
Abstract
For more than a decade, RNA interference (RNAi) has brought about an entirely new approach to functional genomics screening. Enabling high-throughput loss-of-function (LOF) screens against the human genome, identifying new drug targets, and significantly advancing experimental biology, RNAi is a fast, flexible technology that is compatible with existing high-throughput systems and processes; however, the recent advent of clustered regularly interspaced palindromic repeats (CRISPR)-Cas, a powerful new precise genome-editing (PGE) technology, has opened up vast possibilities for functional genomics. CRISPR-Cas is novel in its simplicity: one piece of easily engineered guide RNA (gRNA) is used to target a gene sequence, and Cas9 expression is required in the cells. The targeted double-strand break introduced by the gRNA–Cas9 complex is highly effective at removing gene expression compared to RNAi. Together with the reduced cost and complexity of CRISPR-Cas, there is the realistic opportunity to use PGE to screen for phenotypic effects in a total gene knockout background. This review summarizes the exciting development of CRISPR-Cas as a high-throughput screening tool, comparing its future potential to that of well-established RNAi screening techniques, and highlighting future challenges and opportunities within these disciplines. We conclude that the two technologies actually complement rather than compete with each other, enabling greater understanding of the genome in relation to drug discovery.
Introduction
Functional Genomics and High-Throughput RNAi Screening
Functional genomics, the study of gene/protein function at a genome-wide scale, has provided a wealth of knowledge throughout the past two decades. In particular, functional genomics studies in model organisms, such as yeast, 1 Caenorhabditis elegans, 2 Drosophila,3,4 zebrafish, 5 mice, 6 and numerous mammalian cell lines, have been central to many drug discovery strategies throughout the past decade.7–9 RNA interference (RNAi) has arguably been the most important breakthrough in the field of functional genomics thus far. Prior to RNAi, the techniques used to study the function and role of genes in disease were relatively slow and expensive.10–12 In the postgenomic era, the availability of complementary DNA (cDNA) and genomic sequence data, coupled with the flexibility, specificity, and usability of RNAi, has allowed high-throughput loss-of-function (LOF) screens to be performed at a genome-wide level in a fraction of the time and cost. These screens have provided an unprecedented amount of information on gene function, with the phenotypes associated with gene LOF contributing significantly to a wide variety of fields, including developmental biology, signal transduction, and drug discovery.13,14
RNAi is an endogenous post-transcriptional response to double-stranded RNA (dsRNA) that was originally discovered in C. elegans. 15 RNAi processes have since been demonstrated to control the gene expression in many eukaryotes, including humans, 16 and also to act as an antiviral defense system in many plants and insects. 