
Abstract
Bringing a new drug to the market costs an average of US$2.6 billion and takes more than 10 years from discovery to regulatory approval. Despite the need to reduce cost and time to increase productivity, pharma companies tend to crowd their efforts in the same indications and drug targets. This results in the commercialization of drugs that share the same mechanism of action (MoA) and, in many cases, equivalent efficacies among them—an outcome that helps neither patients nor the balance sheet of the companies trying to bring therapeutics to the same patient population. Indeed, the discovery of new therapeutic targets, based on a deeper understanding of the disease biology, would likely provide more innovative MoAs and potentially greater drug efficacies. It would also bring better chances for identifying appropriate treatments according to the patient’s genetic stratification. Nowadays, we count with an enormous amount of unprocessed information on potential disease targets that could be extracted from omics data obtained from patient samples. In addition, hundreds of pharmacological and genetic screenings have been performed to identify innovative drug targets. Traditionally, rodents have been the animal models of choice to perform functional genomic studies. The high experimental cost, combined with the low throughput provided by those models, however, is a bottleneck for discovering and validating novel genetic disease associations. To overcome these limitations, we propose that zebrafish, in conjunction with the use of CRISPR/Cas9 genome-editing tools, could streamline functional genomic processes to bring biologically relevant knowledge on innovative disease targets in a shorter time frame.
Keywords
Introduction
The past decades have brought huge improvements in the scientific and technological contributions to the drug discovery and development fields. These advances have greatly increased the quality and quantity of the available scientific data, as well as the tools to develop new and better drugs for the patients. For instance, combinatorial chemistry allows nowadays the synthesis of huge and diverse chemical libraries, multiplying the chances of successfully targeting a given chemical space. 1 In the same direction, advances in X-ray crystallography detector technology have enabled the calculation of dynamic 3D protein structures in a wider and more detailed size range. These advances have allowed improved structure-driven strategies—rational drug discovery—required for the identification of promising lead compounds. 2 Then, next-generation sequencing (NGS) has brought new opportunities for understanding genetic disease causality and reducing the costs for identifying potential new drug targets based on genome, transcriptome, proteome, or metabolome alterations specifically detected in patient populations or subpopulations, allowing the advance of precision medicine. In line with that, novel computational methods and innovative high-throughput screening platforms have generated a large volume of high-resolution “omics” data available for researchers. 3 Finally, the advent of genome-editing technologies, such as CRISPR/Cas9, is enabling a broad range of functional genomic applications for understanding gene function in normal physiology and in the context of disease. All these technologies represent an unprecedented opportunity to streamline and enrich the drug discovery process by the identification and/or better understanding of new, improved drug targets.
Despite these advances, likely explaining the increase of approved drugs during the past decade, the pharmaceutical industry is still facing a widely discussed “productivity crisis.” This reduced productivity is explained by the fact that bringing new drugs to the market has escalated in cost at a steeper rate than the number of approved drugs ( Fig. 1 ). The reasons behind this growing rate in the cost per drug ratio (US$2.6 billion) are multiple and complex, and involve scientific factors related to drug research and development (R&D), and regulatory and business aspects, in overall making the whole process highly inefficient. 4,5 Together, the low success rate and high cost have a negative impact on: patients waiting for new or better treatments; health systems spending enormous resources on pharmaceutical treatments; and the pharma industry, which is unable to risk investment on searching treatments for those indications in which the return of investment (ROI) might be low. This is exemplified by Pfizer’s recent decision to stop research on neurodegenerative diseases. 6
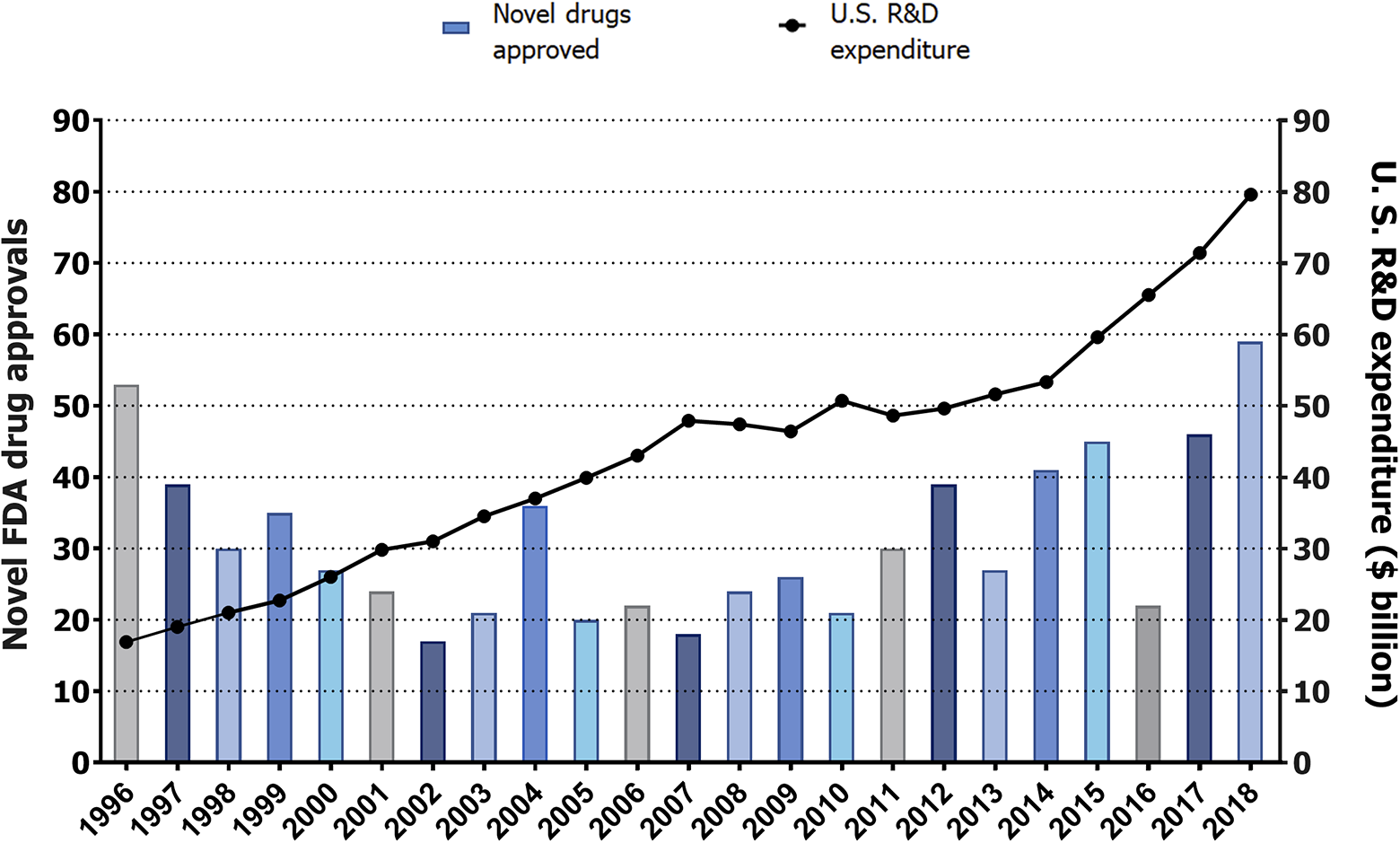
Pharmaceutical research and development (R&D) productivity crisis. From 1996, investment in drug R&D in the United States has grown at a steeper rate than the new number of drugs introduced into the market. The left vertical axis provides the number of novel new molecular entities (NMEs)—small molecules and biologics—approved each year, represented by purple bars. The right vertical axis provides R&D expenditure per year, represented by black dots. The horizontal axis displays the years, ranging from 1996 to 2018. Sources: Drugs@FDA for drug approvals; and Pharmaceutical Research and Manufacturers of America (PhRMA) for R&D expenditure.
