown for the IL-17 antibody.
The proteins we used were all available in a soluble form and thus readily amenable to the development of HTRF assays. However, the technology can be applied to integral membrane proteins using systems such as TagLite (CisBio) that have been developed for activity assessment of G protein–coupled receptors. 33 A wide range of other applications of TR-FRET for antibody drug discovery are outside the scope of this study, such as assays for analysis of activation of downstream signaling molecules 34 and monitoring antibody production during the manufacturing process. 35 Here we have demonstrated that through modification of simple HTRF assay formats measuring protein-protein interactions, it is possible to generate robust assay data for isolation and characterization of antibodies and antibody fragments. These versatile assays can be used throughout the antibody discovery process in both lead isolation and lead optimization, providing key data for selection of a potential therapeutic antibody.
Footnotes
Acknowledgements
We gratefully acknowledge the contributions from MedImmune colleagues to this publication, in particular Protein Sciences, Protein Engineering, and Hybridoma teams for reagent provision and for their overall input into each project.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.