17 The RNAi pathway is triggered by the presence of dsRNA or pre-microRNA, which is cleaved by Dicer (an RNAse II enzyme) into microRNA (miRNA) or small interfering RNA (siRNA) of 20–25 base pairs with an overhang of two nucleotides at the 3′ end. The antisense strand of the miRNA or siRNA duplex is loaded onto the RNA-induced silencing complex (RISC) and used as a guide to identify complementary target mRNA via Watson–Crick base pairing. This results in the cleavage of the target mRNA by Argonaute proteins in the RISC complex, preventing its translation and subsequent protein expression. The RNAi pathway is harnessed experimentally in mammalian cells by the introduction of synthesized siRNA or vectors encoding short hairpin RNA (shRNA; Fig. 1 ). Although shRNAs require processing by Dicer in the cytoplasm, as is the case with miRNAs, siRNAs can bypass this step and bind directly to RISC. Whether provided exogenously (shRNA or siRNA) or endogenously (miRNA), once processed and loaded into the RISC, the method of target messenger RNA (mRNA) recognition and degradation is fundamentally the same. 18
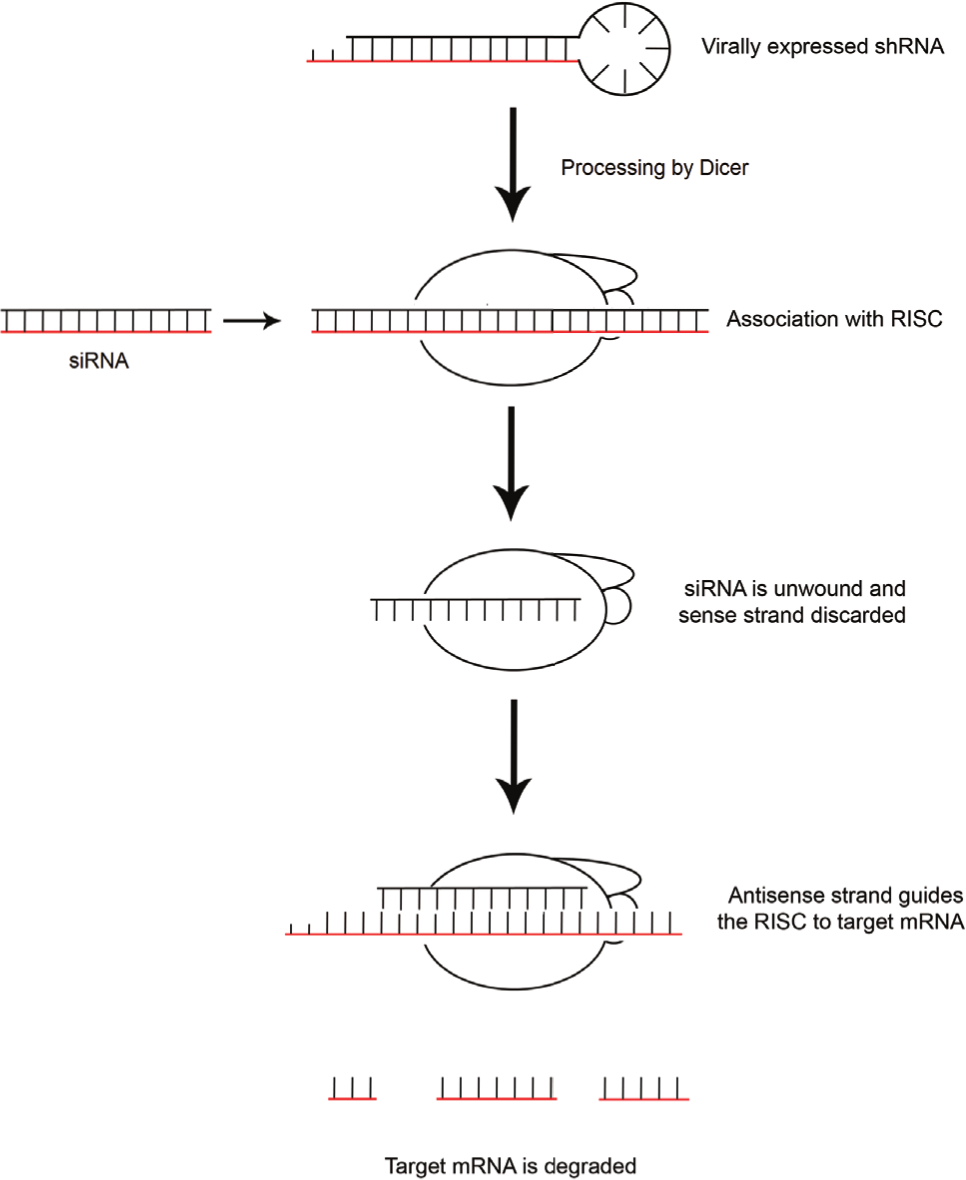
Experimental gene silencing through RNA interference (RNAi). The process of RNAi is initiated by the introduction of double-stranded RNA (dsRNA), such as short hairpin RNA (shRNA) or small interfering RNA (siRNA), to the cytoplasm. shRNAs require cleavage by the RNase Dicer, whereas siRNAs do not require this additional processing. The resulting short dsRNA is subsequently bound by Argonaute and other proteins to form the RNAi-induced silencing complex (RISC). Within the RISC, the siRNA is unwound and the sense strand degraded. The antisense strand then guides the selection of sequence-specific messenger RNA (mRNA), which is subsequently cleaved by the RISC, resulting in its degradation.
The relative ease in which RNAi can be manipulated for large-scale screening has led to the rapid rise in its use across both academia and industry, with RNAi screens typically performed in one of two formats; pooled or arrayed ( Fig. 2 ). Pooled screening is more applicable to shRNA libraries and combines hundreds or thousands of shRNAs into one multiplexed assay. This is usually achieved by introducing pooled vectors into a large population of cells and applying a selective pressure. For example, Tschöp et al. used a pooled lentiviral shRNA screen to identify kinases that regulated the ability of retinoblastoma protein (pRB) to stop cell proliferation. 19 Duplicate sets of cells were infected with lentiviral vectors that each carried a shRNA library targeting 415 human kinases. Cells were subjected to puromycin selection for 4 days before pRB expression was induced in one set, and proliferation and viability were subsequently monitored for a further 4 days. Cells not expressing pRB were used as a control set, and overall they led to the identification of 39 genes that specifically antagonized pRB function and 19 genes that enhanced it. This lentiviral approach has been shown to stably suppress genes effectively in a variety of mammalian cell lines, with screens performed ranging from resistance to oxidative stress to cell detachment and pathway analysis.20–23 ShRNA plasmids can also be directly transfected into cells, and this approach has been successful in many screens, including those involved with HIV 24 and cell death studies. 25 The direct transfection of the plasmid removes the need for a level-2 containment laboratory; however, plasmids generally provide only transient expression, and vector copy number is not as uniform or as well controlled across the cell population when compared to viral infection.