The major causes of failure in clinical phases are lack of safety and efficacy in humans. 7 Indeed, the drug attrition rate is central to the cost explosion and consequent decrease in R&D productivity. Looking backward, clinical failures reflect deficiencies in preclinical data collection, including data from early drug discovery stages. This points to the importance of selecting the best—that is, most predictive and cost-effective—preclinical model for each given indication. 8 In that sense, the success of a potential new therapeutic drug is directly proportional to the accuracy of toxicity and efficacy predictions during the preclinical phase. 7,9 Therefore, the identification and validation of drugs in the most suitable preclinical models are crucial to determining their later success as new therapeutic drugs. 10
A related and important source of clinical failure derives from choosing the “wrong” drug target as the focus of a given therapeutic program. Indeed, a “right” drug target should provide limited on- and off-target toxicity and greater efficacy, likely allowing a reduction in the therapeutic dosage and, hence, reducing further the chances for off-target toxicity. As such, discovering and validating novel drug targets have been singled out as a major opportunity for increasing productivity in drug development. 11 Despite this, the vast majority of annotated proteins have not been exploited as therapeutic targets. In that sense, the Illuminating the Druggable Genome (IDG) initiative, launched by the US National Institutes of Health (NIH), has divided the proteome into four categories, according to their relationship with approved drugs and disease. 12 Of these, only 3% of the annotated human proteins are clinic targets (Tclin)—understood as a protein that binds to at least one approved drug and whose mechanism of action (MoA) is known. The remaining 97% are divided among 6% chemistry targets (Tchem)—a protein that binds to a drug with high efficiency but whose MoA is unclear; 53% biology targets (Tbiol)—a protein known to have a relationship with a disease or some biologic process, but unknown binding efficiency to a bioactive drug; and the remaining 38% correspond to dark targets (Tdark)—a protein with no role or a scarcely known role, but a function that could be inferred by protein homology or that might have been found as a potential disease-associated gene as part of a NGS patient-derived database. Beside this categorization, based on available drug–target–MoA information, not all proteins might be druggable, because this feature depends on protein activity and structure. Even so, when considering the druggability parameter, only 2 out of 22% of the “potentially druggable” human proteins are currently targeted by drugs. 13 Finally, posttranslational modification and alternative splicing produce functional isoforms with different interaction profiles. This may increase further the proteome diversity and represent an additional layer of complexity, thus providing further examples of the importance of discovering and understanding novel drug targets in the context of physiology and disease. 14
In our view, this drug–target knowledge gap provides an enormous therapeutic and business opportunity. Most pharmaceutical companies, however, take advantage of known targets’ biology and available structural data to develop improved and more potent drug candidates, instead of investing resources in looking for drugs against new targets. This approach might show limited applicability and success, since it is intended to enter already-crowded therapeutic areas. Therefore, society might be lacking new treatments, and the pharma industry losing productivity, due to the pharma industry’s aversion to risk. The aim of this perspective article is to explain why and how the use of the zebrafish model, combined with the application of clustered regularly interspaced short palindromic repeats (CRISPR)-based technologies, provides an innovative, lower-risk solution to accelerate the discovery of novel therapeutic targets, while providing a better understanding of the causes of disease and potential drug off-target toxicity.
Improving Pharmaceutical Productivity
As already introduced, the need for more predictive animal models and the low availability of druggable targets might be the most important reasons explaining drug attrition in clinical phases and, consequentially, decreasing pharmaceutical productivity.
Among the current preclinical models, in silico virtual screening is gaining importance for performing initial analyses of drug absorption, target organ concentration, clearance, efficacy and toxicity. The predictive power of these tools is constantly increasing thanks to the aid of machine learning and artificial intelligence. The usefulness of these tools is, however, burdened by a lack of 3D structure data for most protein targets and compounds, and an incomplete back-to-back validation with in vitro and in vivo data. 15 It is expected that databases such as Tox21, which comprises data points on approximately 12 000 chemicals screened in 12 different bioassays, might allow researchers to make such comparisons to refine current in silico tools. 16 Either way, such tools allow the preselection of promising therapeutic compounds, which will still have to be validated later through in vitro and in vivo models.
In vitro systems are always evolving. In addition to classical 2D cell cultures, complex 3D cell cultures, organoids, and organ-on-a-chip technologies have been developed with the aim to mimic and study more faithfully the impacts of drugs on tissues, organ structure, and physiology. 17 Despite these advances, in vitro systems might lack the complex biology of a whole living organism. For that reason, testing drugs in mammals for understanding their toxicity, efficacy, and metabolism before entering clinical phases is required by regulatory agencies and is the most accepted method in the pharmaceutical community. The enormous cost of in vivo drug research, however, implies that just a few compounds can be tested in these models. Thus, in silico and in vitro experimental limitations might have a downstream impact on the quality of those few compounds allowed to be tested later in vivo.
Regarding the discovery of new druggable targets, the advent of NGS-based methodologies has increased the number of identified disease-associated DNA sequence variants. Indeed, whole-exome sequencing (WES) and genome-wide association studies (GWAS) have been crucial in the identification of single-nucleotide polymorphisms (SNPs) at disease-risk loci. 18 More importantly, it has been shown how multiple disease-risk loci overlap significantly with genetic targets of approved drugs, resulting in the duplication of clinical success rates, when the drug target has been genetically associated with the selected disease. 19,20 For example, PCSK9 gain-of-function mutations, causing familial hypercholesterolemia and coronary artery disease, led to the development of alirocumab (Regeneron) and evolocumab (Amgen). 21
This evidence suggests that in-depth analysis of WES and GWAS may improve the accuracy of drug selection by identifying new druggable targets and understanding potential drug off-targets. Unfortunately, the use of conventional preclinical in vivo models, such as rodents, does not provide the necessary throughput to analyze systematically the pathogenic effect of mutations at several genomic loci. The high experimental cost, combined with the low throughput provided by those models, is a bottleneck for discovering and validating novel genetic disease associations at the speed and cost required by the pharmaceutical industry. This remains true even with the discovery of the CRISPR/Cas9 system, which allows a straightforward generation of loss-of-function alleles in all research models. Thereafter, the low efficacy of the target identification process seems to be mainly due to limitations in the phenotypic assessment of gene loss-of-function effects rather than a difficulty in generating new models of gene dysregulation. For this reason, high-throughput cell-based screens have been used as the starting point for target discovery approaches. As stated above, even complex in vitro systems (i.e., organoid-based platforms) 17,22 lack the complexity for mimicking important in vivo phenotypic outputs, such as neurobehavior, metastasis progression, or cardiophysiology, which are crucial for understanding gene function in the context of psychiatric, cancer-related, or cardiovascular-related indications. Hence, a comprehensive understanding of complex indications might remain off-limits in cell-based systems.
The aforementioned limitations in the traditional preclinical pipeline encourage the use of alternative research models. The use of these innovative models is aimed at increasing the experimental throughput and/or biological translatability for target identification, validation, and drug discovery. The zebrafish displays a number of biologic and experimental features that represent a good compromise between current in vitro and in vivo tools and, thus, might help overcome some of the scientific and economical challenges that the pharmaceutical industry currently faces during the preclinical phase of target discovery and drug development.
