
Abstract
For well over a decade, RNA interference (RNAi) has provided a powerful tool for investigators to query specific gene targets in an easily modulated loss-of-function setting, both in vitro and in vivo. Hundreds of publications have demonstrated the utility of RNAi in arrayed and pooled-based formats, in a wide variety of cell-based systems, including clonal, stem, transformed, and primary cells. Over the years, there have been significant improvements in the design of target-specific small-interfering RNA (siRNA) and short-hairpin RNA (shRNA), expression vectors, methods for mitigating off-target effects, and accurately interpreting screening results. Recent developments in RNAi technology include the Sensor assay, high-efficiency miR-E shRNAs, improved shRNA virus production with Pasha (DRGC8) knockdown, and assessment of RNAi off-target effects by using the C9-11 method. An exciting addition to the arsenal of RNA-mediated gene modulation is the clustered regularly interspaced short palindromic repeats/Cas9 (CRISPR/Cas) system for genomic editing, allowing for gene functional knockout rather than knockdown.
Keywords
Introduction
The discovery of RNA interference (RNAi) in mammalian cells 1 has opened up the prospect of modulating gene expression of specific genes and allowed researchers to elucidate their function in disease models and large-scale screens and even explore them as therapeutic agents.
In mammalian cells, the RNAi system exerts its effect via the expression of microRNA (miRNA), which modulates the expression of entire gene families, primarily through targeting the 3′UTR of genes. 2 miRNA is transcribed as long RNAs with one or many embedded hairpins, called primary miRNA (pri-miRNA). pri-miRNA is processed by Drosha/Pasha to yield individual stem-loop hairpins (pre-miRNA), which are exported from the nucleus and processed by Dicer to yield a short double-stranded siRNA. The siRNA guide strand is incorporated into the RISC complex to either cleave or suppress translation of the target mRNA.
The development of RNAi technology has used the endogenous miRNA system as a guide, initially using cytoplasmic components of the miRNA machinery (siRNA loaded directly into the RISC complex) and then working upstream to nuclear components (shRNA generated from virally integrated vectors). At first, siRNA targeting individual genes was delivered to the cell by transfection. Then the development of RNAi stem-loop expression vectors, resembling pre-miRNA, moved to the forefront. More recently, shRNA has been expressed in constructs resembling pri-miRNA, which are processed by the complete miRNA pathway. Today, this is being pushed even further toward mimicking true miRNAs, with the expression of multiple shRNAs within a single construct.
During this review, we discuss the strengths and weaknesses of all the above RNAi systems, with a focus on shRNA, in addition to clustered regularly interspaced short palindromic repeats (CRISPR) genome editing. We will highlight how the technology can be applied to mammalian screening using viral delivery systems and diverse screening formats. Finally, we elaborate on the steps needed to design and interpret screens, select hits, and detect and mitigate off-target effects (OTEs).
Types of RNAi and Their Application
Perhaps the simplest approach to RNAi involves transfection of synthetic siRNA duplexes that resemble Dicer products.1,3 Synthetic siRNA requires the transfection of duplexes into a dividing cell and is therefore not easily applied to slowly or nondividing cells. Transfection of siRNA can result in toxicity due to high siRNA concentration or transfection reagent toxicity, while longevity of the siRNA response is limited by stability in the cell. Despite these limitations, synthetic siRNA has been extremely useful in arrayed screening, due its availability from commercial vendors in whole-genome libraries and its inherent safety as a nontoxic agent. The advent of vector-based stem-loop shRNAs and delivery with retrovirus and lentivirus has been an important development, improving on siRNA in many areas, including long-term gene knockdown, transduction of hard-to-transfect and nondividing cells, and in vivo studies.4–6 Viral vectors are also amenable for both arrayed and pooled libraries, the latter being an area that has expanded significantly in recent years.
One of the first successful examples of these shRNA expression vectors, pLKO.1, uses the U6 RNA polymerase III (pol-III) promoter to drive generation of a stem-loop shRNA, facilitating stable, long-term gene knockdown. 7 This vector has been subsequently improved upon by addition of a WPRE enhancer to improve shRNA production. Although this design is effective in generating high levels of shRNA and suppressing expression of target genes in mammalian cells, 8 it skips the early steps of miRNA biogenesis and may lead to inhibition of endogenous miRNA effects through saturation of Dicer and RISC by supra-physiological shRNA levels.
Embedding shRNA sequences into an endogenous miRNA backbone (e.g., miR30) enables expression from Pol II promoters, leading to efficient processing of shRNA and knockdown with even a single integration per cell, thus making it less likely to interfere with endogenous miRNA processing.9–11 In addition to loss-of-function studies in gene-by-gene or pool-based formats,12,13 miRNA shRNA can be expressed as a poly-cistron and used for combinatorial RNAi studies.11,14
miRNA shRNA constructs can be embedded in the 3′UTR of fluorescent protein reporters, allowing for fluorescence-activated cell sorting (FACS) of cells expressing shRNA without the use of chemical selection.15,16 Fluorescent markers can also be used to ensure an appropriate percentage of cells are infected (20%–30%), resulting in only a single integration per infected cell; this is critical in designing pooled shRNA screens.12,17 To enhance the performance of negative-selection pooled shRNA screens, Zuber et al. 15 have designed and generated a toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. In brief, the green fluorescent protein (Venus) was expressed constitutively from a PGK promoter, along with the reverse Tet-activator (rtTa). Expression of the miRNA shRNA embedded in the 3′UTR of red fluorescent protein was driven from a Tet-regulated promoter ( Fig. 1 ). This system has been used in single and pool-based formats to identify essential genes both in vitro and in vivo. The Tet-regulated expression system has the advantage that essential genes are not depleted prior to the start of the study. 15 Although the Tet-regulated miRNA shRNA system is robust, leakiness from tetracycline response elements (TREs) is still a potential problem. A new third-generation TRE promoter (TRE3G) has been adapted with great success. 16 Table 1 compares the technical aspects and characteristics of the above RNA-mediated gene suppression technologies.