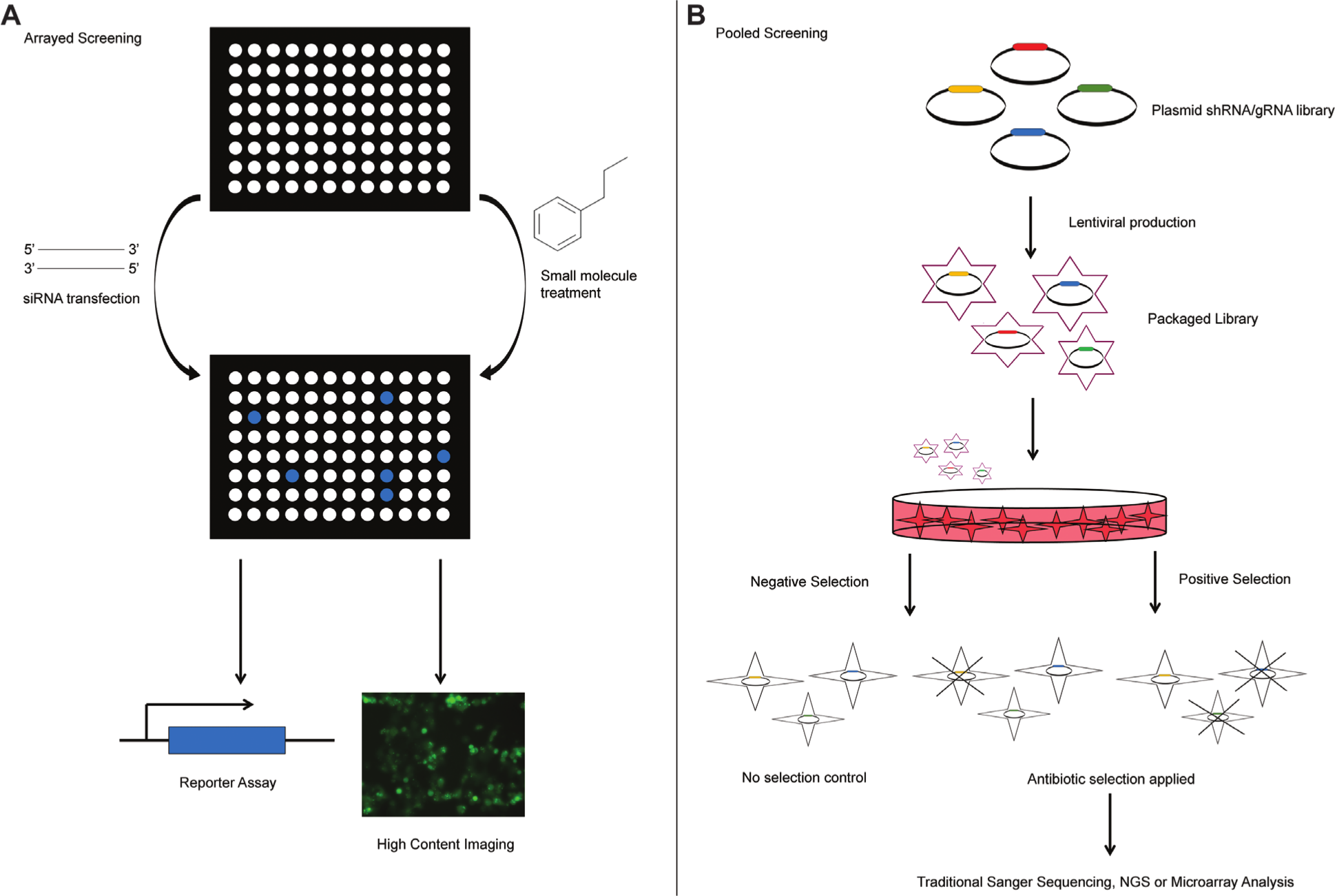
Experimental approaches to whole-genome RNA interference (RNAi) screening. RNAi screens can be performed in (
Arrayed RNAi screening can be a more flexible option, often using siRNAs, with hits from a screen generally requiring very little, if any, deconvolution ( Fig. 2 ). In this assay setup, each well of a microtiter plate contains a siRNA, or pool of siRNAs, directed toward a single gene. The use of a microtiter plate means that the same variety of assay readouts used in compound screening can be applied to RNAi screening. This includes detection methods at the whole-plate level or whole-well level, or high-content techniques measuring individual cells or intracellular events. To date, arrayed siRNA screening has been the most common approach to silence gene expression, with numerous genome-wide screens published in both model organisms and mammalian cell lines. From identifying novel oncogenic signaling pathways 26 to finding new potential targets for influenza 27 and unravelling the mysteries of the cell cycle, 28 high-throughput siRNA screens have contributed greatly to our understanding of biological systems, leading to advancements in therapeutics across a range of disease areas.13,14
The process of arrayed high-throughput RNAi generally requires a certain degree of liquid-handling and automation capability, so for many labs a pooled approach could be more practical. However, if these capabilities are already in place for compound screening, the process of converting a compound-based screen to a siRNA-based screen can be achieved relatively easily. There are many advantages to using an arrayed screen over a pooled screen; these include the duration of the screening process and assay compatibility. Once hits are identified through either screening approach, the validation and follow-up work can be time consuming; therefore, any ways to speed up hit identification are beneficial. Arrayed screens simplify or even eliminate the need for hit deconvolution following the primary screen, with each well containing siRNA targeting one gene, hence speeding up the process. Contrary to this, pooled approaches using shRNAs often require complex or expensive deconvolution strategies to identify which shRNAs have been positively or negatively selected for during the screen; these strategies include microarray profiling or next-generation sequencing (NGS) of DNA. 29 Moreover, with any pooled screen, there is a risk that each gene/shRNA in the full library is not represented uniformly. This can be mitigated by using a high transfection efficiency when creating the virus library, and profiling the representation of shRNAs across the library prior to any selection, 29 again using microarray or NGS techniques; however, this adds yet another lengthy and complicated step to the process. A further advantage of arrayed screens is their compatibility with high-content assays, such as imaging and flow cytometry techniques, to provide a more detailed assessment of cellular phenotypes. Moreover, a high-content approach can identify the siRNA-transfected cells, via co-transfected fluorescent siRNA probes, alongside measuring multiple other parameters such as a cell cycle profile and nuclear area, allowing only these transfected cells to be included in the analysis. 30
As noted, RNAi screening has had a huge impact in the field of functional genomics, and with many RNAi libraries now commercially available for human, mouse, and rat cells, why are we looking for an alternative? The main challenges of an RNAi experiment involve gaining an efficient knockdown of the target gene while maintaining cell viability throughout the experiment and balancing unwanted off-target effects (OTEs). 31 These OTEs can result in the knockdown of target-unrelated genes, leading to confusing results that cannot be confirmed by other methods during follow-up work. For example, OTEs can occur if any base pair mismatch is tolerated between the siRNA-loaded RISC and the target mRNA. This would lead to these additional nontarget genes being knocked down, raising concerns over the specificity of RNAi and the reproducibility of results. 32 OTEs have led many scientists to be skeptical of results from RNAi screens and therefore to seek alternative approaches for their own research.