The Zebrafish Model
The zebrafish model has been traditionally used for understanding the genetic basis of vertebrate embryonic development. 23 Beside basic research applications, the model has been used for understanding and modeling human diseases and performing safety and efficacy drug screenings. 24 Although its exploitation in pharmaceutical research is more recent, the translatability of zebrafish findings has been demonstrated by the different compounds, screened and discovered through the use of this model, that have entered clinical trials in recent years. 25 Examples of molecules currently in clinical Phase I are ORC-13661 (Oricula Therapeutics), a molecule protecting against drug-induced ototoxicity, 26 and trifluoperazine, a molecule repurposed to treat Diamond–Blackfan anemia (DBA). 27 In addition, Epygenix Therapeutics has three different drugs in Phase II for treating Dravet syndrome (https://www.epygenix.com/pipelines/). These successful outcomes are based on the unique biological and experimental features of the zebrafish model. Specifically, zebrafish larvae have small size, embryo development is quick and external, a pair of mating adult fishes produces large progeny (more than 200 eggs), most organs develop completely during the first 5 days post fertilization (dpf), and larvae are transparent. These inherent characteristics have been coupled with available fluorescent transgenic lines, allowing noninvasive live imaging of biological processes from a whole-tissue to subcellular resolution. On top of that, zebrafish larvae up to 5 dpf are considered in vitro systems according to animal welfare regulation of the European Union.
These features allow testing dozens of different drugs, or genetic conditions, in a high-throughput in vitro-like screening setting (i.e., placing single larvae in 96 multiwell plates), 28 which results in the extraction of high translational in vivo outputs. Moreover, using zebrafish in drug development favors the implementation of the 3Rs. This systematic framework aims at reducing, and eventually replacing animal experimentation, but also at refining methods, protocols, and treatments to minimize animal pain and distress while enhancing its well-being. 29 Indeed, using zebrafish larvae suits the 3Rs perfectly, since (as mentioned above) they are not considered in vitro systems up to 5 dpf according to EU animal welfare regulations. 30 As such, the translational value of zebrafish larvae allows the replacement of “higher” vertebrates to evaluate novel compounds. Furthermore, studying the effect of those compounds in a complex organism allows the early and reliable selection of drug candidates, reducing the number of mammals required for subsequent research phases. Finally, noninvasive live imaging, granted by the embryo’s transparency, enables the refinement of the experimental procedures, minimizing animal suffering and distress.
All these features have promoted extensive reviews on the use of zebrafish in the context of safety and efficacy drug screening. 25,31,32 Thus, we will focus on the less traveled path of how zebrafish can be applied to target discovery and validation studies ( Fig. 2 ).
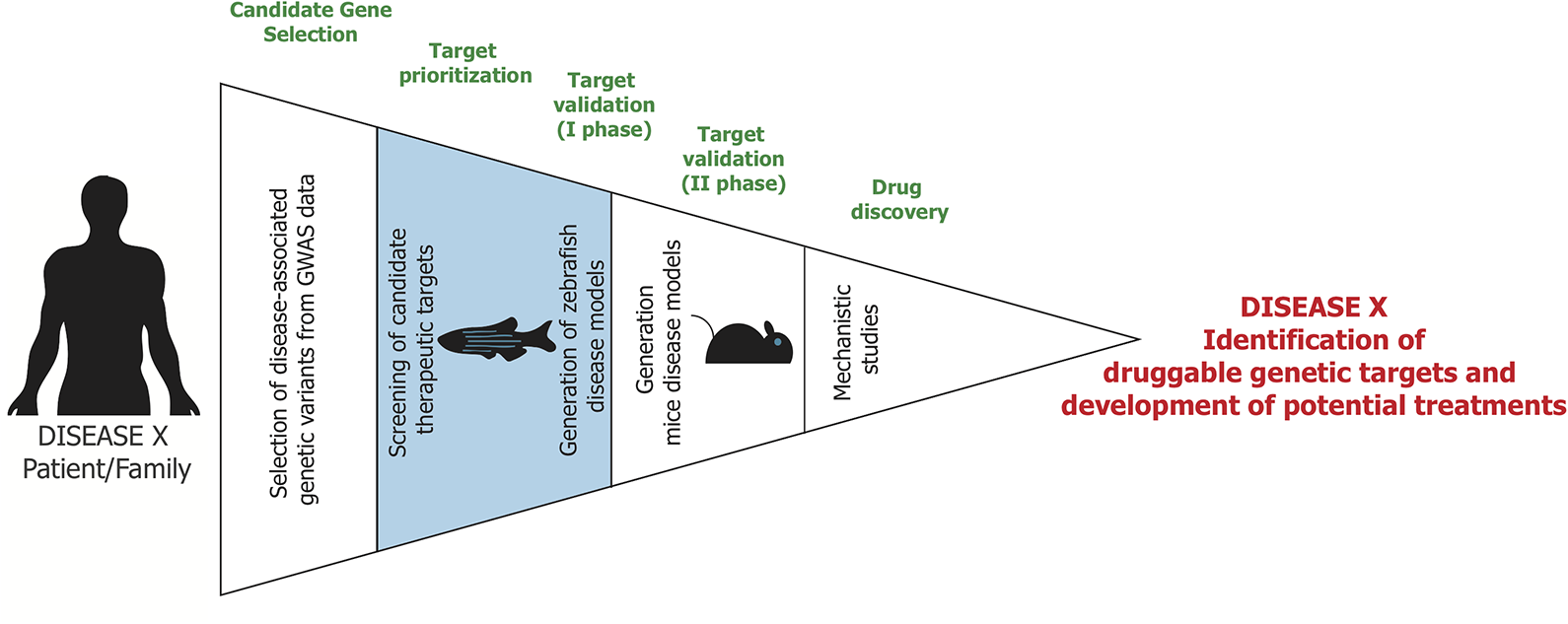
Pipeline for target identification and validation. From left to right, the entire pipeline is divided into different funnel phases. Candidate gene selection: Genome-wide association studies (GWAS) and whole-exome sequencing (WES) from patients reveal genetic variants potentially associated with a certain disease. The genes of interest are selected through in silico methods and comparison with additional datasets [e.g., Online Mendelian Inheritance in Man (OMIM)]. Target prioritization: A first screening enables one to narrow down the number of therapeutic candidates through rapid loss-of-function studies performed with highly efficient CRISPR/Cas9-based methods in zebrafish. Target validation (Phase I): Genetic models carrying SNPs, orthologous to those identified in patients, can be generated in zebrafish to further validate gene function. Target validation (Phase II): Following target validation in zebrafish, selected genetic models of disease can be further validated in more genetically related animal models, such as mice. Drug discovery: The clear mechanism of action of the therapeutic target obtained in the first phases will lead to development of a specific drug suitable for the disease.
Combining Zebrafish and CRISPR/Cas9 for Target Discovery and Validation
Target identification and validation require interrogating the role of disease-associated genes in relevant in vitro and/or in vivo research models mimicking biological processes equivalent to those that cause and promote the progression of the studied pathology. To this end, it is necessary to couple gene manipulation with an experimental setup in which the phenotypic impact of gene disruption can be readily analyzed. The zebrafish experimental advantages outlined above can provide a framework for understanding gene function in most indications. To help with that, target discovery has been accelerated by the appearance of gene-editing technologies, which allow easy manipulation of the genome and thus provide more accessible tools for interrogating gene function. The most powerful technology for genome editing to date, the CRISPR/Cas9 system, has been widely applied to zebrafish. 33 The experimental basics and challenges associated with the generation of knockout, point mutations, or large exogenous insertion alleles have been reviewed before. 34 Therefore, we will focus on applications directly related to target discovery aims.