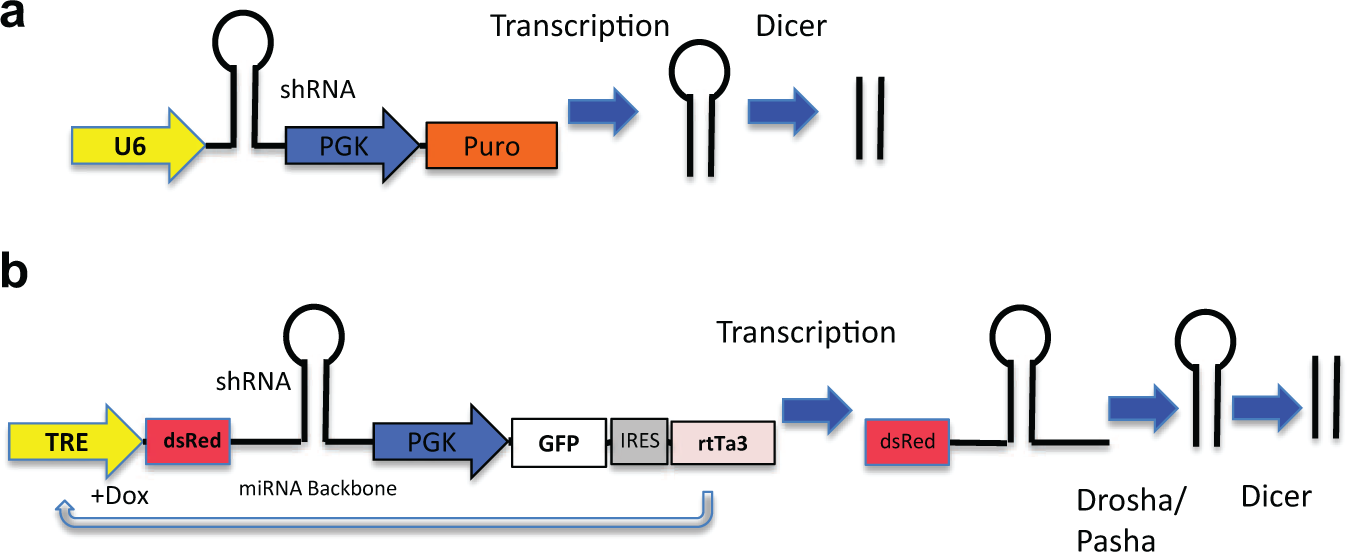
Examples of expression cassettes for (
Comparing Technical Aspects and Characteristics of RNA-Mediated Gene Suppression Technologies.
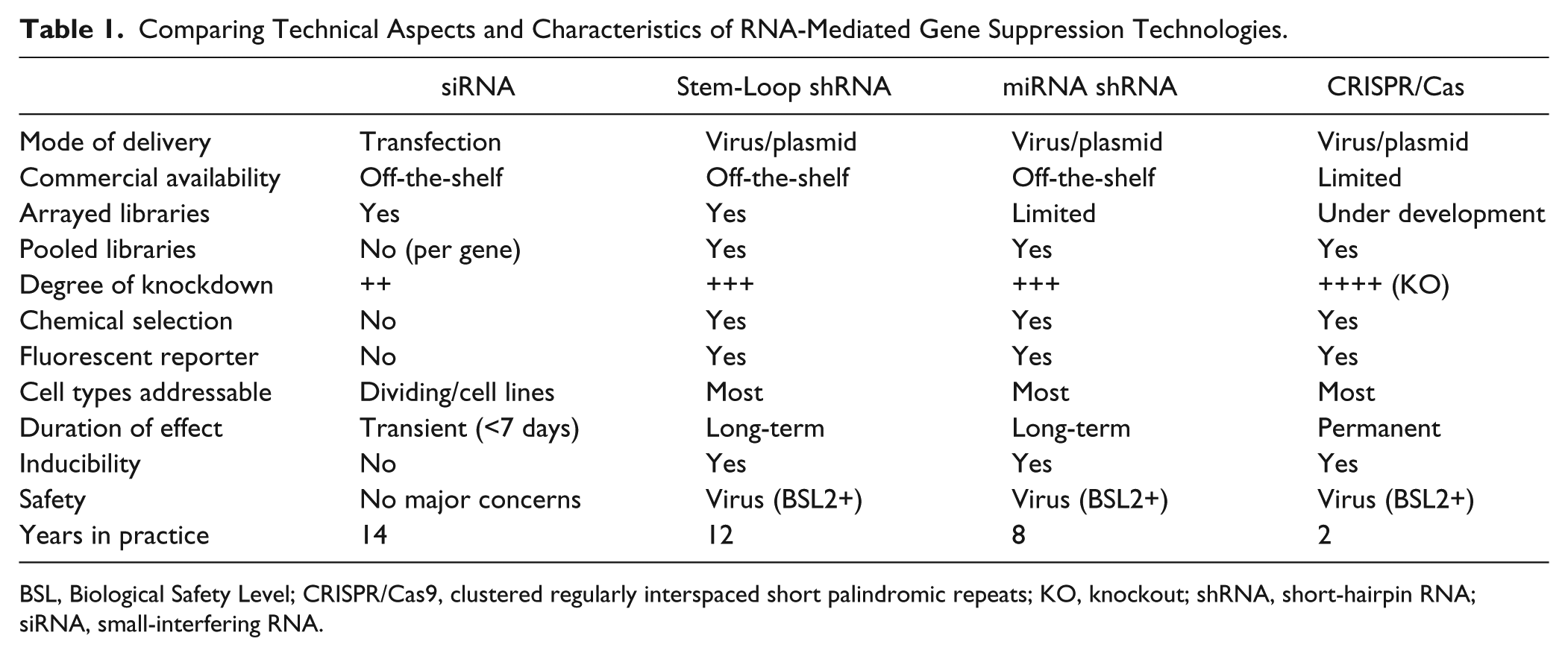
BSL, Biological Safety Level; CRISPR/Cas9, clustered regularly interspaced short palindromic repeats; KO, knockout; shRNA, short-hairpin RNA; siRNA, small-interfering RNA.