The Advent of the CRISPR-Cas Genome-Editing System
Unlike RNAi, which targets the multiple copies of mRNA to knock down gene expression, the RNA-based CRISPR-Cas system directly targets DNA and can lead to complete gene knockout. Clustered regularly interspaced palindromic repeats (CRISPR) work alongside associated Cas genes to form a prokaryotic adaptive immune response to viral invasion. This ingenious way that archea and bacteria have evolved to eliminate the threat of exogenous vectors and phages has been studied sporadically since 1987, 33 but only recently has its potential for genome editing in mammalian cells been explored.34–36 Three types of bacterial CRISPR systems have so far been identified, 37 with type II forming the basis of the CRISPR-Cas9 system that has been developed for mammalian genome engineering. Although prokaryotes rely on two separate pieces of RNA, one that complements the foreign DNA and one that recruits the Cas nuclease to cleave it [i.e., CRISPR RNA (crRNA) and transactivating crRNA (tracrRNA)], 38 CRISPR-Cas9 technology encompasses both these features into just one piece of engineered guide RNA (gRNA). The 5′ end of the gRNA has specificity for a section of target DNA, whereas the 3′ end creates a scaffold to recruit Cas9, the type II specific nuclease. Recruitment of Cas9 and its nuclease activity to the target DNA results in a double-strand break (DSB) in the DNA. In the absence of a template, these breaks are subsequently repaired by error-prone nonhomologous end joining (NHEJ). This process generally leads to small insertion or deletion mutations (INDELs) in the target DNA, normally leading to gene disruption and preventing functional protein expression. 39 Alternatively, if a template is present, then DSBs can undergo homology-directed repair (HDR) to introduce specific mutations into the target DNA. 39
The capacity the CRISPR-Cas9 technology has for PGE is essentially unrivalled. The predecessor technologies, zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), are both protein-based approaches to PGE, which consist of DNA binding domains that can be synthesised to match a target gene sequence, and a nuclease domain to cut the DNA. Both are highly target specific, because two proteins need to bind at specific sites on either strand of the DNA to make the cut;40,41 however, ZFN technology is proprietary 42 and can therefore be expensive and more restrictive, and although TALENs are cheaper to synthesize, their larger size makes them more difficult to transfect and express in cells. Both methods are reasonably difficult to design and have been associated with detrimental off-target DSBs, 43 the repair of which can give rise to nonspecific genome rearrangements. 44
Evolution of CRISPR-Cas as a Screening Tool
The therapeutic value of CRISPR-Cas has been reviewed in great detail, and exciting developments in mouse models45–47 have given hope for future therapies for genetic diseases. However, the arrival of CRISPR-Cas as a large-scale screening tool for use in drug discovery has been more understated in the mainstream media. The developments in this area are just as exciting and have led many scientists to wonder whether there is a continued need for RNAi screening.
To date, three versions of the CRISPR-Cas system have been used for mammalian cell screening ( Fig. 3 ), the first and most common being the wild-type humanized Cas9 (hCas9; Fig. 3 ). hCas9, guided by a specific gRNA, introduces DSBs and activates the cell’s DNA repair pathway as described above. NHEJ then results in INDELs within the target gene sequence, ultimately disrupting gene expression. In 2013, data from four screens were published almost simultaneously,48–51 and although the screens were essentially carried out in isolation, the methodologies were surprisingly similar. Each screen used a pooled lentiviral approach similar to that used for pooled shRNA screening, with three studies using a genome-wide library and the fourth using a more focused library of gRNAs. Cells were transduced with lentiviral vectors containing the gRNA libraries, then positively selected for resistance to cell death induced by chemotherapeutics, 48 vemurafenib, 51 anthrax and diphtheria toxins, 49 or Clostridium septicum alpha-toxin and 6-thioguanine. 50 Most gRNAs will confer no resistance to the cell for these various challenges and will subsequently be lost through the selection process, whereas those that do confer resistance will be enriched. For example, Zhou et al. observed the enrichment of all gRNAs that targeted the same known anthrax receptor gene, ANTXR1. 49 In addition to proving that CRISPR-Cas9 could be used as a high-throughput screening tool through revealing known biology, these studies also commented on how the system could be improved through better gRNA design 48 and the inclusion of novel validated genes 49 in the libraries, and by directly comparing the technology to shRNA. To this end, Koike-Yusa et al. tested the comparison to shRNA. 50 Validated shRNA sequences for six genes identified as potential hits during the initial CRISPR-Cas9 screen were cloned into a lentiviral vector and directly compared to the equivalent gRNAs. Although all gRNAs produced resistant cells using the CRISPR-Cas9 system, only one shRNA produced a comparable number of resistant cells.