For the analysis of gene function, the use of isogenic mutant lines remains the standard method in the zebrafish community. 35 In addition, the continuous optimization of CRISPR/Cas9-based technologies has allowed the achievement of up to a 100% mutagenesis rate in embryos injected with Cas9–guide RNA complexes. 36 This feature enables the induction of biallelic gene loss-of-function larvae (CRISPANTs), which can be analyzed phenotypically in the first generation (F0; ∼1 week) without waiting for the multiple generations required for obtaining genetically equivalent homozygous mutant larvae (≥F2; ∼7 months). Importantly, the high rate of egg production offers the possibility to test several therapeutic targets in a reduced time. Then, coupling this rapid gene inactivation with high-throughput phenotypic platforms available to the zebrafish community results in a more effective (in terms of better translatability toward human biology than in vitro systems) and less time-consuming and costly (when compared to mammal research) target identification process. If the phenotype expected from gene disruption can be readily analyzed, the use of CRISPANTs might help narrow down the number of disease-associated genes identified by WES and GWAS for most indications ( Fig. 2 ). The main limitation of isogenic mutants is the larger cost and experimental time to reach phenotypic evaluation. However, once F2 generation is reached, isogenic homozygous mutants display a comparable phenotypic penetrance among individuals. In that sense, CRISPANTs’ main drawback derives from the fact that phenotypic penetrance correlates linearly with double-strand break rate variability. In our experience, this phenomenon requires correlating genotype and phenotype individually and increasing the sample size to increase statistical significance. Luckily, obtaining large sample sizes is perfectly possible when performing research with zebrafish larvae.
Besides validating potential disease-associated genes via loss-of-function approaches (isogenic knockout or CRISPANT), the evolutionary conservation in amino-acidic sequence allows studying the impact of rare genetic variants by introducing human pathogenic mutations into zebrafish gene orthologs. During recent years, different CRISPR/Cas9-based technologies have been developed to this aim. 34 To date, the preferred method for creating point mutations consists of inducing a double-strand break at the genomic target locus and providing an exogenous DNA donor template for homology-directed repair (HDR) mechanisms. Through this method, recapitulation of human pathogenesis has been possible in zebrafish models for different diseases. For example, disease-causing human SNPs have been introduced into the zebrafish tardbp and fus genes to generate models of amyotrophic lateral sclerosis through the use of single-stranded oligodeoxynucleotide (ssODN) sequences. 37 An important drawback in HDR methods is the low efficacy in generating the desired gene modification. To overcome this issue, double-strand-break-independent methodologies have been developed. A precise single “base editing” (BE) method, implemented in human cells in the first place, 38 has been rapidly set up in the zebrafish model. 39 This strategy is based on the fusion of a cytidine deaminase to a Cas9 nickase (nCas9). This protein complex is able to induce conversion of a targeted cytidine to adenine, guanine, or thymine, expanding the toolbox required for the introduction of human polymorphisms. 40 BE’s principal limitation is the impossibility to convert any desired base into a different base along the entire zebrafish genome. To help in this matter, prime editing, a breakthrough CRISPR/Cas9-based approach, has been implemented very recently by Anzalone and colleagues. 41 This method is based on a novel genome-editing tool composed by a chimeric engineered Cas9 nickase–reverse transcriptase fusion protein and a prime-editing guide RNA (pegRNA). The pegRNA has the dual function of pairing to the chosen target site in the genome and providing an RNA template with the desired modifications at the locus of interest. Therefore, after binding to the target site via pegRNAs, the Cas9 induces a single-strand break (nick) with only one active catalytic domain, and edits the target sequence with the reverse transcriptase domain, by using the template contained in the pegRNA. Such elegant technology brings several advantages compared to previously published methods: It (1) does not require a double-strand break of the DNA or an exogenous DNA template, (2) induces lower off-target editing, and (3) allows all 12 possible base-to-base conversions plus insertions and deletions of different lengths. Although not yet implemented for the zebrafish, this technology is already revolutionizing the genome-editing field and pushing it toward an increasingly accurate recapitulation of human disease in biomedical research models. Table 1 summarizes the CRISPR/Cas9 approaches available in zebrafish research for target identification and validation pipelines.
CRISPR/Cas9 approaches used in zebrafish for target identification and validation.
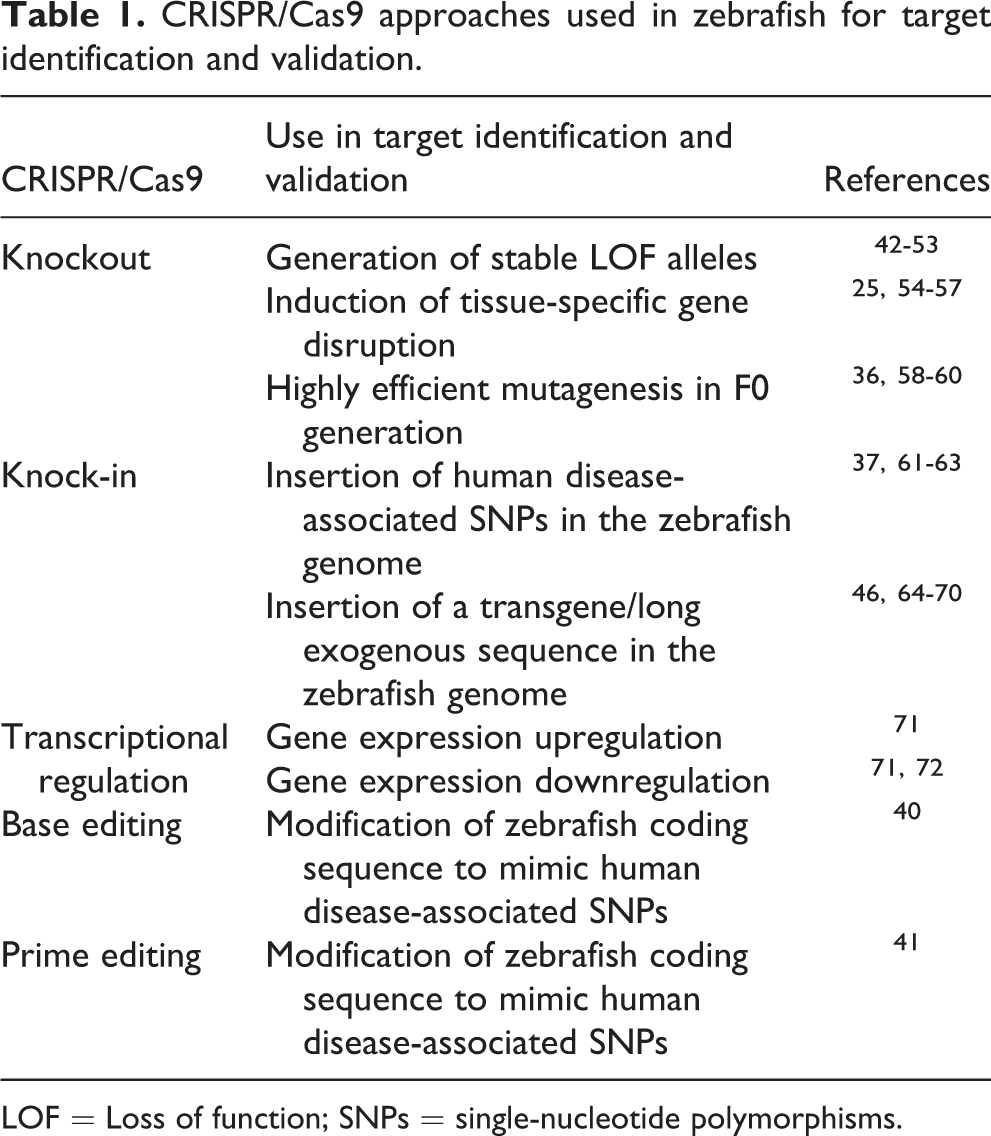
LOF = Loss of function; SNPs = single-nucleotide polymorphisms.