Improving the Utility of shRNA: The Sensor Assay, miR-E, and Pasha
Some shRNAs may fail to suppress their targets due to inefficient biogenesis, leading to false negatives in RNAi screens. To identify highly effective miRNA shRNA, an RNAi “Sensor” assay was recently established for measuring the potency of thousands of miRNA shRNA 18 ( Fig. 2 ). In brief, a high-throughput parallel approach was established to validate 20,000 miR-30 shRNAs at single-copy conditions in a pooled format against nine genes. 18 These shRNAs were placed under the control of a Tet-inducible promoter in a single reporter vector (pSENSOR), also containing the shRNA cognate target sequence in the 3′UTR of a constitutively expressed fluorescence reporter (Venus). Effective shRNAs reduce the Venus reporter expression by RNAi-mediated mRNA cleavage, resulting in a fluorescence signal decrease. Repetitive rounds of doxycycline induction and withdrawal, or “ping-pong,” accompanied by FACS, enrich for the best shRNA and target site combinations for each gene. 18
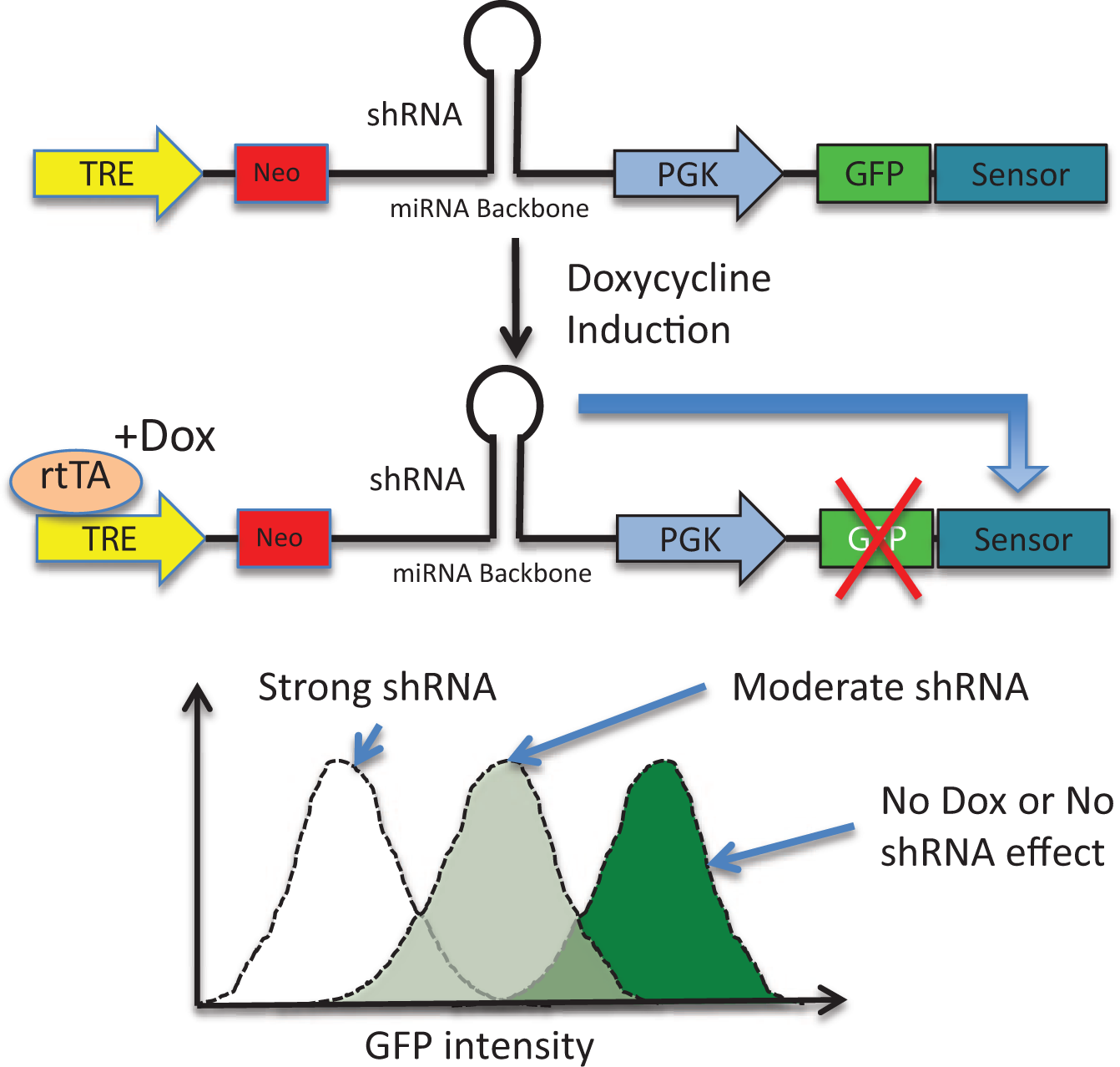
Sensor assay performed in NIH3T3 cells stably expressing rtTA. Cells are infected at a low multiplicity of infection with a single integration per cell using a pool of virus expressing Tet-regulated short-hairpin RNA (shRNA) against the target gene, along with constitutive green fluorescent protein (GFP)–Sensor expression. The Sensor region has a cognate target for shRNA in each virus. Efficient suppression of the target by shRNA leads to GFP messenger RNA degradation and GFP-low populations that can be sorted by fluorescence-activated cell sorting (FACS). Strong shRNAs are enriched by successive rounds of Tet-induction and FACS sorting, followed by next-generation sequencing to quantify enriched shRNA.