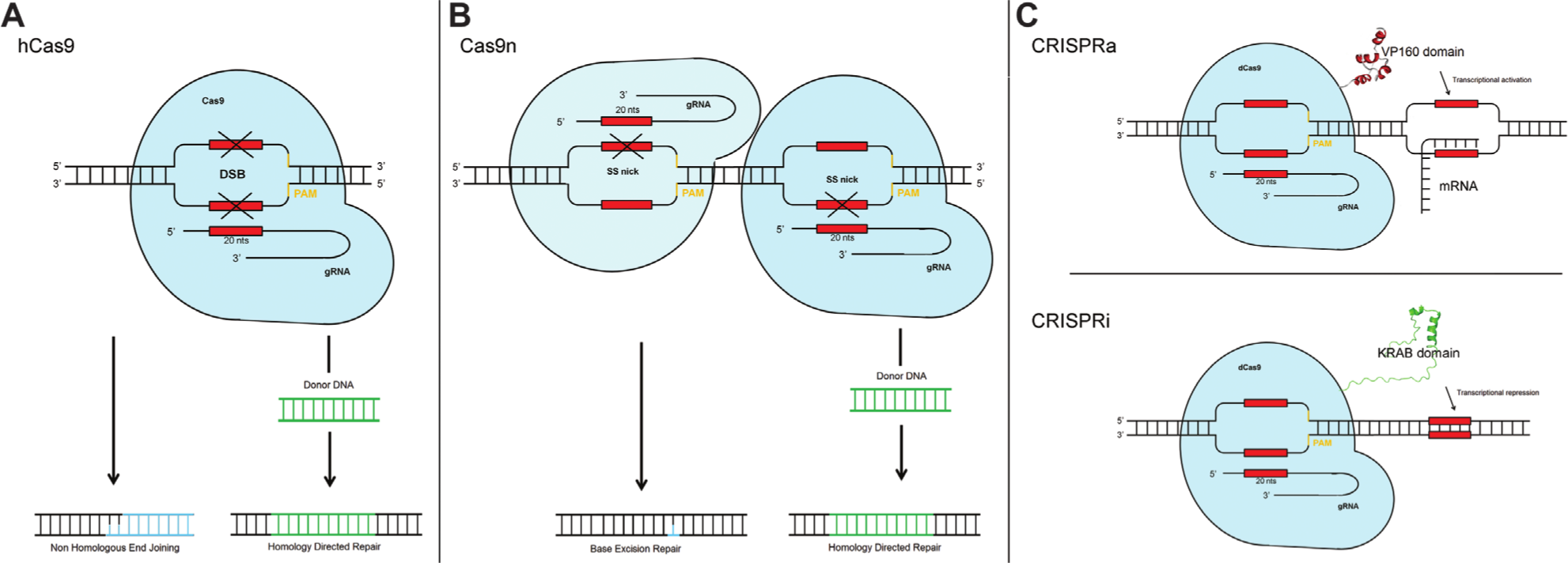
Experimental clustered regularly interspaced palindromic repeats (CRISPR)-Cas systems for screening. Three CRISPR-Cas9 systems are currently used for precise genome editing (PGE) and screening in mammalian cells: (
Although all these screens reported high targeting efficiency of gRNA, some OTEs were discernible. Thus, a second-generation Cas9 system was quickly developed by researchers using a mutated Cas9 nickase that in isolation, instead of creating a DSB, only creates a “nick” in one strand of the DNA (Cas9n; Fig. 3 ). 52 The system requires independent binding of two separate gRNA–Cas9n complexes to complementary strands of the same target genomic region. This results in the induction of single-stranded “nicks” on each strand of the DNA and hence a DSB similar to that of wild-type Cas9. Because any single-strand nicked genomic DNA away from the target site will be efficiently repaired with high fidelity through a variety of single-strand break repair pathways,53–55 this new Cas9n approach has been suggested to reduce off-target mutagenesis effects by 50- to 1000-fold. 52 Cas9n shows analogous targeting efficiencies to the wild-type Cas9, whereas its nickase functionality mimics the dimerization requirements, and therefore the precision, of TALENs and ZFNs. This system can also be used to generate both knockout and knock-in genotypes using recombination cassettes; however, the overall technical intricacy of the process makes it less feasible for application in high-throughput screening. 56
Qi et al. developed an alternative approach specifically for use in gene regulation. 57 This system uses a catalytically inactive Cas9 enzyme (dCas9; Fig. 3 ) that, when coexpressed with a gRNA, specifically and strongly silences the gene of interest without editing the genome. 58 Moreover, this modified CRISPR system allows for the suppression (CRISPRi) or activation (CRISPRa) of genes without the need for error-prone NHEJ or reliance on the efficiency of DNA repair pathways.57–59 For example, dCas9 has been fused to repressor or activator proteins to enhance gene silencing or activation potential by Gilbert et al. 60 This work was followed up by an elegant paper using both CRISPRi and CRISPRa to provide complementary information on genes essential for K562 cell proliferation. 61 The authors describe the use of two enhanced dCas9 proteins, dCas9-KRAB for suppression and dCas9-SunTag for activation ( Fig. 3 ). The SunTag protein tag 62 was fused to catalytically dead Cas9, and once expressed the SunTag recruits multiple copies of a coexpressed transcriptional activator, leading to enhanced transcription. Results from the SPI1–GATA1 and CEBPA–CEBPG gene pairs in particular demonstrated the complementarity of the screen. 61 In both gene pairs, one member inhibits the function of its partner, and, accordingly, SPI1 and CEBPA were strong hits in the CRISPRa screen, and their partners GATA1 and CEBPG were hits in the CRISPRi screen. This approach was then used to elucidate a more complex pathway and also to screen for genes that control sensitivity to the cholera–diphtheria (CTx-DTA) toxin. The results further highlighted the complementary nature of the CRISPRi and CRISPRa screens, and gave insight into how CRISPRa can be used to understand the upregulation of pathways. In addition, the suppression of genes by CRISPRi was found to be inducible and reversible, with minimal OTEs.