Considerations When Using Zebrafish in Target Discovery and Validation
Despite the several advantages provided by the use of the zebrafish in disease modeling, there are some potential drawbacks to be examined to design successful strategies for the analysis of disease-associated genes. Importantly, zebrafish are members of the Teleostei infraclass, which likely arose around 340 million years ago from a common ancestor 73 that, compared to other vertebrate species, underwent a further round of whole-genome duplication (WGD). 74 Thereafter, a consistent number of human genes have two orthologs in zebrafish. The most accepted models explaining the evolutionary significance of genomic loci duplication suggest that, from a unique ancestral gene, the two novel paralogs have evolved new functions (neofunctionalization) or partitioned the original function (subfunctionalization). 75 While comparative analysis of the models based on genomic, phylogenetic, and spatiotemporal expression and protein sequence data suggests that neofunctionalization might have had a greater impact than subfunctionalization on gene evolution, 76 other studies support the opposite scenario. 77 Inquiring about the evolution of functional divergence of potentially disease-associated genes is crucial from a target validation perspective. For example, as a result of subfunction partitioning, two domains of an ancestral protein would be expressed in different tissues in zebrafish, whereas the mammalian ortholog would have a more widespread expression pattern. If, on one hand, this makes the recapitulation in zebrafish of the effect of a global loss of function challenging because both paralogs should be inactivated, on the other hand it offers an advantage in terms of drug discovery. Indeed, a therapeutic compound has target proteins that, in different tissues, can interact with different tissue-specific factors. Therefore, a compound designed to act on the function of the target protein in a specific tissue is highly likely to affect the function of the same protein in all other expression domains of the protein, presumably leading to side effects. Studying the effect of loss of function of zebrafish paralogs can help to develop drugs that specifically target tissue-specific domains and biological interactions, allowing increases in the efficacy and safety of the treatment.
In addition, there might be an overlap in the function of two zebrafish paralogs. Here, the generation of a loss-of-function allele for only one paralog might lead to two possible scenarios: On one hand, the knockout of a single paralog might induce the expected disease phenotype. This result would suggest that disease etiology in humans is caused by gene haploinsufficiency. In this case, the generated zebrafish knockout model can help identify specific, disease-causing molecular mechanisms. On the other hand, the mutation on one paralog might lead to a milder phenotype than expected because of genetic redundancy or compensation by the other paralog. In this case, a double knockout mutating both paralogs would recapitulate the phenotype expected from the global loss of function.
Another important aspect is the fact that 82% of human genes potentially associated with disease display a zebrafish ortholog. 78 This feature has allowed, in recent decades, the generation of data on vertebrate gene function and involvement in disease. For genes without a clear genetic correspondence, it is likely that a different zebrafish protein exerts the same biological function. This is the case for the BRCA1 tumor suppressor gene. Even if BRCA1 has no zebrafish ortholog, the BRCA1-associated BARD1 gene has a zebrafish ortholog and encodes for a protein that displays a similar function. 78 Importantly, among the 20 genes that have been associated with human Fanconi anemia, a rare syndrome characterized by a defective response to DNA damage, all of them, except BRCA1, display a zebrafish ortholog. 79,80 This case study exemplifies how zebrafish could still be modeled to study pathogenesis either by targeting genes encoding for functionally analogous proteins or by targeting different proteins associated with the disease of interest. It is also possible that the human gene of interest is present in the zebrafish genome, but sequence conservation might be too low for a targeted mutagenesis approach. One possible approach to tackle this issue would take advantage of CRISPR/Cas9-mediated knock-in methods. The simultaneous disruption of the zebrafish locus and insertion of a human-mutated complementary DNA (cDNA) would allow the study of disease-associated mutations, even for nonconserved genomic loci.
Finally, in addition to using zebrafish in studying pathophysiology generated by loss or gain of function of genes of interest, the applications can be extended beyond the protein-coding genome, representing approximately 1% of the transcribed human genome. 81 Indeed, in recent years, functional noncoding RNAs (ncRNAs) have emerged as key regulators of biological processes. More than 200 diseases have been associated with the most abundant class of ncRNA, the long noncoding RNAs (LncRNAs). 82 Nevertheless, lncRNA inactivation is more challenging than coding gene disruption, since it might result in unintended deletion of regulatory DNA motifs or usage of cryptic promoters or alternative transcription starting sites. Importantly, in a very recent report, a strategy based on insertion of a transcription termination sequence into lncRNA loci by CRISPR/Cas9-mediated knock-in has been implemented in zebrafish. 83 This method allows a straightforward and highly effective method for inactivation of lncRNA and the study of potentially disease-associated noncoding RNAs.
Zebrafish-Based Phenotyping Platforms
Besides a straightforward generation of mutated alleles allowed by the CRISPR/Cas9 system, the main advantage of using the zebrafish model for target validation and drug discovery comes from the experimental accessibility to phenotypes mimicking human pathological phenotypes. To prove this point, we will discuss examples of zebrafish phenotyping platforms in the context of three disease fields that represent major burdens for society: neurological disorders, cancer, and cardiovascular diseases. We will also discuss how all these features might be applied to implementing precision medicine approaches.
Neurological Disorders