A medium-throughput version of Sensor assay was also designed to directly quantify single-copy shRNA knockdown. 16 A reporter construct expressing the fluorescent protein dTomato harbors the target sites of established control shRNAs, as well as an array of shRNA target sites to be tested (up to 20), and was driven from the PGK promoter. This construct was retrovirally transduced in single copy into murine embryonic fibroblasts to produce stable reporter cell lines. In a second round of infection, a vector expressing the shRNA to be tested, coupled to a green fluorescent reporter, was introduced. Upon shRNA expression, the decrease in red fluorescence, quantified by flow cytometry, represented the shRNA knockdown level and was normalized to that of control shRNAs, measured in parallel. This assay has been shown to be particularly useful to quickly evaluate individual shRNAs as strong, immediate, or weak.
Recent efforts to identify highly effective miR-30 shRNA led to an optimized miRNA shRNA reagent, termed miR-E, for “enhanced microRNA-based hairpin.” 16 This enhancement involved changing the ACNNC motif 3′ of the basal stem in the existing miR-30–based RNAi backbone. The newly developed miR-E reagents enhanced mature shRNA production and knockdown potency following single-copy integration, critical for effective pooled shRNA screens.
Of note, when generating potent shRNA (such as miR-E in retro- or lentivirus), capable of a high degree of target knockdown, there is also a risk that essential gene suppression in viral production “packaging” cells will result in cell death and underrepresentation in the library. Suppressing the expression of Pasha (DRGC8), a component of the miRNA processing machinery, with siRNA or shRNA 19 has been shown to be an effective approach in increasing viral titer when using the modified mir30 backbone, miR-E, 16 and also increases the RNA transcripts available for viral packaging.
RNAi Design
Many siRNA and shRNA reagents and libraries are available off-the-shelf, so the end user might not consider design. However, pooled approaches to library synthesis and the desire to screen more focused gene sets, or to probe deeper coverage per gene, once again elevate the topic of shRNA design considerations to the forefront. Design methods have improved considerably; we will highlight some of the main advancements.
In the development of siRNA, it was apparent that certain sequences were more potent than others. Features such as the thermodynamic bias at the 5′-antisense end of miRNAs and siRNAs, as well as guanine/cytosine (GC) content, nucleotide position, stability, and secondary structure, were important indicators of potency. Learning algorithms were rapidly developed as the size siRNA libraries tested grew from 180 to over 2000.20–22
As the shRNA field unfolded, algorithms designed to predict potent siRNA were used to develop the first shRNA reagents, but it soon became apparent that shRNAs have additional processing rules. In a study of the knockdown of 27 shRNAs on 11 genes (and 38 independent shRNA), siRNA prediction algorithms could not explain shRNA efficacy results, due to the processing steps by Drosha and Dicer. 23
A new set of rules was created for prediction of potent shRNA from a pooled loss-of-function screen by the RNAi Consortium (TRC), in which 130,000 21-mer shRNA targeted 27,000 genes (human and mouse). 24 These rules included 30% to 50% GC content, with higher GC at the 5′ end and lower on the 3′ of the antisense, among other position-specific rules. Several years later, the Sensor assay was applied to a large-scale screen, with 20,000 shRNAs targeting nine genes, using the design of small interfering RNA (DSIR) algorithm, as described earlier. 25 Unbiased analysis of this study revealed more refined potency rules (Sensor rules), which include nucleotide and position combinations: U1, A/U 13/14, G 20/21, and AU richness in the first two-thirds of the shRNA. Training on this large set demonstrated that addition of thermodynamic rules improved on the sequence and position-only rules. 26
Until recently in our hands, DSIR filtered by shRNA Sensor rules was the algorithm of choice for the prediction of potent shRNA for miR30 shRNA. This combination yielded 20% precision at 10% recall (i.e., one to two of six shRNAs tested were potent). SplashRNA, a new shRNA potency prediction algorithm, 27 shows double the precision at 10% recall in 10-fold cross-validation, effectively halving the number of shRNA that need to be tested against a new target gene. This boost in predictive performance was achieved by combining an 8-degree spectrum kernel with a weighted degree kernel in a support vector machine (SVM) learning framework. This work is to be published soon, and predictions will be publicly available.