All three Cas9 systems use cells that either express Cas9 stably or coexpress Cas9 alongside the gRNA from a single vector. The latter method allows for more flexibility in cell type, and these vectors are commercially available. Studies show that continuous Cas9 expression is not cytotoxic;35,63 furthermore, Platt et al. have shown that constitutive Cas9 expression has no effect on basal function in mouse neurons when parental and Cas9-expressing cells were compared. 64 Chronic expression of Cas9 may yet prove to amplify OTEs and possibly have other unforeseen consequences; however, this is not expected to be an issue for screening.
The overall aim of functional genomics studies is to link specific genes to specific functions during the primary screen, and validate these gene–function relationships in follow-up work. A gold-standard approach to such gene validation is to rescue the detected phenotype by re-expression of the gene of interest. In the case of RNAi screens, this process has added complications: Either the RNAi needs to target outside the cDNA rescue construct (e.g., the 3′ UTR), or, more generally, the rescue cDNA construct needs to be modified to become resistant to the relevant RNAi sequence.65,66 Contrary to this, because CRISPR-Cas knockout screens lead to permanent deletion of the gene, rescue experiments are generally more straightforward, using commercially available cDNA constructs and potentially leading to rapider gene validation. In both cases, it is also possible to gain vital structure–function information for the gene of interest by performing rescue experiments with mutated rescue constructs (e.g., those that are catalytically inactive). 65
Challenges of the CRISPR-Cas System for Screening
Cleavage by Cas9 not only is determined by the complementarity of the gRNA for the target gene sequence but also requires additional component sequences to be included, increasing the technical difficulties of designing and making CRISPR libraries. For example, the process of creating a single gRNA that incorporates both crRNA and tracrRNA requires the expression of this fused gRNA from a polymerase III promoter that can effectively transcribe short sequences. One example that has been used extensively for gRNA expression is the human U6 polymerase III promoter (U6), but this restriction means that target sequences must initiate with the base “G” for transcription to occur. 35 Moreover, the CRISPR mechanism itself requires a specific protospacer adjacent motif (PAM), a short 3–5 base pair sequence at the 3′ end of the target sequence, for acquisition of the gRNA and subsequent Cas9 cleavage. 67 The PAM sequence differs from the bacterial species from which Cas9 was derived, with the most commonly used Cas9 taken from Streptococcus pyogenes (SpCas9). SpCas9 requires the PAM sequence “NGG” (where N represents any base); thus, together, these two caveats mean that target sites are limited to those containing the sequence “GN20GG.”
Computational analysis of the occurrence of GN20GG in the human genome shows that the predicted targeting space of SpCas9 is restricted to ~40% of exons. 35 Further studies have, however, since demonstrated that SpCas9 will also cleave with reduced efficiency at the NAG PAM sequence, increasing target possibilities 68 yet introducing a measure of doubt in the targeting specificity of CRISPR-Cas. Ranganathan et al. also have expanded the potential targeting space of SpCas9 by using a H1 polymerase III promoter to initiate gRNA expression, which has less restrictions when compared to the U6 promoter, allowing the targeting of sites that occur ~15% more frequently in the genome. 69 Of course, the use of other Cas9 species with alternative PAM sequences would enable a wider coverage of the genome, and although the identification of functional Cas9 from different bacterial species has been identified,70–72 these have not yet been extensively characterized and validated for PGE.