Neurological disorders are diseases of the central and peripheral nervous system that are increasingly recognized as one of the most prevalent disorders with a high burden to the patients, their families, and society. For example, currently 50 million people have epilepsy, and there are 47.5 million people with dementia, a disease that grows at a rate of 7.7 million new cases every year. 84 During the past two decades, the genomic revolution has led to the identification of thousands of genes known to be associated with neurological disorders. This is exemplified by epilepsy, in which close to 1000 genes have been linked. 85 Zebrafish could be a promising model to mimic such disorders for target discovery and validation, because in vitro models lack the complexity needed to model complex neurologic processes, and in vivo mammalian models require labor-intensive and time-consuming behavioral and morphological assays. 86 By 5 dpf, zebrafish larvae respond to all stimuli—vision, olfaction, taste, tactile, balance, and hearing—as well as complex CNS-driven behaviors like memory and learning. 87 Moreover, zebrafish present homologous neural pathways with humans and conserved neurotransmitter systems such as dopamine, serotonin, histamine, glutamate, γ-aminobutyric acid (GABA), acetylcholine, and noradrenalin, although differences in expression patterns are observed. 88,89 When compared to mammalian models, zebrafish larvae show advantages due to their small size, enabling high-throughput neurotoxicity and neurobehavioral screenings in 96-well microplates ( Fig. 3 ). Furthermore, the combination of zebrafish transparency with the vast number of available fluorescent transgenic lines allows the functional imaging of the entire nervous system in living larvae. 90
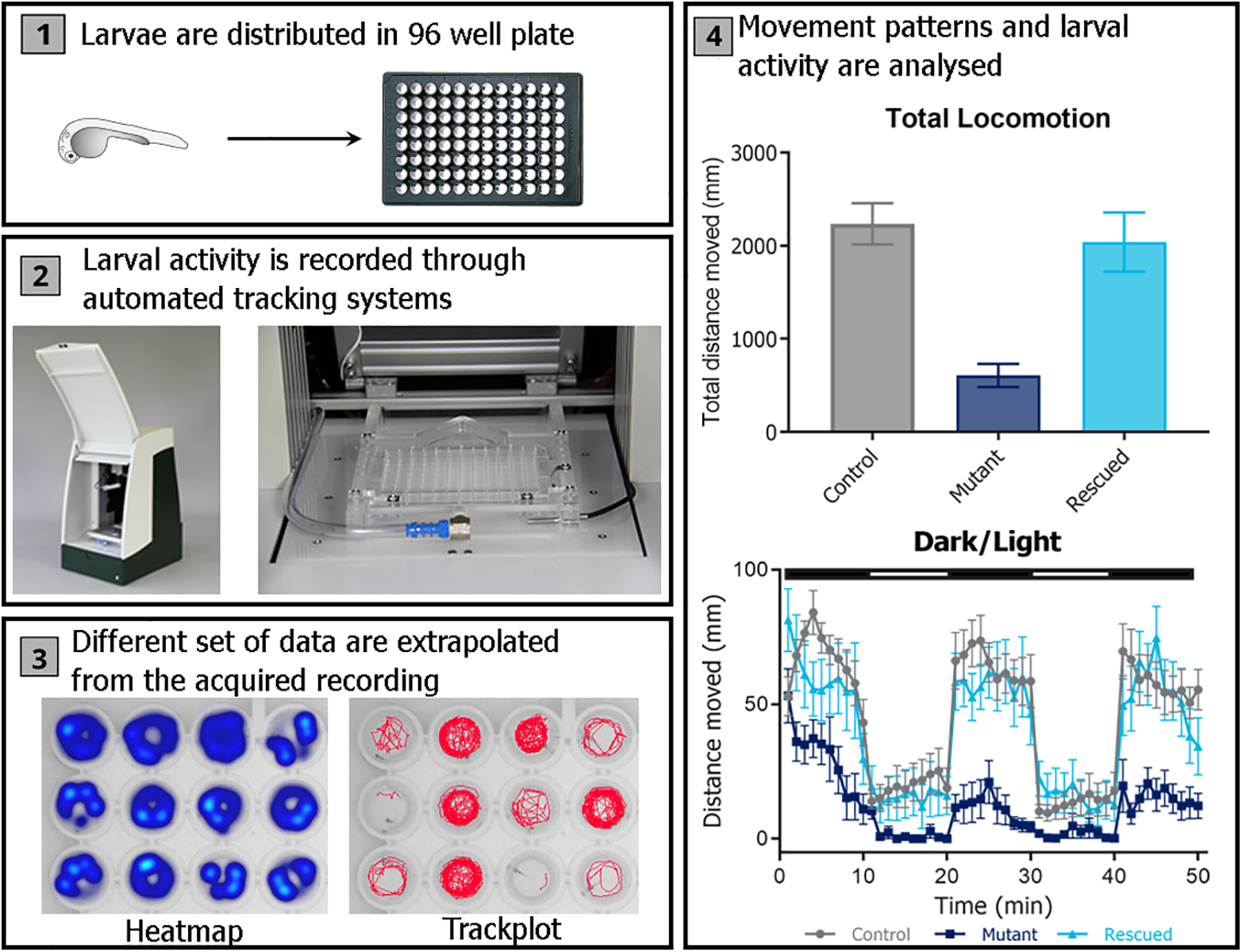
Zebrafish larvae behavioral-screening platform. (
Based on these features, forward and reverse genetic strategies have been used to discover and validate genes involved in CNS processes and later associated to neurological disorders. The majority of these studies have used morpholinos (MOs), 91,92 which are modified antisense oligonucleotides that cause a transient “knockdown” of target gene expression by blocking messenger RNA (mRNA) splicing or translation. 93 The popularity of MOs has decreased, however, since the development of gene-editing tools and especially CRISPR/Cas9 technology. Although a limited number of studies to date have used CRISPR/Cas9 to generate target discovery studies in the neurological field, it is likely that a growing number of studies will harness it in the near future, similar to what is happening in the cardiovascular field (explained in detail in the following section). As an example, a study has used this technology to generate C3orf70 knockout zebrafish mutants, which showed impaired circadian rhythm and altered light–dark neurobehaviors. The C3orf70 gene was found to be a common target of Neurog1/2 and Asc11 during neurogenesis. Furthermore, expression patterns of C3orf70 have been shown to be specific to the midbrain and hindbrain of zebrafish larvae. Thus, defects in C3orf70 may be associated with neurodevelopmental and neuropsychiatric disorders related to these brain areas, and therefore might be used as therapeutic targets. 42 A more recent publication shows the combined effort led by Harvard University and Novartis to use zebrafish for tackling psychiatric disorders. Harvard’s Fishman Lab has generated mutants for 90 genes associated, through GWAS studies, with autism, schizophrenia, and other disorders in humans. By looking at their collective behavior, they have been able to categorize them in different categories according to velocity, direction, or group spacing. This work represents a huge step toward the understanding of social behavior and how its interpretation can serve for understanding the genetic causes of psychiatric disorders. 94
Regardless of which technology has allowed the generation of mutants, the tools currently available to the zebrafish community enable the analysis of neural-specific phenotypes such as visual impairment, ototoxicity, alterations in locomotor activity, developmental neurotoxicity, olfactory toxicity, anxiety, seizures, social behavior, and impaired memory and learning. 87,95 -99 More subtle and complex stimuli, such as those produced by psychotropic drugs, can also be assessed. 100 -102 An interesting example of the power of zebrafish in modeling complex behavior is the study done by Hoffman et al. using zebrafish mutants of the autism risk gene contactin-associated protein-like 2 (CNTNAP2). 103 They performed a high-throughput quantitative behavioral and pharmacological screening of 14 psychoactive compounds, and identified that biochanin A, an estrogenic compound, reverses the mutant behavioral phenotype. These results open the door to new targets and pharmacological pathways that can be tackled to counteract autism, and reinforce the use of zebrafish in modeling such complex neurological behaviors.
Finally, one of the most successful examples of the potential of zebrafish in this field is represented by research led by the Baraban Lab at the University of California, San Francisco, which has built a drug discovery platform based on the exploitation of the scn1a-deficient zebrafish model of the severe childhood epilepsy Dravet syndrome, which presents spontaneous seizures. Using this platform, they have conducted a phenotype-based screen of 320 compounds and found that clemizole inhibited convulsive behaviors and electrographic seizures. 104 As a result, Epygenix Therapeutics, a spinoff founded by Dr. Baraban, is currently recruiting patients for testing clemizole (under the name of EPX-100) in clinical Phase II. 105
Cancer Research
Cancer is responsible for an estimated 9.6 million deaths in 2018, being the second leading cause of death worldwide after cardiovascular disease. 106 Despite the advances in antitumor therapies, cancer incidence and deaths are continuously growing year after year. The search for biologically relevant, faster, and more affordable methods to discover more precise, safe, and efficacious therapies is a medical priority. Zebrafish have been found to spontaneously develop tumors that are similar to human malignancies in genetics, morphology, histology, and signaling pathway. 107 -110 Moreover, the vast majority of human cancers can be reproduced in zebrafish through chemical treatments, gene editing, and tumor cell transplantation. 111 -113 More importantly, the zebrafish model can be used to identify novel driver genes in cancer, which is crucial for the development of targeted therapies. A successful approach toward this direction is the recent work by the group of Leonard Zon. 24 In their study, an extensive screening for mutations in cancer-related genes in 43 primary or metastatic human mucosal melanomas was performed. In almost 40% of the tumors analyzed, a negative regulator of mitogen-activated protein kinase (MAPK), SPRED1, was found inactivated. Furthermore, SPRED1 loss of function was associated with concomitant mutations in the KIT-protooncogene, indicating functional cooperation in oncogenesis. To understand the effects of SPRED1 loss in multiple genomic backgrounds harboring patient mutations, the authors developed a rapid and robust method for simultaneous expression of oncogenes and inactivation of tumor suppressor genes via a tissue-specific CRISPR/Cas9 approach. Through this strategy, it was found that SPRED1 loss acts as a driver of mucosal melanoma in the contest of KIT mutations. The method, called MAZERATI (Modeling Approach in Zebrafish for Rapid Tumor Initiation), represents an extremely useful tool that can be used for tumor modeling and to study genetic interactions in cancer.