The Advent of CRISPR/Cas
In comparison to RNAi, the CRISPR/Cas system is an RNA-guided genome editing tool used to generate permanent mutations in the genome, which can result in either a loss of function or a gain of function.28–30 Two other genome editing nuclease technologies are available for use in mammalian cells: zinc-finger nucleases (ZFNs) 31 and transcription activator-like effector nucleases (TALENs). 32 Although ZFNs and TALENs have been shown to be successful genome editing tools, the relatively high cost and time-consuming efforts to engineer de novo gene-specific nucleases have rendered them less facile for large-scale screens. The CRISPR/Cas system, on the other hand, represents a more cost-effective, fast, and efficient choice for screening.
The CRISPR/Cas system, found in many bacteria and most archaea, plays a role in acquired immunity against viruses. CRISPR are repeated sequences in the genome of prokaryotes, which integrate genomic sequences from invading viruses. Following infection, the CRISPR loci undergo transcription to form CRISPR RNA (crRNA) and a trans-activating crRNA (tracrRNA), which then directs the Cas-nuclease to target the foreign DNA, producing double-stranded breaks, which are then followed with error-prone nonhomologous end joining (NHEJ) repair. This process results in genomic mutation of the invading genetic element in a sequence-specific manner. 33
The CRISPR/Cas system has recently been manipulated to edit the genomes in mammalian cells.28–30 The type II CRISPR/Cas system, most frequently used for genome editing technology, is the simplest of three CRISPR/Cas systems and requires only one CRISPR-associated (Cas) protein, the Cas9 endonuclease, to target and cleave DNA. 28 The use of a 20–base pair single-guide RNA (sgRNA) has been found to be capable of bypassing the need for crRNA and tracrRNA sequences to allow for hybridization to the gene of interest and subsequent cleavage by Cas9. 34
The CRISPR/Cas system holds much promise for large-scale genetic screens, previously dominated by RNAi. This is due to its relatively simple requirements, durability of knockout, and ability to target multiple gene loci at one time via the introduction of multiple sgRNA sequences. Analogous to RNAi, the CRISPR/Cas system can be scaled up and applied to large-scale screening. In just 1 year since the CRISPR/Cas system was first used in mammalian cells, several large-scale screens have been reported.35–38 These studies used similar design algorithms to create sgRNA, and synthesis of oligonucleotides was done either individually (for more focused libraries) 37 or by using DNA microarrays (for pooled genome-wide libraries).35,36,38
In a full-genome, pooled, negative selection screen for essential genes, an sgRNA library was transduced into two separate human cell lines, A375 and HUES62, with known essential genes being identified as hits. 35 Another genome-wide, pooled, positive selection screen was performed looking for resistance to the protein kinase inhibitor vemurafenib in the human cancer cell line A375. Hits from this study correlated well with a previous shRNA screen. 35 These studies provide emerging evidence that CRISPR/Cas is a powerful tool for genome-scale screening, with minimal OTEs and a high validation rate for hits. As such, the CRISPR/Cas system provides for a complementary method to study gene function and adds to already available RNAi tools by generating gene knockouts rather than suppressing mRNA levels.
Since shRNA can produce partial and reversible gene suppression, while CRISPR/Cas produces gene knockout, RNAi may be better at mimicking the effects of small-molecule therapeutics, while the CRISPR/Cas system leads to complete loss of function and is therefore invaluable for genetic studies. The CRISPR/Cas system can address a wider range of possible target classes, including noncoding regions of the genome. Despite its promise, the CRISPR system, in common with RNAi, can also have OTEs, 39 a situation that might be improved with Cas-nickase. 40 Another caveat is that mutations caused by CRISPR seem to be random and can be loss of function, gain of function, or neutral. Mutations producing survival and/or proliferate advantages will be enriched, which could impair the power of CRISPR/Cas for negative selection screening.
Screening Technologies
In our discussion of RNA-mediated genomic screening, it is important to discuss the various methods used in arrayed and pooled-based screening, as this can be as crucial to the end result as is the choice of reagent. Here, we will discuss high-content screening (HCS) for arrayed plate-based screens (although non-HCS, plate reader–based approaches are still in use) and highlight FACS and next-generation sequencing (NGS) for pooled screens.