Contrary to the CRISPR-Cas9 system, RNAi silencing methods rely on targeting mature mRNA and have no PAM restriction. Engineered siRNAs can theoretically target >99% of human mRNA sequences, 58 yet OTEs continue to hamper the technology. Commercial suppliers of siRNAs use numerous techniques to mitigate OTEs, the most common being chemical modifications of the siRNA duplex to increase specificity and stability. Chemically modified siRNAs have been shown to reduce off-target silencing by up to 66%. 73 In addition, the seed region, a short 2–8 base pair sequence at the 5′ end of the siRNA, has been found to be complementary to many mRNAs and so produce a myriad of OTEs. 74 This complementarity can be analyzed after screening using genome-wide enrichment of seed sequence (GESS) matches 75 and Haystack 76 software (Haystack, Cheshire, CT). Both programs are freely available online to analyze large-scale data sets and to identify potential seed region OTEs. Various siRNA technologies, such as Dharmacon SMARTpools (Dharmacon, Lafayette, CO), offer additional protection against OTEs for arrayed RNAi screening. Four siRNAs targeting the same gene are mixed in one reagent. The total working concentration of the pool is, for example, 25 nM, yet each individual siRNA is at a lower concentration of ~6 nM. Because each of the siRNAs is unlikely to induce the same OTE, the low concentration of the individual siRNAs decreases the chance of OTEs while providing additive on-target gene knockdown. 77
Although OTEs of CRISPR-Cas systems have been reported as being significantly less than those of RNAi, OTEs do exist, and with hCas9 systems they potentially can be more problematic due to the potential to induce dsDNA breaks. There is disagreement on the specificity of CRISPR-Cas, with some studies displaying little or no OTEs48,78,79 and others reporting a high tolerance for mismatches by gRNA that could lead to OTEs.80,81 Fu et al. further showed that CRISPR-Cas induces a high frequency of OTEs in human cells by conducting an in-depth, large-scale assessment of the effect of gRNA mismatches in U2OS:EGFP cells. 82 The quantitative reporter assay rapidly assessed inactivating frameshift INDEL mutations, concluding that single and double mismatches were highly tolerated, with a mean rate of mutation at verified off-target sites of 40%. 82
The specificity of CRISPR-Cas systems, although not entirely understood, is determined by the gRNA. gRNAs are typically ~20 bases long, yet target specificity is thought to be conferred by a 12-base seed region adjacent to the PAM. 83 Mismatches introduced at the seed region have been shown to arrest DNA cleavage, whereas mismatches furthest from the PAM were tolerated. Two more recent studies have shown that Cas9 can bind with seed regions as small as five bases in length,84,85 although this does not seem to cause off-target editing, suggesting that additional base pairing is required for cleavage in tandem with seed region specificity. Moreover, although the tolerance of different PAM sequences by spCas9 (e.g., NAG) is beneficial for genome-wide screening, by increasing the possible target sequences, it also increases the number of potential off-target sites. 86 Exploring other Cas orthologs could identify those that require PAMs with a higher degree of specificity and therefore reduce OTEs; however, this may further limit the targetable space.
The prevalence of OTEs in RNAi throughout the past decade is actually beneficial for the CRISPR-Cas system. Bioinformaticians have built up a store of algorithms and software to characterize OTEs, 31 rationally design siRNAs, 87 and predict efficacy, 88 and all of this computational power has been available to CRISPR-Cas from the outset. This has allowed gRNA design technology to be shared quickly via open-access software, and the development of scoring algorithms and other gRNA validation applications. More hands-on approaches also have been developed to reduce CRISPR-Cas OTEs. In addition to the second-generation nickase system described previously, Fu et al. found that by shortening the length of the targeting region of the gRNA from 20 to 17 base pairs, OTEs are reduced by 5000-fold or more while retaining the targeting efficiency of their longer counterparts. 89 The CRISPRi system has had less data published on OTEs, yet studies show no discernible OTEs in E. coli 57 and minimal OTEs in mammalian cells.60,90 Like RNAi, OTEs are less damaging in the CRISPRi system because there is no DNA cleavage. Moreover, because inactivation by CRISPRi is inducible, any damaging phenotypes can be reversed and analyzed in more detail if necessary.
A further challenge when planning a CRISPR-Cas knockout screen is that all copies of the gene must be edited. Although in many mammalian cell lines, this is limited to two gene copies, the inherent genome and chromosomal instability of many cancer cell lines can require tens of copies to be edited per gene to generate a complete knockout, a good example being HeLa cells. 91 Therefore, if a particular cancer cell line is essential, ensuring a consistent knockout throughout the screen could prove to be a significant task. This is an emerging area of study and as such has not yet been fully evaluated; however, there are numerous ways to validate the efficiency of gene knockout by CRISPR-Cas. These include fluorescence in situ hybridization (FISH), digital PCR, and NGS; however, their relative throughput would need to be carefully considered when analyzing gene knockout efficiency alongside a CRISPR-Cas screen editing of thousands of genes. An alternative CRISPR-Cas approach for cell lines with a high gene copy number could be to use the CRISPRi system; this only requires binding of the Cas/gRNA complex to the target site on DNA to induce its effects on gene expression, and as such should be more robust to changes in gene copy number. 58
Efficiency and Delivery of the CRISPR-Cas System
Another potential benefit of CRISPR-Cas over RNAi could be in the area of on-target efficiency. Although CRISPR-Cas has been shown to have a higher targeting efficiency than other PGEs, the efficiency of CRISPR-Cas is highly dependent on the cell type and the complementarity of the gRNA. Most commercial siRNAs now guarantee a knockdown of at least 70%, yet this type of guarantee has yet to be matched by gRNA suppliers for the CRISPRi system. Furthermore, CRISPR-Cas PGE efficiency has shown to vary widely, as exemplified by Mali et al., who reported efficiencies of 10–25% in 293T cells, 13–38% in K562 cells, and 2–4% in induced pluripotent stem (iPS) cells. 35
Efficiency in both technologies can also be decreased by an ineffective delivery system and variations in cell biology. For example, a delivery efficiency of 25% compared to 50% effectively means that double the amount of cells would need to be screened to find the same number of targeting events. Although RNAi and PGE technologies currently require either the transfection of nucleic acids or the transduction of viruses into the cell for gene suppression or knockout to occur, all practicable methods of transfection and transduction confer a certain degree of toxicity. 92 Although physical transfection techniques such as electroporation are highly efficient, their impact on cell health can be detrimental.93,94 Nanoparticles exploit natural biological pathways to deliver siRNA and PGE components into cells, and cationic liposomes are the most widely used in vitro transfection method due to their higher transfection efficiency. 95
There are many commercial transfection products available that are rigorously tested for toxic effects on the more popular cell lines, such as HeLa, Jurkat, and HEK; however, data are often lacking for cells that are traditionally hard to transfect. In addition, as the field of personalized medicine expands and cell culture techniques improve, the use of primary cell lines will increase. Although specialized reagents exist for both primary and hard-to-transfect lines, meticulous optimization of reagent volumes, cell density, and other factors must take place. Without specialized screening equipment and protocols in place, assay development and optimization often can be difficult and time-consuming. The use of biological, noninvasive transfection methods such as cell-penetrating peptides (CPPs) has shown promise in the delivery of siRNAs and CPP-CRISPR and CPP-gRNA conjugates into human cells. 96 The cytotoxicity of CPPs is undetectable at concentrations needed for delivery, 97 although the chemistry involved and the cost could prove restrictive for higher-throughput screens.