Although zebrafish tumor genetic models have been shown to be useful for the study of tumor initiation and development, as well as the interactions between malignant and normal cells and tissues, they have also some associated limitations such as the time required for tumor formation and variability in cancer incidence and growth. Some of these drawbacks can be overcome through xenotransplantation studies of human tumor cells in zebrafish. Indeed, larval xenografts are the most common strategy to assess cancer progression in this model. They offer great potential for evaluating proliferation, migration, and neovascularization and to test drugs affecting these cancer hallmarks ( Fig. 4 ). 114 In addition, the possibility to generate zebrafish patient-derived xenografts (PDX) rapidly enough to obtain results within the time window between a patient’s cancer diagnosis and the initiation of treatment is of striking importance. Along this line, Fior et al. have shown comparable responses to FOLFOX and FOLFIRI chemotherapeutic treatment and biological therapies (Cetuximab) between patients with colorectal cancer and their corresponding zebrafish PDXs. 115
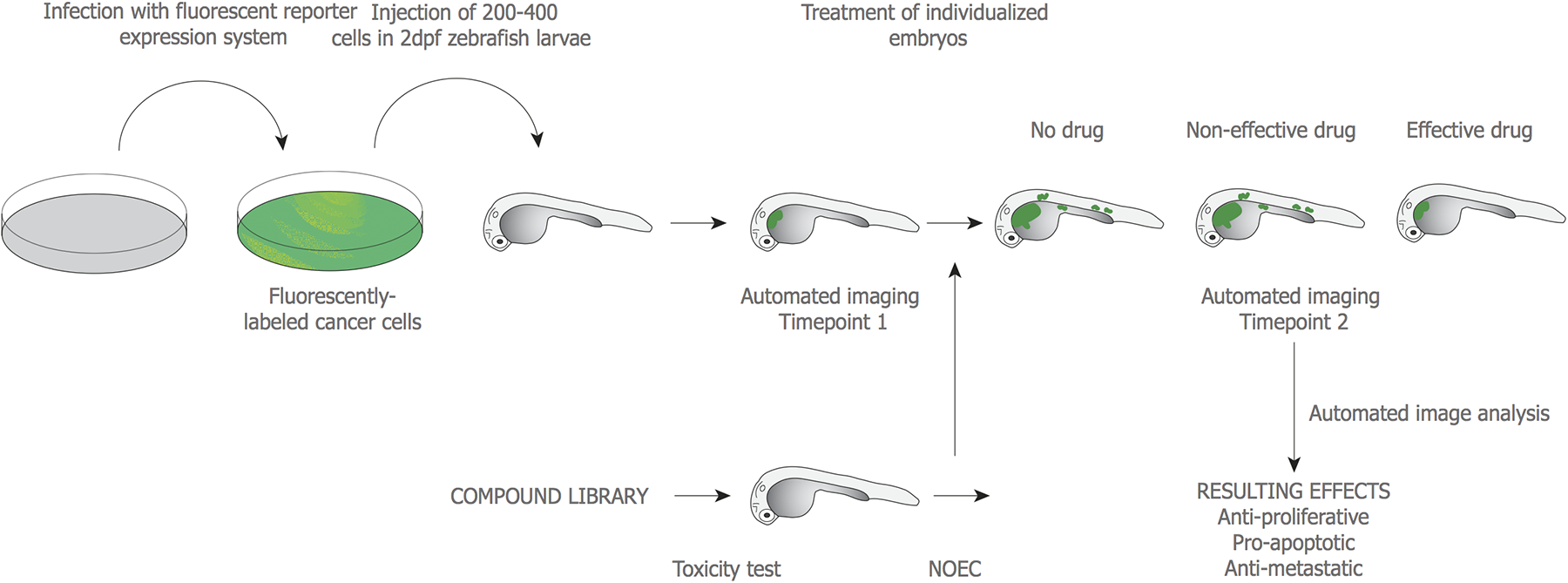
Pipeline for a drug-screening platform based on zebrafish xenotransplantation of human tumor cells. From left to right: Cells, immortalized or derived from patient biopsies, are fluorescently labeled, injected into 48 h post fertilization (hpf) zebrafish larvae, and incubated at a temperature (32–35 °C) spanning between zebrafish optimal (28.5 °C) and human cells optimal (37 °C). Subsequently, injected larvae are imaged at time point 1 (tp1) to measure initial tumor volume and dispersion. After, injected larvae are treated with a no-observed-effect concentration (NOEC) of candidate drugs, which was previously calculated. A second imaging is performed at tp2 to measure the potential variation throughout time in tumor volume and cell dispersion. Drugs can be evaluated by their impact on tumor growth and metastatic potential through the calculation and comparison of tumor mass and dispersion of cells at the two time points, in treated and untreated animals. (Adapted from Ref. 114 .)
Cardiovascular Disease
The World Health Organization (WHO) estimates around 18 million deaths per year due to cardiovascular diseases (CVDs), accounting for the 31% of global deaths worldwide. 116 Large-scale population studies have identified thousands of rare and common genetic variants and revealed more than 70 causative loci potentially involved in the pathophysiology of coronary artery disease (CAD), 117 the leading form of CVD. Although zebrafish have been extensively used for research in heart and blood vessel development, 118 in less than a decade the escalated use of gene editing has converted zebrafish into an ideal model for human CVD. This is due to the fact that the zebrafish heart displays multiple features that are similar to human cardiac tissue. The zebrafish heart rate [150–200 beats per minute (bpm) at 72 h post fertilization (hpf)] is closer to the human heart rate (60–100 bpm) than to the rodent heart rate (500 bpm), and cardiac electrophysiological properties are similar. 119 -121 In addition, the availability of tissue-specific transgenic lines labeling heart, 122 blood cells, 123 and vessels 124 with fluorescent reporters allows in-depth analysis of cardiovascular phenotypes in vivo. A growing number of zebrafish lines carrying mutated alleles for GWAS-derived genetic targets has been generated to establish causality between gene inactivation and different types of cardiomyopathies such as dilated cardiomyopathy 125 and congenital heart defects. 126 More importantly, a recent report has shown that efficient gene disruption with almost complete penetrance can be induced via the CRISPR/Cas9 system to assess cardiovascular phenotypes in F0 zebrafish larvae. 127 This enables the targeting of multiple candidate genes and the analysis of resulting null phenotypes in a short window of time, overcoming one of the major limitations of mutant-lines generation. Lately, we have developed a fully automated high-throughput microfluidic system coupled with integrated software for analysis of cardiac and blood flow phenotypes: ZeCardio ( Fig. 5 ). The system has been developed to study drug-induced cardiotoxicity in zebrafish larvae, 128 but has now been optimized for understanding gene function and performing efficacy drug screenings. We hope that, once new therapeutic targets are identified, the same high-throughput system can be used to screen for compounds rescuing pathological phenotypes identified in genetic and pharmacological zebrafish disease models.