In HCS, cells are labeled with multiple fluorescent markers (fluorescent proteins, dyes, and fluorophore-linked antibodies), which are measured in multiple channels (and possibly phase-contrast or transmission light microscopy images) and recorded in a highly automated fashion. As with other forms of cytometry, intensity information is captured from each of the labels, and, in addition, morphology-based information about the fluorescent markers is derived and then used to build complex experimental end points such as cell shape, intracellular protein localization, translocation events, co-localization of proteins, and cell-cell interactions.41,42 Multiwell plates also make it possible to capture statistically significant data from a relatively small number of cells compared with flow cytometry.43,44 Typically in HCS, adherent or bottom-settled cells are queried in multiwell (96, 384, 1536) plates in a 2D landscape, although a series of two or more optical slices through the Z dimension are also possible. A growing area of interest in the high-content imaging field are assays involving the use of 3D imaging to capture information from tissues, organoids, or other multicellular complexes.45,46
A recent example of the power of HCS was demonstrated in a genome-wide siRNA screen in HeLa cells. Regulators (accelerators and inhibitors) were identified of Parkin translocation to damaged mitochondria, in an effort to better understand mitochondrial quality control in relation to the neuropathogenesis of Parkinson disease. 47 Mitochondrial membrane potential was first depleted via chemical means, and then candidate genes were selected after filtering out siRNAs, which were nonspecifically cytotoxic or had depleted the absolute number of mitochondria. A thorough validation of siRNA hits was corroborated on several levels. These include the use of additional siRNA reagents, measuring mRNA levels, lentiviral shRNA knockdown in human iPS cells, Western blots, TALEN-based genome editing approaches, phenotype rescue using candidate gene overexpression, and C9-11 mismatch controls (see below). This extensive postscreening validation yielded a high confidence level in the hits and helped to mitigate the possibility of OTEs. In another study, HCS was used to screen siRNA libraries and measure poxvirus infection and replication in HeLa cells. 48 This study demonstrates the power of identifying a large number of candidate genes, able to positively and negatively regulate a complex biological process, and cluster based on gene pathway and function, leading to a better understanding of disease mechanism. A common aspect of both these studies is the use of siRNA libraries from two commercial vendors (Ambion [Foster City, CA, USA], one siRNA per well, and Dharmacon [Lafayette, CO, USA], siRNA multiplexed in well per gene) in the initial screen, thereby increasing the number of independent reagents targeting a single gene product. Such an approach gives the opportunity for greater target validation at an early stage and, with multiple reagents giving a similar phenotype, reducing the possibility of pursuing an OTE from a single siRNA.
HCS has also been successfully applied to viral expression of shRNA. An array of about 5000 hairpins targeting 1028 genes was applied to tumor cell lines using a multiparameter assay for mitotic index. This study identified several novel candidates as well as identifying known modulators and was compared with previous screens performed in other backgrounds: Drosophila and human fibroblasts. 49 The appropriateness of imaging assays and lentiviral shRNA systems has also been demonstrated in cell-based assays that require long-term culture, need differentiation, or use difficult-to-transfect cells. In a study using SH-SY5Y neuronal cells, lentiviral shRNA was used to infect dividing cells that were subsequently differentiated, with multiple assays applied to the resulting neurons, including neurite-outgrowth and mitochondrial function. 50
Pooled shRNA screening has been increasingly popular in recent years, driven in part by not requiring the expensive and bulky automation needed for genome-scale arrayed screens. 51 Pooled screens also have a distinct advantage over arrayed screens when studying genes affecting cell proliferation over multiple doublings (and lasting perhaps weeks in length) due to space constraints imposed for cell growth by well geometry and the difficulty in refeeding cells in the plates.52,53 Pooled shRNA screens can be scored for a decrease in shRNA species through dropout or an increase in shRNA through some selective advantage conferred to the cells. To score proliferation (i.e., the “flip-side” cell death assays), genomic DNA is collected, and then the relative abundance of each shRNA is measured and quantified by NGS or microarray.43,52 Pooled screening can also be used for assay end points that do not involve cell death or proliferation. For this approach, cells may be sorted into populations using FACS, based on a fluorescent reporter.25,54 An excellent example of this approach is a study where human embryonic stem cells (hESCs) were infected with a pooled lentivirus TRC shRNA library and the infected cells subsequently enriched using puromycin selection. The pooled population was expanded before undergoing neural differentiation and then sorted into three groups by FACS based on their marker PS-NCAM expression, followed by DNA extraction and NGS. In addition to finding known neurodevelopmental targets, hits were also identified that had previously been shown to play roles in neurodegeneration. 55 Technically, when the cell population does not go through multiple population doublings, or there is a high degree of assay variability, it may be necessary to increase the shRNA representation (1000–2000 vs 300–500 for a proliferation screen), which might limit the number of genes screened in a single pool. 15 In addition, pooled approaches have made it possible to easily pursue high coverage of individual genes with multiple (>30) unique shRNA reagents to the same target genes, making it easier to infer a decreased likelihood of OTEs. 56 This deep-tiling approach has resulted in multiple potent shRNA per target, increasing the overall veracity of RNAi screens. A comparison of arrayed and pooled screening strengths and weaknesses is outlined in Table 2 .
Comparison of Arrayed vs. Pooled Screening Approaches.
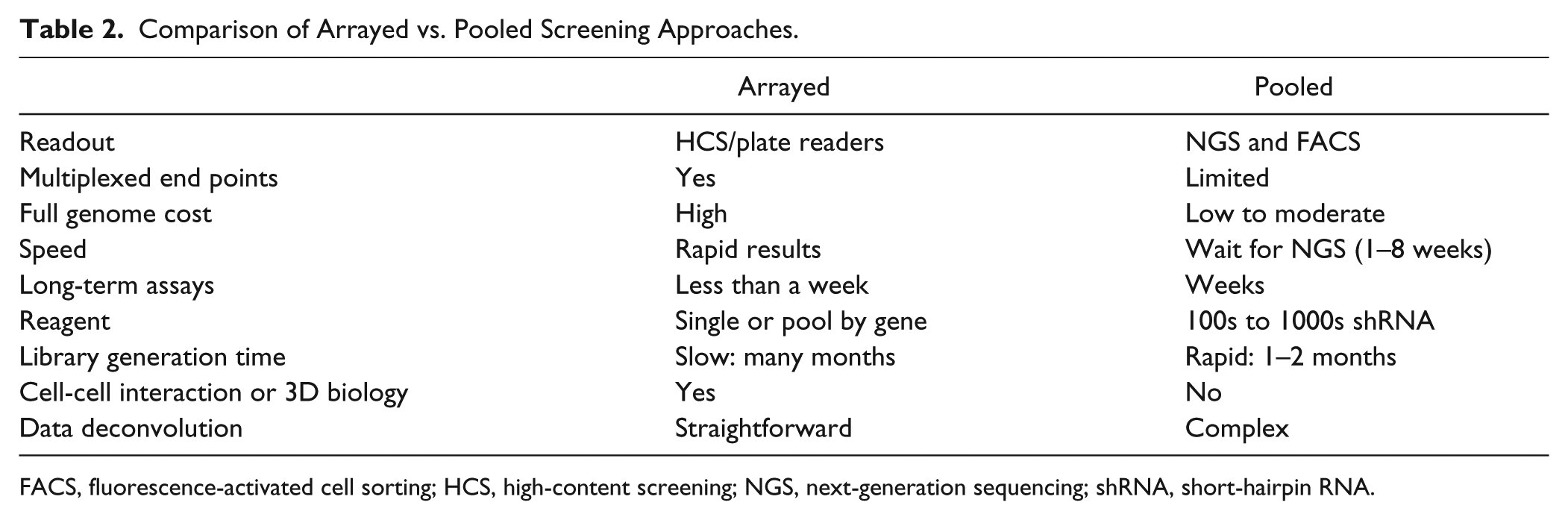
FACS, fluorescence-activated cell sorting; HCS, high-content screening; NGS, next-generation sequencing; shRNA, short-hairpin RNA.