The challenges associated with introducing oligos or plasmids into cultured cells is frequently overlooked in reviews and largely ignored in methodologies, yet often it consumes a large proportion of resources and time during the screening process. A successful experiment needs to strike a careful balance between the toxicity of the delivery reagent to the cell and the efficiency of nucleic acid or viral delivery to enable gene knockdown or knockout. Lengthy assay optimization procedures and a robust assay design will go a long way to mitigating problems in these areas.
Conclusions
Well-validated human whole-genome RNAi libraries and sublibraries are commercially available for high-throughput LOF screens; yet the low cost and reported minimal OTEs of the CRISPR-Cas technology, alongside the promise for novel applications, mean that many researchers are changing tact. CRISPR could be a good alternative and overtake RNAi among laboratories new to the field of functional genomics screening, with the dCas9 CRISPRi system most analogous to RNAi screening, avoiding potentially destructive DSBs yet suppressing gene expression more efficiently. Its versatility holds promise that it could surpass the capabilities of RNAi in the future, especially for increasing genomic knowledge of nonmodel organisms, 98 identifying important evolutionary events, 99 and enhancing industrial food biotechnology. The editing potential of CRISPR systems has already been used for exploring the wheat genome, 100 and CRISPR screening could be used for discovering mutations that aid resistance to pathogens or improve nutritional value. 101 Furthermore, groups already running pooled lentiviral shRNA screens could simply modify protocols to incorporate lentiviral CRISPR libraries. Successful, more focused screens have been reported,48,49,102 and as the technology grows in sophistication, a greater range of pooled and arrayed libraries will undoubtedly become commercially available in the near future.
The use of the CRISPR-Cas system as a genome-engineering tool will continue to grow in popularity for creating robust, well-controlled assays in cell lines and model organisms. CRISPR-Cas has clear benefits over other PGE techniques when it comes to introducing disease-associated mutations, gene knockout, gene knock-in, and in-frame fusions of reporter genes. Yet, as the technologies stand, RNAi still has a future in a screening setting. Although for many experiments, complete knockout using CRISPR-Cas may be a cleaner approach, transient and incomplete gene knockdown via RNAi could provide a more realistic indication of potential small-molecule inhibition103,104 and disease phenotypes. This is partly due to the fact that most therapeutic agents directly target the functional protein, rather than DNA or RNA, often having effects within minutes of addition. RNAi has been shown to have effects on protein levels within 4 h, with maximal knockdown between 24 and 48 h, whereas CRISPR-Cas experiments are generally run during a 3–10-day timescale. Obviously, this has ramifications in experimental design, and it has the potential to cause issues when screening rapidly dividing cells in an arrayed format, because cells would overgrow the well within this timescale. Furthermore, the partial knockdown effect of siRNA is also beneficial when assessing the phenotypes of target genes that are essential for cell survival,105,106 in which a complete gene knockout approach is unviable.
In terms of ultra-high-throughput genome-wide screening, siRNA technology is currently able to integrate well with the arrayed format that is widely used in the pharmaceutical industry and contract research organizations for compound screening. Through this integration, valuable functional genomics data can be generated quickly, and relatively cheaply, using existing screening expertise and limited extra resources. The potential of CRISPR-Cas to be used downstream of RNAi screens is vast, to confirm hits by creating equivalent knockout cell lines. Using arrayed siRNA screening in conjunction with the PGE functionality of CRISPR-Cas would currently exploit the best of both worlds, producing rapid results with a high degree of confidence. Although OTEs remain an issue in both technologies, reducing the OTEs of RNAi has long been a priority in functional genomics. The lessons learned from RNAi have already gone a long way to help CRISPR-Cas overcome OTEs, with new findings in CRISPR-Cas also likely to aid RNAi improvements. In the near future, it will be key to perform further direct comparisons between RNAi and CRISPR-Cas in terms of screening, particularly assessing results from a range of assay formats, and carefully monitoring OTEs for both technologies. Presently, using CRISPR–Cas to knock out nonessential genes could quickly become the routine method of validating RNAi screening hits; the marriage of these two technologies would create a complementary, robust screening platform, enabling the elucidation of more complex pathways and the identification of novel targets for drug discovery.
Footnotes
Acknowledgements
We thank Brett Litten for help with generating the figures, and Kirsty Rich, Mark Wigglesworth, Lorenz Mayr, and Steve Rees for critical reading of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.