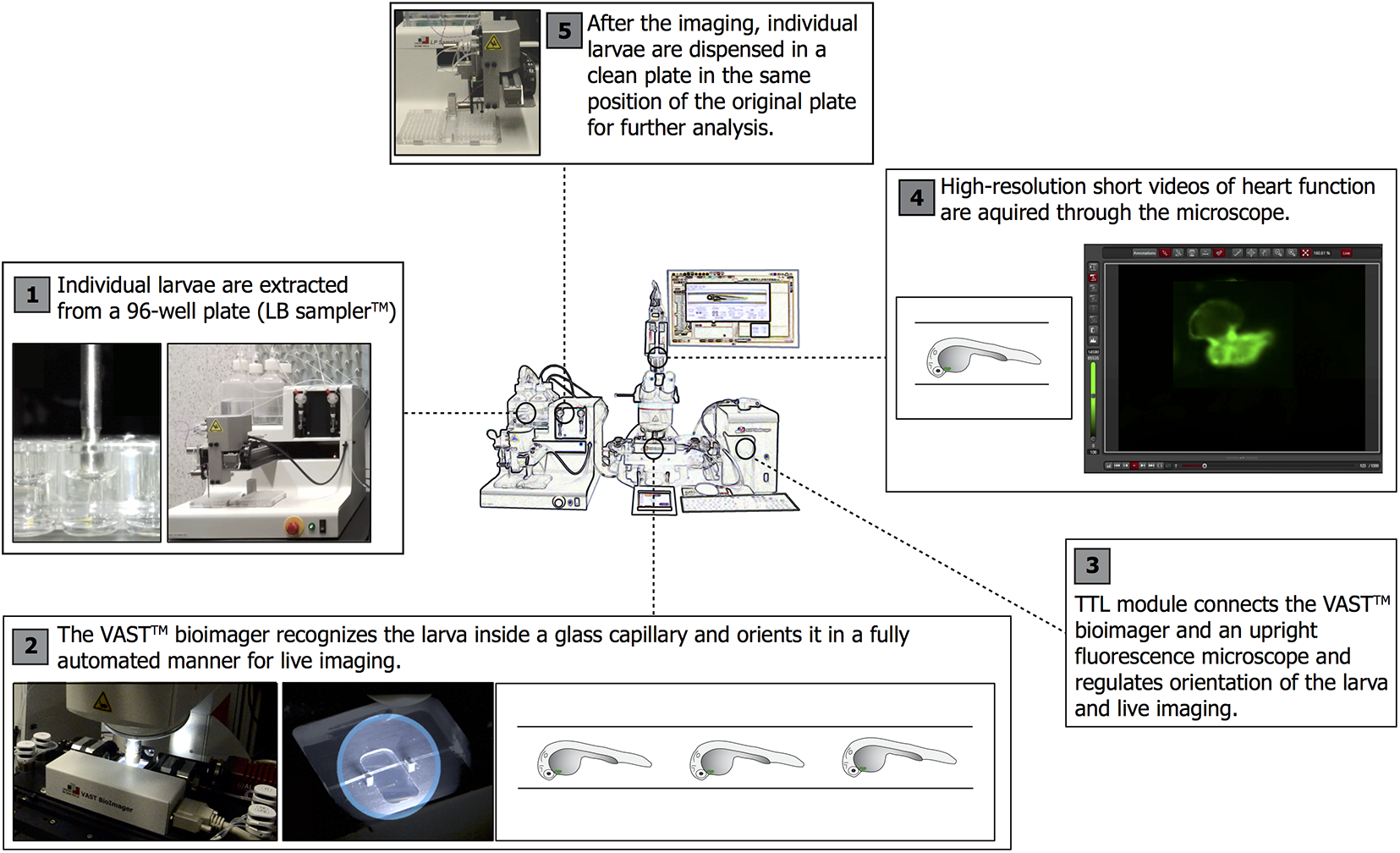
Platform for high-throughput cardiovascular analysis in zebrafish larvae. From left to right: (
In addition, zebrafish, unlike mammals, have an inherent capacity to regenerate their cardiac tissue throughout life. 129 Heart regeneration is granted in zebrafish by genetic programs promoting cardiomyocyte (CM) dedifferentiation and proliferation after injury. 130 The same genetic regenerative programs are present albeit dormant in the adult mammalian heart, where postmitotic CMs fail to reenter cell division to replace damaged cells after infarction. 131 This makes zebrafish a powerful model to unravel key molecular pathways that might induce heart regeneration in humans after myocardial infarction. In fact, the identification of master regulators of functional and morphological recovery of the cardiac tissue represents a promising and novel avenue in the fields of CVD and regenerative medicine.
Complementary to the in vivo setup for high-throughput genetic and drug screenings, in vitro systems, based on three-dimensional (3D) cell aggregates forming spontaneously from enzymatically digested zebrafish larvae, have been developed. 132,133 These zebrafish heart aggregates (ZFHAs) further expand the toolbox for cardiovascular research, disease modeling, and drug discovery provided by the intact larvae.
Additional Uses of Zebrafish-Based Target Discovery: Personalized Medicine
To increase R&D productivity, the pharma industry is pursuing additional approaches that could also benefit from the use of zebrafish for discovering new drug targets. One clear example could be the personalized medicine approach. Personalized or precision medicine aims at identifying and understanding the causes of a disease in individual patient segments, to define more specific and efficacious treatments. Therapeutic areas such as neurologic disorders and cancer, which include a wide range of different subcategorized diseases, are more prone to enter the precision medicine framework. In those specific therapeutic areas, drug companies have found a golden opportunity to overcome the productivity crisis. 134 On one hand, the identification of patient cohorts, defined by efficacy and toxicity biomarkers and a better understanding of the pathology, might lead to the discovery of new druggable targets specific for each subcohort or the reposition of commercially available drugs. On the other hand, personalized medicine treatments are specific to a small proportion of patients. This is important given that regulatory agencies are collaborating with research institutions, clinicians, and biotech and pharma companies to design streamlined clinical trials for rare diseases, reducing their sample size and duration. This results in a more efficient drug development process and might have a beneficial economic impact for all stakeholders. 135,136 Moreover, the validity of precision medicine has been demonstrated by the improved survival and prognosis of patient groups under personalized treatment, when compared with those not treated through personalized approaches. 137 Despite these successes and potential benefits, it will be necessary to have a better understanding of the differences among patient subcohorts, their specific biomarkers, and potential drug targets to fully exploit this promising venue. We believe the use of the zebrafish model could have a great impact in this context.
In that sense, patient stratification is one of the critical points of personalized medicine. The contribution to the disease of a single gene or genetic mutation widely varies depending on the monogenic or polygenic etiology of the disease and the individual genetic background. CRISPR/Cas9 genome editing could potentially grant the rapid generation of numerous zebrafish mutant lines mimicking single or combined mutations that would allow to identify their contribution to pathogenesis. This would help researchers understand the sources of the disease phenotypic variation and understand better patient groups and subcohorts. Next, based on the data obtained, zebrafish cohort-specific models could be generated offering a tool for high-throughput drug screening. In addition to preclinical screenings, this could also have a great impact for clinicians, since those tests can be performed in a time frame compatible with clinical decisions. In oncology, pioneering studies on zebrafish avatars, patient-derived tumor xenotransplants in larvae, show the possibility to investigate inter- and intratumor heterogeneity and use that model as a predictive tool for co-clinical assays. 138,139 It would be interesting to study if combining this approach with CRISPR/Cas9 genome editing could further enhance the predictive ability of the model. As an example, the disruption of genes in the zebrafish host mimicking patient genetic background would provide insights into its potential involvement in tumor progression and resistance to therapy, improving the therapeutic treatment selection. The data and possible applications of the CRISPR/Cas9 strategy presented here show that this approach could improve and accelerate the discovery of cohort-specific targets and the development of specific treatments, and have a great impact on the personalized medicine field.
Conclusions
In this perspective article, we have emphasized to great lengths how the discovery of new druggable targets offers major therapeutic and business opportunities for the pharma industry. Today, generating mutated alleles in every in vitro or in vivo model is an accessible task thanks to the CRISPR/Cas9 technology. Thus, a competitive edge in target discovery and validation provided by any research model might result from the balance between biologic translatability and phenotyping accessibility. In vitro systems provide easy access to cellular and organ behaviors, but they might lack the complexity—behavior, interplay among different organs, metabolic and immune systems, and so on—to mimic entirely the biology of a given human disease. On the contrary, mammals show a highly comparable physiology to humans, but analyzing gene function (phenotyping) in these models is a long and costly process, hampering the study of multiple variants in a timely fashion. Indeed, target discovery and validation might be hampered by the high cost of mammalian research and lack of biological translatability of in vitro models. Due to these limitations, the zebrafish model might represent a good solution for performing target discovery studies. On one hand, it displays more complex in vivo phenotypes than in vitro systems, and, on the other hand, it provides easier and more economic access to phenotyping than mammalian models. Indeed, the combination of a conserved genome and physiology with humans, the fast gene inactivation provided by CRISPR/Cas9, and medium- and high-throughput phenotyping offers a good chance for increasing the understanding of human disease and accelerating the discovery of novel drugs and targets for most indications and also in the context of personalized medicine.
Footnotes
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are currently employed by ZeClinics S.L.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Carles Cornet was funded with an Industrial Doctorate Fellowship (MINECO; DI-14-06969), and Vincenzo Di Donato was funded by a Marie Skłodowska-Curie Individual Fellowship (UE; IF-845713).