Pooled proliferation-based screens are well suited for studying “synthetic-lethal” interactions, where the shRNA library is screened in the presence of any factor that by itself might not have a significant effect or in which a cell may have acquired resistance to that treatment. Examples of this type of screen are chemical inhibitors, which target a particular pathway, that are lethal only when a compensatory pathway is knocked out by a shRNA.57,58 The therapeutic importance of these screens is in the identification of treatments targeting the tumor without killing normal somatic cells. Another example is the use of a pooled library of shRNAs (4500) targeting signaling and cancer genes to look for sensitizers to a chemical inhibitor of PLK1. It was found that knockdown of the retinoid receptor RXRA was able to confer resistance of the four cell lines used to the PLK1 inhibitor, GSK461364. It was subsequently shown that retinoic acid could sensitize the tumor cells to GSK461364, which could be therapeutically important. 59
Assay Performance, Hit Identification, and Statistics in Plate-Based Screening
When it comes to screening RNAi libraries in an arrayed format, there is an extensive literature on the quality and normalization of results within individual plates and screens, using statistical measures to identify hits.43,44,47 Examples of standard tests for data quality and assay window are Z′ and strictly standardized mean difference (SSMD). The Z′ calculation has a time-honored history in both antagonist compound and RNAi screening and is ideal if the screening window is large and positive controls are available with robust activity. However, this is not always the case with RNAi reagents and phenotypic screens. The SSMD is a less conservative approach and is recommended for RNAi screens with moderate controls that have moderate effect. It is defined as the ratio of the difference between the means and the square root of the sum of the squares of the standard deviations of the two populations. Hits are typically identified by comparison to a negative control or to the population of samples on the plate. Comparing with a control RNAi can be complicated by the need for a large number of control wells in arrayed screens and by potential OTEs. A more useful approach is the z score (number of standard deviations), which compares each sample with the mean of the plate (or screen), or the robust z score (number of median absolute deviations), which relates each sample to the plate median and is comparatively insensitive to outliers. Both these methods have the advantage of taking into account the inherent variability in the assay (for review, see Birmingham et al. 44 ).
In both arrayed and pooled screening, hits are often identified as those that have a high level of statistical significance. Multiple (more than two) reagents identifying a hit to a single gene decrease the likelihood of hits resulting from OTEs. Hits can be identified in pooled screening using similar approaches to arrayed screens as outlined above.8,43,44 In addition, using a pooled approach, it is relatively straightforward to sample the cells at different time points to measure the kinetics of shRNA depletion or enrichment.8,52,60
NGS for Pooled shRNA Screen Deconvolution
The recent emergence of low-cost NGS technology has given researchers a more facile approach than hybridization readouts for the deconvolution of pooled RNAi screens. To quantify the relative abundance of different shRNAs in the pooled population, genomic DNA is extracted and amplified for the integrated variable DNA regions associated with the shRNA. The PCR primers are barcoded to multiplex PCR products from different samples in a single NGS lane. 15
In quantifying pooled screens by NGS, three parameters are important in measuring shRNA (for RNAi) or sgRNA (for CRISPR) activity: first, the fold-change of representation between experimental and control groups is examined; second, statistical variation of shRNA representation between biological or technical replicates is assessed; and third, the relative abundance of shRNA in the library is also an important factor, as over- or underrepresented shRNAs are more prone to random noise in screening. Traditionally, NGS data have been analyzed by the fold-change first, followed by a reproducibility check in paired replicates. Although this method has proved to be successful in identifying biologically important genes in either positive or negative screening,17,61 the criteria and threshold for hit calling are arbitrary, variable, and nonstatistical. In the case of nonpaired replicates, other methods are needed to evaluate variation within a group, and several have been described. 44 Most of these methods consider a single parameter (e.g., mean ± k standard deviation) or two parameters (SSMD for hit identification). It is also possible to use Bayesian approaches, which could cover all three parameters and assign a false discovery rate (FDR). Some of these approaches have been heavily used in RNAseq to identify differentially expressed genes and are reviewed elsewhere. 62 For pooled shRNA screening with NGS readouts, we recommend the most commonly used and freely available differential-expression software packages: edgeR, DESeq, Cuffdiff, PoissonSeq, baySeq, and limma for RNAseq. 62 Depending on the nature of the screening and the readouts of positive controls, threshold of fold-change and FDR (or adjusted p-value) can be used to determine the list of hit candidates in the pooled shRNA screen.
Typically, three to six shRNAs (or more) are designed per gene in a screening library in the hopes of finding multiple species, which can knock down the target gene. Since the effectiveness of shRNA design by a state-of-the-art algorithm is still not guaranteed, the appearance of a lone active shRNA does not exclude the possibility of it being a bona fide on-target hit. However, genes with more than two associated hairpins on the hit list are more likely to be true hits and should have priority in subsequent validations. To reduce the possibility of false positives, computational methods (RSA, RIGER), which can also be used for arrayed screens,8,63 consider the rankings of all shRNAs against one gene and assign a single score to each, generally reducing the false-positive rate by shRNAs, although potentially missing novel genes. It should be noted that there is no single best method for shRNA pooled screening deconvolution in every setting.
Mitigating Off-Target Effects in RNAi Screens
Some degree of OTE in RNAi screening is a common and unavoidable by-product of genomic assays, due to several factors. A recent study of HCS screens for pathogen infection of mammalian cells concluded that most individual siRNA hits identified were in fact false positives due to OTEs. 64 The primary cause in this and most cases is due to the interaction of the RNAi seed region (bases 2–8) with the 3′UTR of numerous genes, in a miRNA-like manner. 2 In RNAi screens, this effect leads to the knockdown of multiple genes that are not the designated target for the RNAi reagent, leading to their designation as a false positive.65,66 In designating an RNAi regent as a hit, this again highlights the need to have multiple positive reagents per gene with different seed sequences. Other factors affecting OTE include poor library design, passenger strand loading into RISC (which can be limited by chemical modification of siRNA 67 ), and reagent toxicity due to high siRNA and shRNA concentration.65,66 To minimize OTE, the algorithm must account for miRNA homology, as well as unique sequences, lacking homology to other genes and the presence of multiple gene isoforms. 68
The workflow to determine if an RNAi is a true hit or a false positive due to OTEs can be time-consuming and expensive, particularly following genome-scale screens.43,66 Typically, hits are evaluated by quantitative PCR to ensure that the observed phenotype correlates with target gene mRNA levels. Measurement of protein expression by Western blot is easy to perform on a limited scale; however, good antibody reagents are not always available. One approach to determine if an RNAi is targeting the anticipated gene is to rescue the phenotype by adding back a mutated form of gene, to which the RNAi reagent can no longer bind. 43 This tends to be a very time-consuming and technically difficult approach and in practice does not appear to be in routine use.
Common seed analysis (CSA) 69 and genome-wide enrichment of seed sequence matches (GESS) 70 are methods of comparing the seed region (bases 2–8) of hits with those of the entire RNAi population, identifying seed sequences that are highly enriched and therefore likely to be false positives due to OTEs. Both computational methods are available to RNAi users through open access. While both CSA and GESS are excellent approaches to “weed out” false positives from screens, true positives can be lost if there are not a sufficient number of RNAi reagents per gene or if most reagents are not potent. A bench-level approach to mitigate OTEs and eliminate false positives is to confirm hits with a new set of RNAi reagents of different sequence. 69 Another bench-level method that has been used to identify false-positive hits in siRNA screens is termed C9-11. This method takes advantage of changes made to bases at positions 9 through 11 with their complement, thereby only allowing target binding in the seed region. In this scenario, true-positive hits lose activity, whereas a false positive, driven by seed region OTEs, will still have activity. 71 Compared with CSA and GESS, C9-11 can be tested experimentally for small studies and large alike.
Rather than eliminating false positives from a screen, a recent computational approach, termed Haystack, combines prediction of seed region binding to the 3′UTR of genes with OTEs observed in published screens. This software is also available through open access to the RNAi user community. 72 This method has been successfully used to identify true hits from published screens, even when siRNAs against a gene were not present in the library. 72
Discussion
In conclusion, like any new pioneering technology, we have witnessed the start of the field of RNAi launch with unabashed enthusiasm and promise, only to be tempered years later by the practical caveats that come with addressing complex biological systems with ever-improving tools. Ironically, the tools of RNAi have evolved in a retrograde manner with respect to the physiological mechanism, beginning first with our understanding of the determinants of cytoplasmic siRNA and then progressing back through the nuclear membrane for a better appreciation of miRNA-based gene regulation. Clearly, the renaissance of RNAi is upon us, and rather than being competed into obscurity by upstart gene editing technologies such as CRISPR/Cas, we envision the two technologies as being complementary. The rapid ascent of CRISPR-based platforms can be partially attributed to lessons learned with RNAi. Undoubtedly, the unprecedented ability to quickly knock out genes will, in some cases, serve to elucidate mechanisms that were inaccessible with incomplete target knockdown via RNAi. Notwithstanding, we are in the earliest days of understanding CRISPR off-target effects. While the nascent components for CRISPR-based research continue to show progress, the more well-developed tools of RNAi will continue to call our attention to novel drug targets and to pathway interconnections of which we had not been aware.
Footnotes
Acknowledgements
The authors thank Scott W. Lowe and members of his laboratory for their useful discussions regarding the present work.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
C.-H.H. and C.-C. C. are funded by the Ministry of Education of Taiwan. M.F., Q.X., A.H., and R.J.G. are funded by the Geoffrey Beene Cancer Research Center at MSK.